
Abstract
Aims:
Downregulation of nuclear factor erythroid 2-related factor 2 (Nrf2) contributes to doxorubicin (DOX)-induced myocardial oxidative stress, and inhibition of mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) increased Nrf2 protein level in rat heart suffering ischemia/reperfusion, indicating a connection between MALT1 and Nrf2. This study aims to explore the role of MALT1 in DOX-induced myocardial oxidative stress and the underlying mechanisms.
Results:
The mice received a single injection of DOX (15 mg/kg, i.p.) to induce myocardial oxidative stress, evidenced by increases in the levels of reactive oxidative species as well as decreases in the activities of antioxidative enzymes, concomitant with a downregulation of Nrf2; these phenomena were reversed by MALT1 inhibitor. Similar phenomena were observed in DOX-induced oxidative stress in cardiomyocytes. Mechanistically, knockdown or inhibition of MALT1 notably attenuated the interaction between Nrf2 and MALT1 and decreased the k48-linked ubiquitination of Nrf2. Furthermore, inhibition or knockdown of calcium/calmodulin-dependent protein kinase II (CaMKII-δ) reduced the phosphorylation of caspase recruitment domain-containing protein 11 (CARD11), subsequently disrupted the assembly of CARD11, B cell lymphoma 10 (BCL10), and MALT1 (CBM) complex, and reduced the MALT1-dependent k48-linked ubiquitination of Nrf2 in DOX-treated mice or cardiomyocytes.
Innovation and Conclusion:
The E3 ubiquitin ligase function of MALT1 accounts for the downregulation of Nrf2 and aggravation of myocardial oxidative stress in DOX-treated mice, and CaMKII-δ-dependent phosphorylation of CARD11 triggered the assembly of CBM complex and the subsequent activation of MALT1. Antioxid. Redox Signal. 42, 115–132.
Introduction
Doxorubicin (DOX), an anthracycline antibiotic, is widely used to treat breast cancer, leukemia, and many other types of malignancies. However, the long-term application of DOX is largely limited because of its cumulative dose-dependent cardiotoxicity, including irreversible degenerative cardiomyopathy and congestive heart failure (Singal and Iliskovic, 1998; Swain et al., 2003; Young et al., 1981). DOX-induced cardiotoxicity can be acute, occurring during and within 2–3 days of its administration (Chatterjee et al., 2010). Multiple factors contribute to the pathogenesis of DOX-induced cardiotoxicity, including oxidative stress, mitochondrial damage, apoptosis, and necrosis. Among these factors, overproduction of reactive oxygen species (ROS) because of disturbance in the balance of redox-cycle between DOX and semiquinone DOX plays a fundamental and indispensable role in DOX-induced cardiotoxicity, resulting in oxidative mitochondrial damage, cardiomyocyte apoptosis, and necrosis (Carvalho et al., 2014; Wallace et al., 2020; Yu et al., 2018). Therefore, elucidating the mechanisms underlying DOX-induced oxidative stress could identify novel targets for alleviating DOX-induced cardiotoxicity.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key regulator of the cellular defense mechanisms against oxidative stress. Generally, the protein level of Nrf2 is controlled by the Kelch-like ECH-associated protein 1 (Keap1)-cullin 3 (Cul3) complex. Under homeostatic conditions, Nrf2 is bound to Keap1, which facilitates Nrf2 for Cul3-mediated ubiquitination and subsequent proteasomal degradation. In response to oxidative stress, electrophilic agents (such as quinone) inactivate Keap1 to block the connection between Keap1 and Cul3, resulting in a decrease in the ubiquitination of Nrf2 and an increase in Nrf2 stabilization, which promotes the translocation of Nrf2 to the nucleus to initiate the transcription of antioxidative genes, including glutathione (GSH), superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px) (Eggler et al., 2005; Eggler et al., 2009; Itoh et al., 1999; Roberts et al., 2023; Tonelli et al., 2018; Zhang et al., 2004). However, there are reports that the protein level of Nrf2 in mouse heart or cultured cardiomyocytes is decreased in response to DOX-induced oxidative stress, which compromises the antioxidative function of Nrf2 and thus aggravates DOX-induced myocardial oxidative stress (Cheng et al., 2022; He et al., 2023; Hu et al., 2023; Jiang et al., 2022; Yang et al., 2023; Zhou et al., 2023). These reports indicate that other mechanisms but not the Keap1-Cul3 complex may account for the downregulation of Nrf2 in DOX-induced cardiotoxicity.
Recently, we have demonstrated for the first time that inhibition of mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) increased the protein level of Nrf2 in rat heart suffering from ischemia/reperfusion (Jiang et al., 2023). This finding reminds us to connect MALT1 with the downregulation of myocardial Nrf2 in DOX-treated animals. As a multifunctional protein, MALT1 either functions as an adaptor protein to assemble and recruit proteins such as caspase recruitment domain-containing protein 11 (CARD11) and B cell lymphoma 10 (BCL10) to form the so-called CARD11-BCL10-MALT1 (CBM) complex or acts as a paracaspase to cleave specified substrates such as A20, HOIL1, and CYLD (Coornaert et al., 2008; Gehring et al., 2018; Klein et al., 2015; Staal et al., 2011). In addition to the functions of scaffolding and proteolytic activity, paracaspase/MALT1-dependent ubiquitination of nuclear factor kappa B (NF-κB) essential modulator (NEMO) has been identified (Zhou et al., 2004), suggesting that MALT1 also possesses a ubiquitin ligase function. Owing to its multifunction, it is likely that MALT1 accounts for the downregulation of myocardial Nrf2 protein level via its proteolytic activity and/or E3 ubiquitin ligase activity in DOX-treated animals.
It is well accepted that activation of MALT1 heavily depends on the assembly of the CBM complex. Next, we sought to identify the underlying mechanisms for initiation of the CBM complex formation in heart after DOX treatment. There is evidence that the activated calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylates CARD11 to promote the assembly of the CBM complex in T cell receptor (TCR)-induced NF-κB activation (Ishiguro et al., 2007; Oruganti et al., 2011). It is not known, however, whether activation of CaMKII is able to initiate the formation of CBM complex in heart suffering from DOX toxicity via phosphorylation of CARD11. Among the four isoforms of CaMKII (α, β, γ, and δ), CaMKII-δ is highly expressed in the heart (Mayer et al., 1995). A number of studies have reported that activation of CaMKII-δ was responsible for ischemia/reperfusion and DOX-induced myocardial injury (Zhang et al., 2022; Zhang et al., 2016). Based on these studies, we hypothesize that activation of CaMKII-δ increases the phosphorylation of CARD11, which facilitates the assembly of the CBM complex and activation of MALT1, leading to downregulation of Nrf2 protein level and the subsequent aggravation of oxidative stress in heart suffering DOX toxicity.
There are three purposes for this study: (1) to evaluate whether MALT1 contributes to myocardial oxidative stress in DOX-treated mice, (2) to determine it is MALT1-dependent proteolytic activity or MALT1-dependent ubiquitin ligase activity responsible for the downregulation of myocardial Nrf2 protein level in DOX-treated mice, and (3) to verify whether CaMKII-δ triggers the CBM complex-dependent activation of MALT1 in DOX-treated mice.
Results
Inhibition of MALT1 alleviated myocardial oxidative injury and mitochondrial damages in DOX-treated mice
A single injection of DOX for 4 days led to cardiac dysfunction and myocardial damage, as evidenced by decreases in ejection fraction (EF) and fractional shortening (FS) (Fig. 1A–C), whereas increases in the activities of serum creatine kinase (CK) and lactate dehydrogenase (LDH) as well as a decrease in the ratio between heart weight and tibia length (HW/TL) (Fig. 1D–F), suggesting that the DOX-induced cardiotoxicity model was successfully established. Expectedly, the DOX-induced cardiotoxicity was evidently attenuated in the presence of MALT1 inhibitor (MI-2) (Fig. 1A–F).
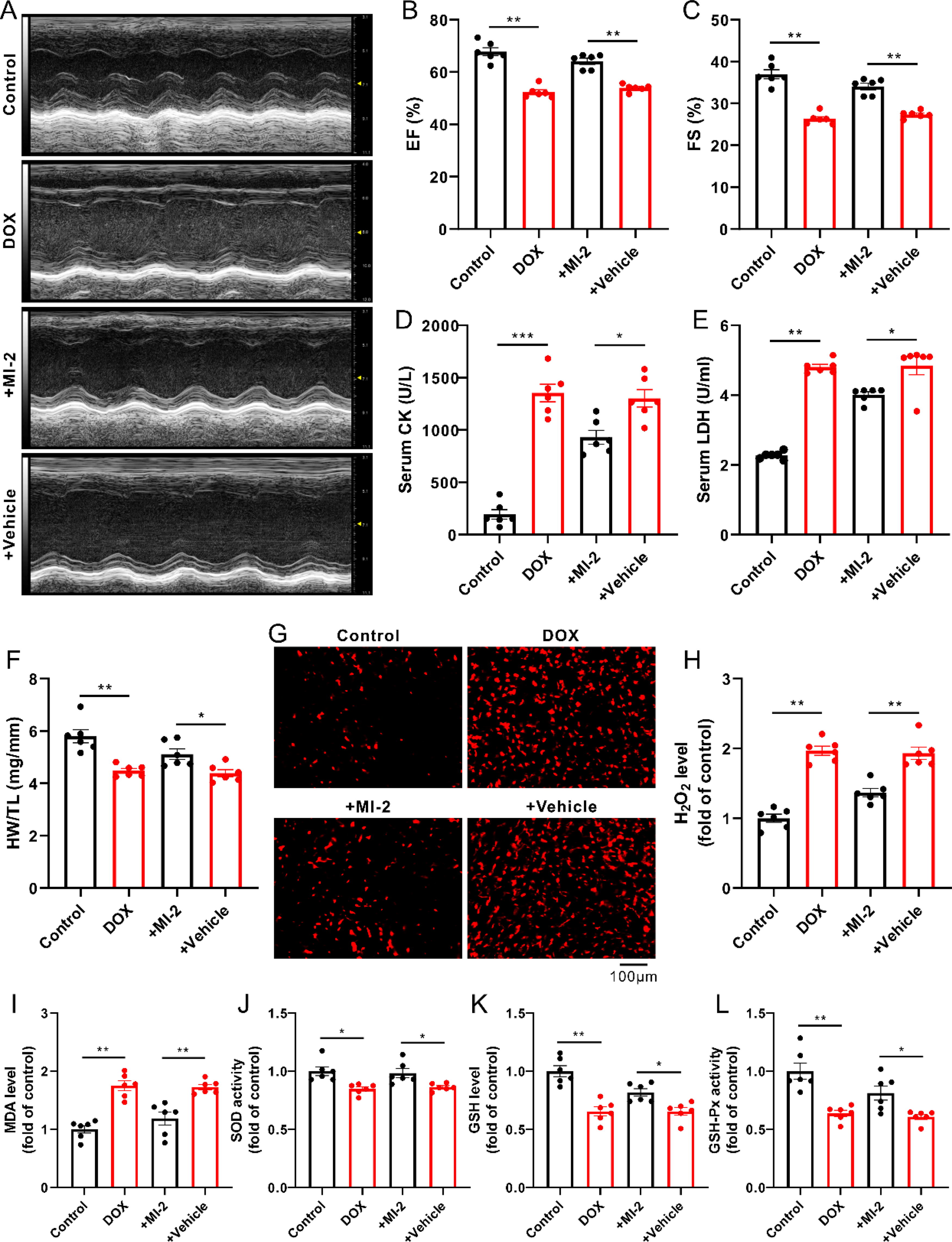
In agreement with previous reports (Cheng et al., 2022; Hu et al., 2023; Zhou et al., 2023), in the present study, DOX-treated mice displayed an increase in myocardial oxidative stress, reflected by increases in the levels of ROS (such as H2O2 and malondialdehyde [MDA]) as well as decreases in the level of GSH and the activities of SOD and GSH-Px (Fig. 1G–L). Interestingly, DOX-induced myocardial oxidative stress was reversed by MI-2, the MALT1 inhibitor (Fig. 1G–L).
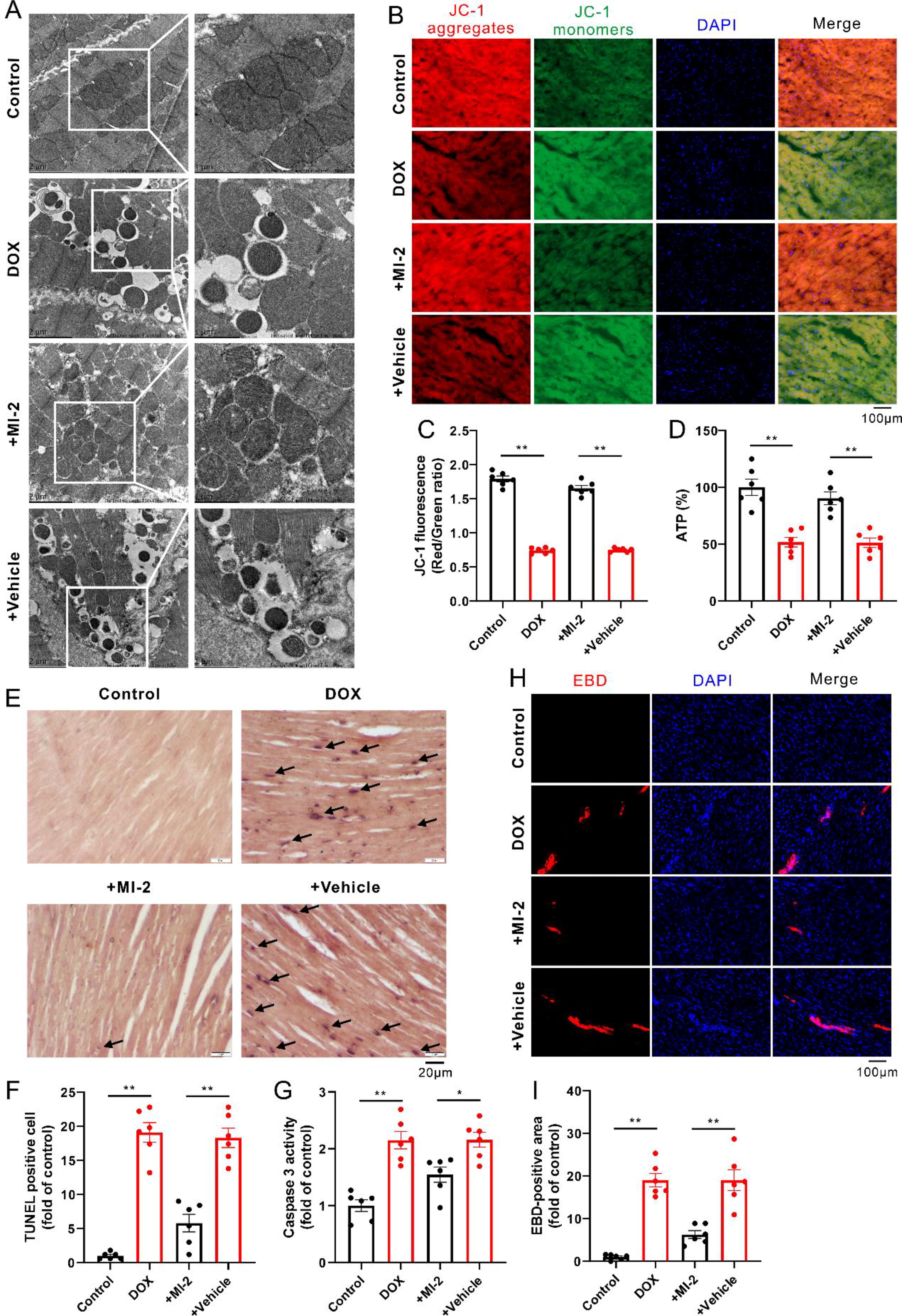
As expected, DOX damaged the mitochondrial morphology and function, as evidenced by the swelling, vacuolar degeneration, and the increased density of mitochondria with vague and fractured cristae, whereas the decreased mitochondrial membrane potential (ΔΨm) and adenosine triphosphate (ATP) production (Fig. 2A–D), concomitant with the increased cellular apoptosis and necrosis, as indicated by increases in the transferase-mediated dUTP nick end labeling (TUNEL)-positive cells (Fig. 2E and F), the activity of caspase 3 (Fig. 2G), and the Evans blue dye (EBD)-positive staining cells (Fig. 2H and I); these phenomena were notably reversed in the presence of MI-2 (Fig. 2A–I).
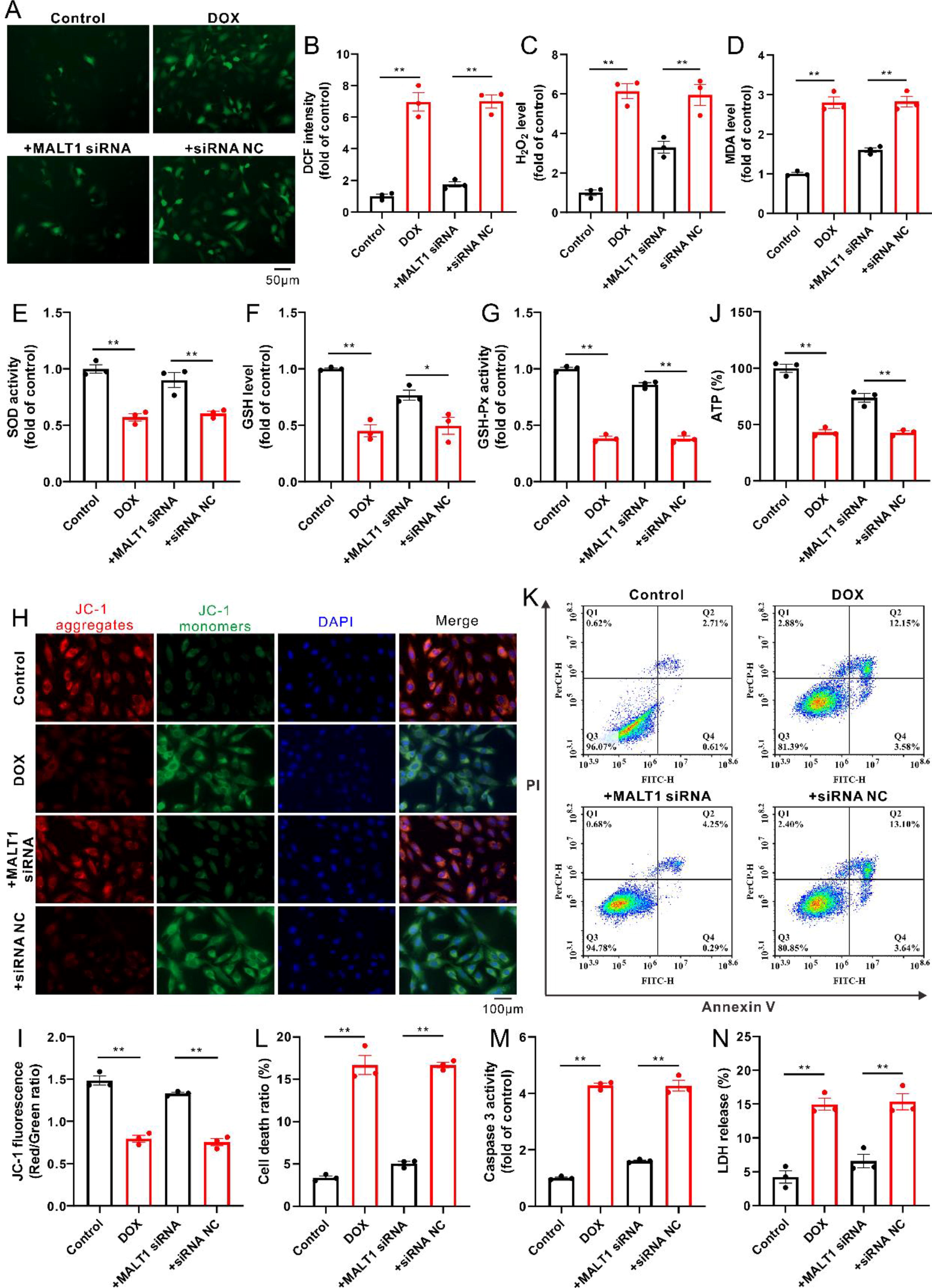
Knockdown of MALT1 suppressed oxidative damages in DOX-treated H9c2 cells
In line with the results in mice, incubation with DOX also induced oxidative stress in the cultured cardiomyocytes, as evidenced by increases in the levels of ROS (such as H2O2 and MDA) as well as decreases in the level of GSH and the activities of SOD and GSH-Px; these phenomena were attenuated in the presence of MALT1 small interfering RNAs (siRNAs) (Fig. 3A–G). Expectedly, incubation with DOX also resulted in mitochondrial dysfunction in the cultured H9c2 cells, reflecting the decreased ΔΨm and ATP production (Fig. 3H–J), accompanied by increased apoptosis and necrosis because of the increases in Annexin V+/PI− and Annexin V+/PI+ cells, the activity of caspase 3, and LDH release (Fig. 3K–N); these phenomena were evidently attenuated in the presence of MALT1 siRNAs (Fig. 3H–N).
MALT1-dependent k48-linked ubiquitination but not MALT1-dependent proteolytic activity was responsible for the downregulation of Nrf2 in DOX-treated mice and H9c2 cells
As shown in Figure 4A–F, the protein level of MALT1 was elevated, whereas the protein level of Nrf2 was decreased in DOX-treated mice or H9c2 cells; these phenomena were notably reversed in the presence of MI-2 or MALT1 siRNAs. In addition to full-length Nrf2 (∼95 kDa), administration of DOX resulted in a likely truncated Nrf2 in the cultured H9c2 cells (∼55 kDa). However, neither MALT1 siRNAs nor the proteasome inhibitor (MG132) impacted the likely truncated Nrf2 (Supplementary Fig. S1A and B), ruling out the possibility of MALT1-dependent cleavage of Nrf2.
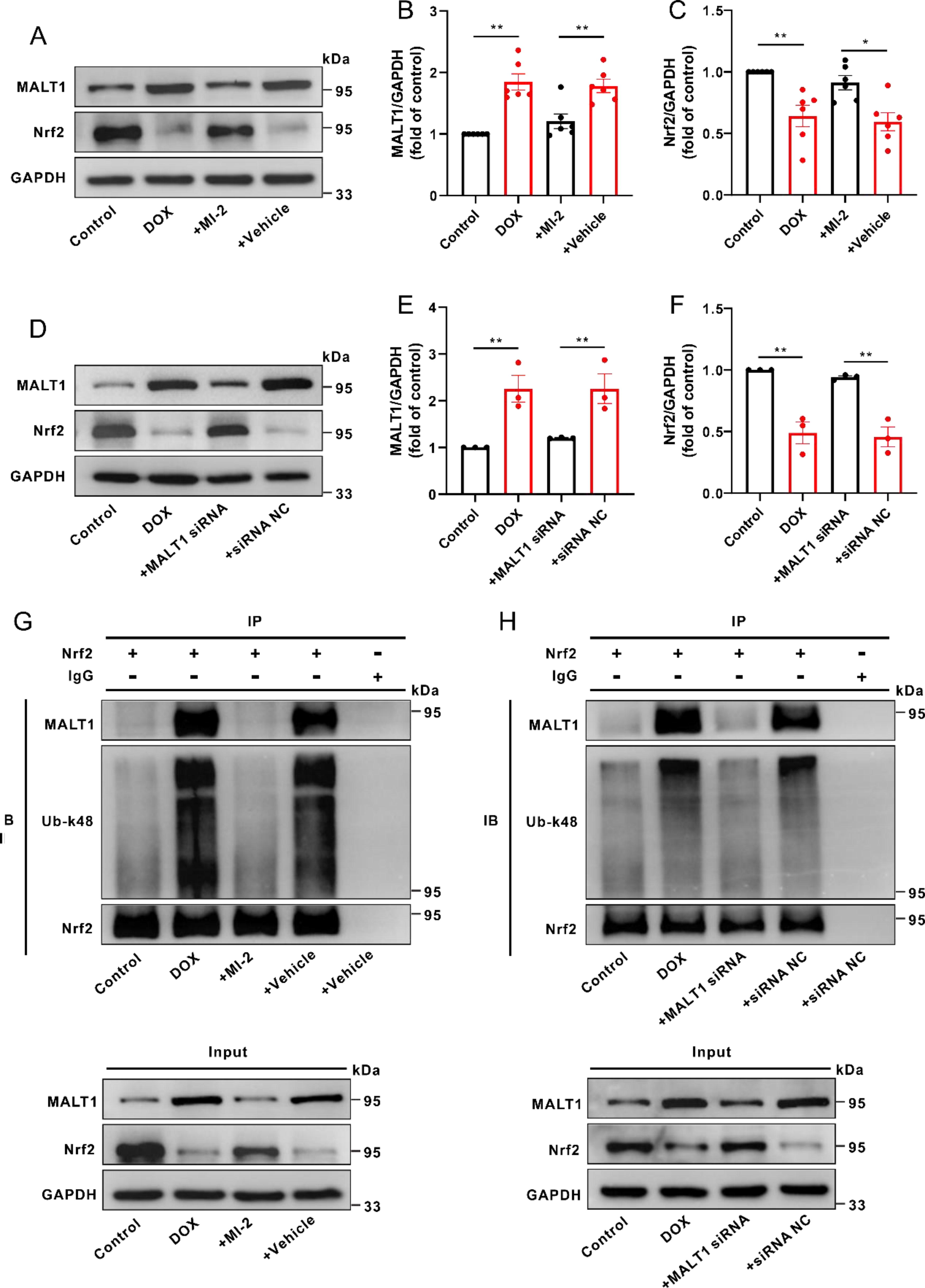
Interestingly, the k48-linked ubiquitination of Nrf2 was increased, whereas the interaction between Nrf2 and MALT1 was enhanced in DOX-treated mice or H9c2 cells. Inhibition or knockdown of MALT1 notably attenuated the interaction between Nrf2 and MALT1 and decreased the k48-linked ubiquitination of Nrf2 (Fig. 4G and H). Furthermore, knockdown of MALT1 promoted the nuclear translocation of Nrf2 in DOX-treated H9c2 cells (Supplementary Fig. S2A). These results supported that MALT1-dependent k48-linked ubiquitination of Nrf2 is responsible for the decreases of protein level and nuclear translocation of Nrf2.
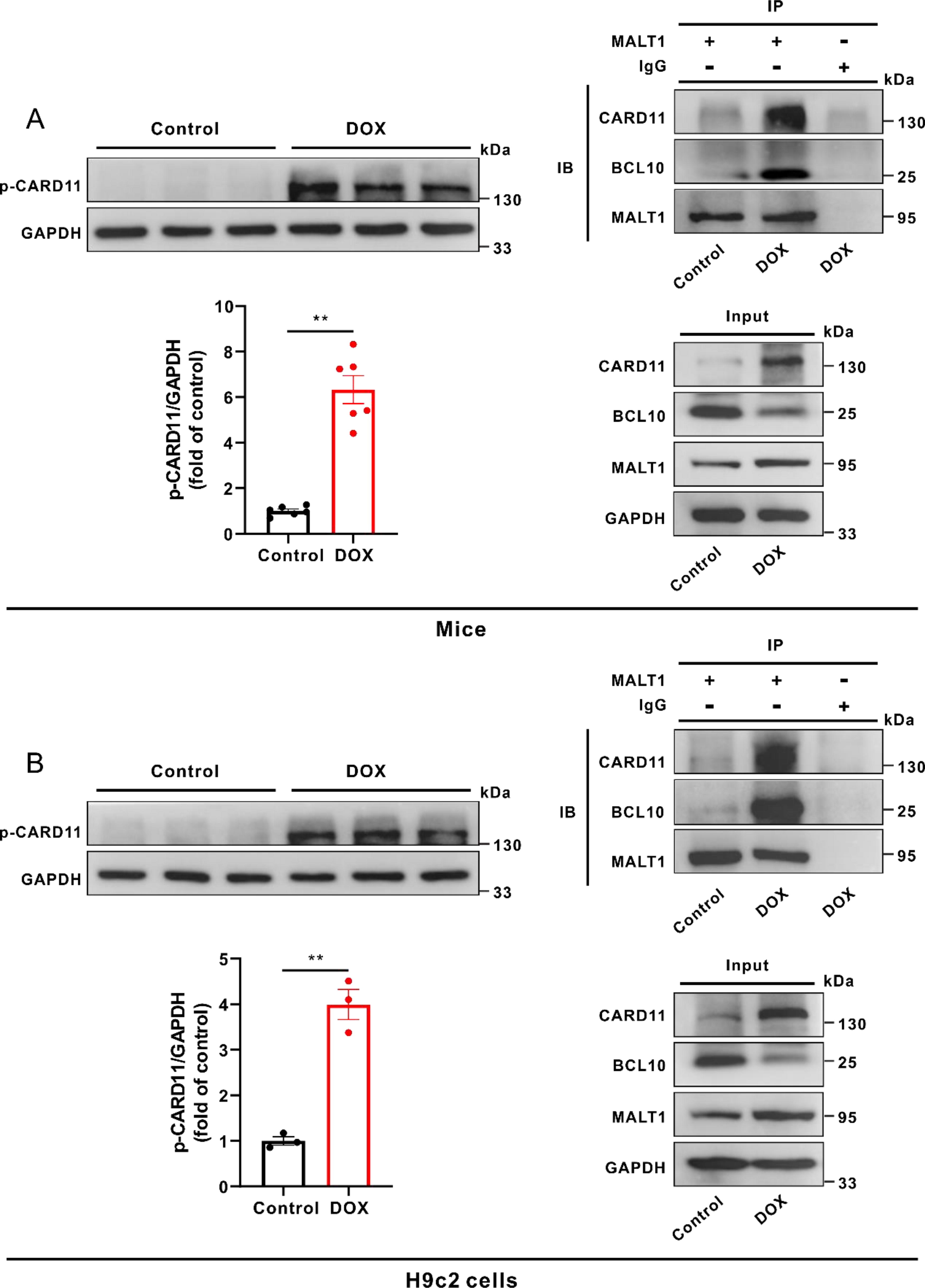
The increased phosphorylation of CARD11 triggered the assembly of CBM complex in DOX-treated mice and H9c2 cells
As shown in Figure 5A, the phosphorylation level of myocardial CARD11 was notably increased in DOX-treated mice, concomitant with the enhanced interactions between CARD11, BCL10, and MALT1. Similar phenomenon was observed in DOX-treated H9c2 cells (Fig. 5B). Knockdown of MALT1 did not impact the phosphorylation level of CARD11 but prevented the formation of the CBM complex in DOX-treated H9c2 cells (Supplementary Fig. S2B and C). Moreover, knockdown of CARD11 disrupted the assembly of CBM complex and reduced the MALT1-dependent k48-linked ubiquitination of Nrf2 in DOX-treated H9c2 cells (Supplementary Fig. S3A–C). These results suggested that certain protein kinases promote the CBM complex-dependent MALT1 activation via phosphorylation of CARD11.
Inhibition or knockdown of CaMKII-δ attenuated oxidative stress and restored mitochondrial function in DOX-treated mice or H9c2 cells
DOX-induced oxidative stress was attenuated in the presence of hesperadin, an inhibitor of CaMKII-δ, as indicated by the reduced levels of ROS (such as H2O2 and MDA) as well as the increased level of GSH and activities of SOD and GSH-Px (Fig. 6A–F). Consistently, the DOX-induced damage of mitochondrial morphology and function was restored after inhibition of CaMKII-δ, as evidenced by the recovery of mitochondrial morphology, and ΔΨm and ATP production (Fig. 6G–J).
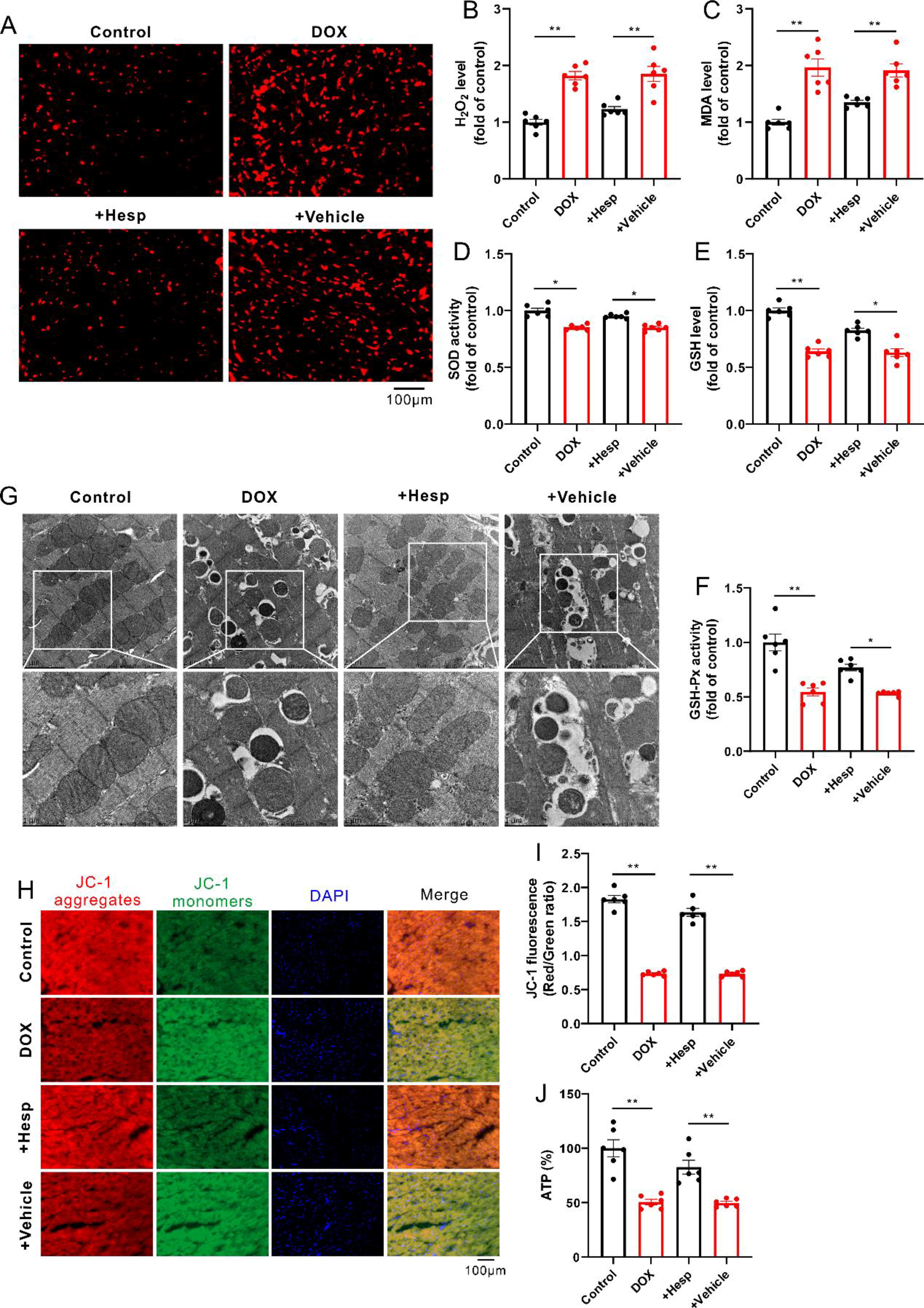
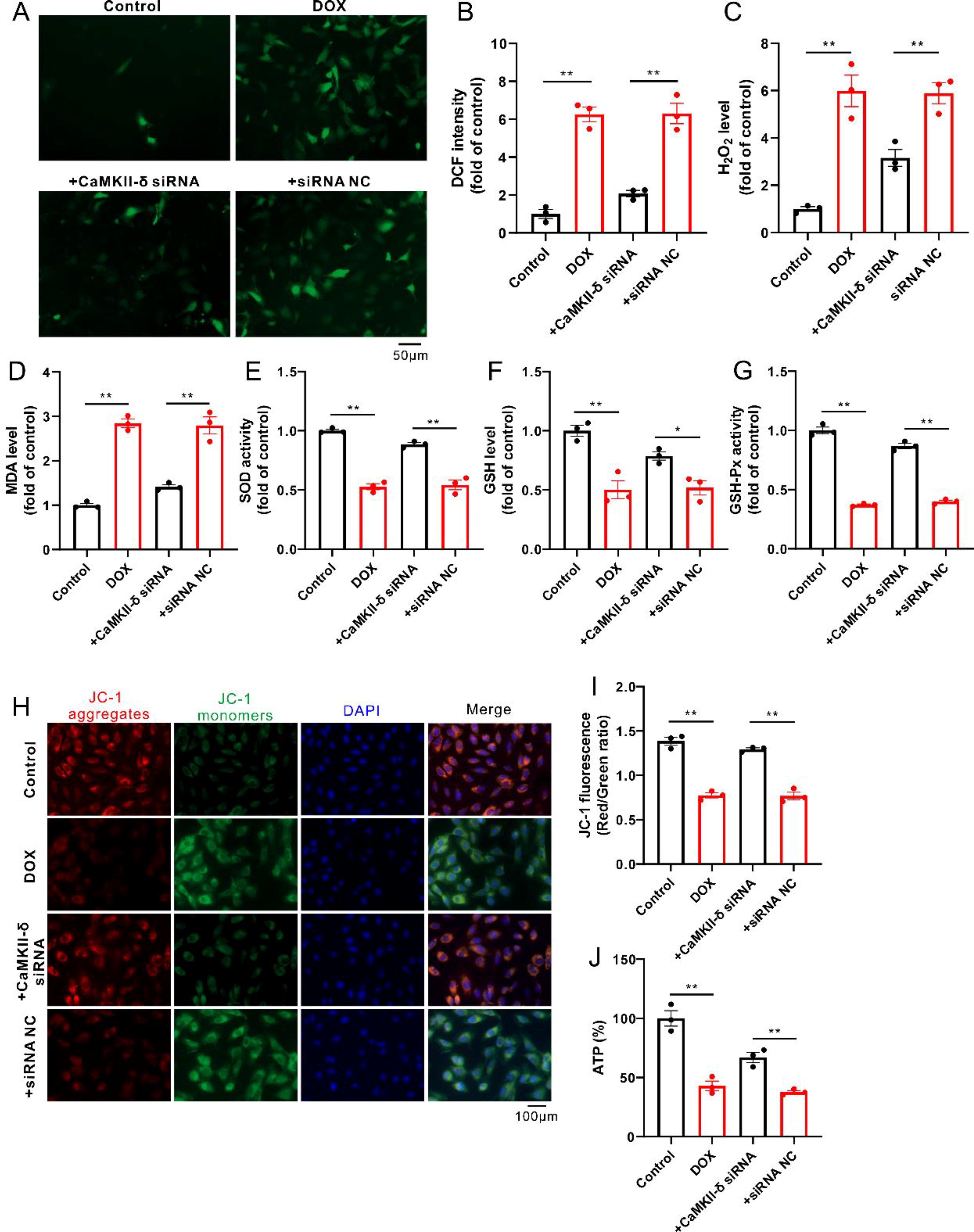
Similar to the results in an animal study, knockdown of CaMKII-δ reduced oxidative stress in DOX-treated H9c2 cells, reflected by decreases in the levels of ROS (such as H2O2 and MDA) as well as increases in the level of GSH and the activities of SOD and GSH-Px (Fig. 7A–G). As expected, knockdown of CaMKII-δ significantly attenuated DOX-induced mitochondrial dysfunction, as indicated by the restored ΔΨm and ATP production in H9c2 cells (Fig. 7H–J).
Inhibition or knockdown of CaMKII-δ disrupted the assembly of CBM complex and reduced the MALT1-dependent k48-linked ubiquitination of Nrf2 in DOX-treated mice or H9c2 cells
As shown in Figure 8A–E, the phosphorylation level of myocardial CaMKII-δ was decreased in the presence of its inhibitor (hesperadin) in DOX-treated mice, accompanied by decreases in the protein levels of p-CARD11 and MALT1 as well as an increase in the protein level of Nrf2. Interestingly, DOX-triggered formation of the myocardial CBM complex in mice was evidently attenuated by CaMKII-δ inhibitor (hesperadin), concomitant with a decrease in k48-linked ubiquitination of Nrf2 (Fig. 8F and G).
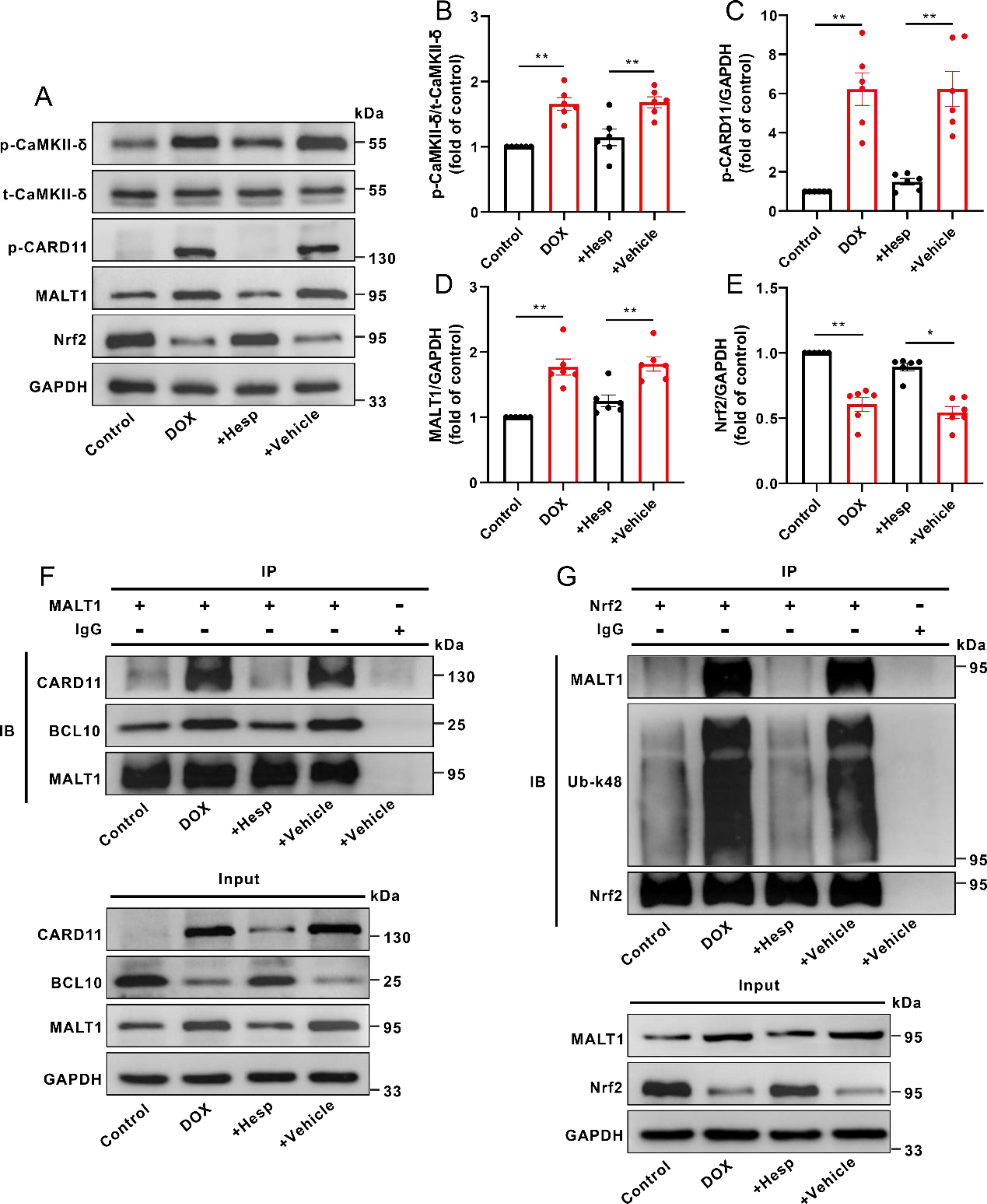
In agreement with the results in an animal study, CaMKII-δ siRNAs also reduced the protein level of p-CaMKII-δ in DOX-treated H9c2 cells, together with decreases in the protein levels of p-CARD11 and MALT1, whereas an increase in the protein level of Nrf2 (Fig. 9A–E). Similarly, knockdown of CaMKII-δ also blocked the assembly of CBM complex in DOX-treated H9c2 cells, accompanied by a reduction in k48-linked ubiquitination of Nrf2 (Fig. 9F and G). In addition, overexpression of MALT1 counteracted the inhibitory effect of CaMKII-δ inhibitor on Nrf2 ubiquitination in DOX-treated H9c2 cells (Supplementary Fig. S4A).

Discussion
There were three major findings for this study: (1) MALT1-mediated downregulation of Nrf2 protein contributed to myocardial oxidative stress, mitochondrial damage, and cell death in DOX-treated mice; (2) MALT1-dependent k48-linked ubiquitination but not MALT1-dependent proteolytic activity was responsible for the downregulation of Nrf2; and (3) CaMKII-δ-dependent phosphorylation of CARD11 triggered the assembly of CBM complex and the subsequent activation of MALT1. To the best of our knowledge, it is the first study to demonstrate that the E3 ubiquitin ligase activity of MALT1 accounts for the downregulation of myocardial Nrf2 and oxidative stress in DOX-induced cardiotoxicity.
The semiquinone DOX resulting from the metabolic process of DOX is capable of quickly transferring its unpaired electrons to the molecular oxygen to generate superoxide anion. During this process, the semiquinone DOX is rapidly reoxidized to the parent DOX, and the superoxide anion converts to other ROS such as hydrogen peroxide, hydroxyl radical, and hypochlorous acid (Young et al., 1981). This redox-cycle between DOX and semiquinone DOX induces constant production of ROS, which directly attacks biological macromolecules, including lipids, proteins, and nucleic acid, resulting in oxidative stress, mitochondrial dysfunction, and ultimate cell death in the heart (Carvalho et al., 2014; Wallace et al., 2020; Yu et al., 2018). As we know, it is the DNA intercalation and inhibition of topoisomerase II but not ROS that plays a major role in the anticancer effects of DOX. However, it is DOX-induced overproduction of ROS that plays a fundamental and indispensable role in cardiotoxicity because the cardiac cells possess a higher number of mitochondria compared with most of the other cell types and mitochondrion is a key organelle to generate ROS. For this reason, it is particularly susceptible for the cardiac cells (but not other cell types, including most cancer cells within the indications of DOX) to overproduce ROS after DOX treatment, leading to oxidative damage of cardiac cells (Hortobagyi, 1997 Wallace et al., 2020). Thus, proper clearance of ROS is an effective strategy to alleviate DOX cardiotoxicity but has little impact on the anticancer effects of DOX.
The redox-sensitive transcription factor Nrf2 is able to clear ROS by promoting the transcription of antioxidative genes, including SOD, GSH, and GSH-Px (Tonelli et al., 2018). Unfortunately, the protein levels of Nrf2 were significantly decreased in DOX-treated mice and H9c2 cells, which compromised the antioxidative function of Nrf2 and subsequently aggravated the oxidative stress in DOX-induced cardiotoxicity (Cheng et al., 2022; He et al., 2023; Hu et al., 2023; Jiang et al., 2022; Yang et al., 2023; Zhou et al., 2023). Therefore, elucidating the mechanisms underlying the downregulation of myocardial Nrf2 may help to alleviate DOX-induced cardiotoxicity. Keap1 was identified as an adaptor for Cul3-based E3 ubiquitin ligase to promote Nrf2 ubiquitination and its subsequent proteasomal degradation, which keeps a baseline level of Nrf2 to balance the redox reaction under homeostatic condition. Numerous studies have found that the binding of Keap1 with Cul3 was released and Keap1 is no longer a target for Nrf2 for ubiquitination in response to oxidative stress, although Keap1 retains its affinity for Nrf2 (Eggler et al., 2005; Eggler et al., 2009; Itoh et al., 1999; Zhang et al., 2004). Consistent with these reports, our results showed that the interaction between Nrf2 and Cul3 was significantly weakened in DOX-treated mice and H9c2 cells, confirming that Cul3 lost the control in Nrf2 ubiquitination. Theoretically, in such situations, Nrf2 levels should be elevated because its Cul3-mediated ubiquitination is blocked. However, in this study, we found that Nrf2 levels were still decreased, and knockdown of Keap1 did not impact the k48-linked ubiquitination and protein levels of Nrf2 (Supplementary Fig. S5A–C). Therefore, it is not the Keap1-Cul3 complex but other unknown mechanisms that account for the downregulation of myocardial Nrf2 in DOX-treated mice.
Recently, we have found that inhibition of MALT1 increased the protein level of Nrf2 in rat heart suffering ischemia/reperfusion (Jiang et al., 2023). We thus focused on the connection between MALT1 and Nrf2 in this study. To date, at least two major functions have been identified for MALT1: scaffold and protease. As a protease, the cleaved forms of its substrates (the truncated substrates) can be seen, and they are subsequently degraded by the proteasome. Based on the reports, BCL10 is a substrate of MALT1, and the assembly of CBM complex can activate MALT1 to cleave and degrade BCL10 (Hailfinger et al., 2011; Rebeaud et al., 2008). Consistent with the reports, in the present study, DOX facilitated the CBM complex assembly, leading to activation of MALT1 and the subsequent degradation of BCL10. Inhibition or knockdown of MALT1 can efficiently block the cleavage of substrates, as evidenced by a decrease in the cleaved forms. MG132, a proteasome inhibitor, led to an increase in the cleaved forms (Coornaert et al., 2008; Hailfinger et al., 2011; Jeltsch et al., 2014; Staal et al., 2011). In the present study, we actually observed the likely truncated Nrf2 in DOX-treated H9c2 cells. However, knockdown of MALT1 or administration of MG132 had no effect on the likely truncated Nrf2, ruling out the possibility that the protease activity of MALT1 accounts for DOX-induced downregulation of myocardial Nrf2.
Besides the protease activity, MALT1 also possesses the E3 ubiquitin ligase activity. It has been reported that BCL10 promoted NF-kB activation through inducing the MALT1-dependent ubiquitination of NEMO in lymphoma cell lines, and knockdown of MALT1 attenuated BCL10-induced NEMO ubiquitination (Zhou et al., 2004). Consistent with this study, our results showed that inhibition or knockdown of MALT1 notably blocked the k48-linked ubiquitination of myocardial Nrf2, concomitant with increases in the protein levels of Nrf2 in DOX-treated mice and H9c2 cells. Thereby, it is likely that the E3 ubiquitin ligase activity of MALT1 accounts for DOX-caused downregulation of myocardial Nrf2. Interestingly, NEMO was increasingly ubiquitinated when coexpressed with CARD11-BCL10. As MALT1 is a component of the CBM complex, which consists of CARD11, BCL10, and MALT1, it is likely that the assembly of the CBM complex could activate the E3 ubiquitin ligase function of MALT1 (Zhou et al., 2004). However, the mechanisms behind initiation of the myocardial CBM complex formation in DOX-treated mice remain largely unknown. In the present study, our results showed that the phosphorylation levels of myocardial CARD11 were evidently increased in DOX-treated mice and H9c2 cells, accompanied by the enhanced interactions between CARD11, BCL10, and MALT1. Moreover, knockdown of CARD11 disrupted the formation of CBM complex and reduced the MALT1-dependent k48-linked ubiquitination of Nrf2 in DOX-treated H9c2 cells, indicating that phosphorylation of CARD11 initiates the assembly of CBM complex and the subsequent activation of MALT1. Next, we focused on determining the protein kinase responsible for the phosphorylation of CARD11.
There is evidence that the activation of CaMKII-δ contributes to ischemia/reperfusion- and DOX-induced myocardial injury, and inhibition of CaMKII-δ is able to alleviate cardiomyocyte death and the subsequent myocardial injury (Zhang et al., 2022; Zhang et al., 2016). In the immune synapse, activation of CaMKII phosphorylated CARD11 and then promoted the assembly of the CBM complex to induce NF-κB activation (Ishiguro et al., 2007; Oruganti et al., 2011). These studies hinted that activation of CaMKII-δ is likely to facilitate the formation of the myocardial CBM complex through phosphorylating CARD11 in DOX-induced cardiotoxicity. According to our results, DOX treatment indeed activated CaMKII-δ, subsequently phosphorylated CARD11, and triggered the formation of the CBM complex, which activated the E3 ubiquitin ligase function of MALT1 and then induced the k48-linked ubiquitination of Nrf2; these phenomena were attenuated after inhibition or knockdown of CaMKII-δ.
As we mentioned before, proper clearance of ROS can alleviate DOX cardiotoxicity without compromising its anticancer effects. Actually, in addition to reducing DOX-induced oxidative damage of cardiac cells, prevention of CBM complex formation and inhibition of MALT1 may offer additional benefits for clinical application in the patients receiving the treatment of DOX. There is evidence that the formation of CBM complex and MALT1 activation is able to initiate the canonical NF-kB pathway, which promotes the survival of malignant lymphomas (Oeckinghaus et al., 2007; Zhou et al., 2004). Differently, NF-kB activation has been reported to facilitate the transcription of inflammatory genes in cardiac cells (Yao et al., 2022), which induces heart inflammation. Thus, prevention of CBM complex formation and inhibition of MALT1 may enhance the anticancer effects of DOX and attenuate heart inflammation at the same time.
Innovation and Conclusion
In summary, our present study has demonstrated for the first time that it is the E3 ubiquitin ligase and not the protease activity of MALT1 responsible for the downregulation of Nrf2 and aggravation of myocardial oxidative stress in DOX-treated mice, and CaMKII-δ-dependent phosphorylation of CARD11 triggered the assembly of CBM complex and subsequent activation of MALT1 (Fig. 10). Therefore, MALT1 may be used as a potential target to treat DOX-induced cardiotoxicity.

Limitations of the Study
There are two major limitations that need to be acknowledged and addressed regarding the present study. First of all, we did not provide direct evidence to clearly demonstrate that MALT1 itself is an E3 ubiquitin ligase although the paracaspase domain of MALT1 has been reported to possess intrinsic E3 ubiquitin ligase activity (Zhou et al., 2004). Further study using an in vitro reconstitution system can verify that MALT1 possesses E3 ubiquitin ligase activity. Second, although the paracaspase domain of MALT1 possesses both the E3 ubiquitin ligase activity and the protease activity, and MI-2 inhibits the protease activity of MALT1 via directly binding to its paracaspase domain (Fontan et al., 2012; Zhou et al., 2004), it does not mean that MI-2 also inhibits the E3-ubiquitin ligase function of MALT1. More studies are needed before drawing a firm conclusion.
Materials and Methods
Electronic laboratory notebook was not used.
Animal experiments
Male C57BL/6 J mice (8 weeks old, 25 ± 2 g) were obtained from the Laboratory Animal Center, Xiang-Ya School of Medicine, Central South University, China. All animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals,” published by the National Institutes of Health (The Eighth edition, 2011) and the Animal Researcher: Reporting In Vivo Experiments (ARRIVE) guidelines. The experiments were approved by the Institutional Animal Care and Use Committee of Central South University.
Mice received a single injection (i.p.) of DOX (MedChemExpress, Shanghai, China, 15 mg/kg body weight dissolved in saline) or saline. To assess the role of MATL1 in DOX-induced cardiotoxicity, daily injection (i.p.) of its inhibitor MI-2 (Selleck Chemicals, Shanghai, China, 20 mg/kg body weight dissolved in vehicle [5% DMSO + 30% PEG300 + 5% Tween 80 + 60% ddH2O]) or an equivalent volume of vehicle was started on the day of DOX treatment. MI-2 irreversibly inhibits MALT1 via directly binding to the paracaspase domain of MALT1 (residues 329–728), and it is nontoxic to animals at a dose of 25 mg/kg (Fontan et al., 2012). To assess the role of CaMKII-δ in the assembly of CBM complex and activation of MALT1, daily injection (i.p.) of its inhibitor hesperadin (MedChemExpress, 5 μg/kg body weight dissolved in vehicle [10% DMSO + 40% PEG300 + 5% Tween 80 + 45% saline]) or an equivalent volume of vehicle was also started on the day of DOX treatment. After injection of DOX for 4 days, all mice were sacrificed under deep anesthesia, and blood samples and heart tissue were collected for morphological, biochemical, and molecular analysis.
Cell experiments
The rat cardiomyocytes (H9c2 cells) were purchased from the Cell Bank of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China, and the cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, USA) supplemented with 10% fetal bovine serum (ExCell Bio, South America) in a humidified atmosphere of 5% CO2 and 95% air at 37°C.
The cells were incubated with DOX (1 μM) for 24 h to establish a DOX-induced cardiotoxicity model in vitro. To verify the role of MALT1 in DOX-induced cardiotoxicity and oxidative stress, cells were transfected with MALT1 siRNAs (RiboBio Co., Ltd., Guangzhou, China, 50 nM) at 24 h before DOX treatment. To verify the role of CARD11 in DOX-induced MALT1 activation, cells were transfected with CARD11 siRNAs (RiboBio Co., Ltd., 50 nM) at 24 h before DOX treatment. To verify the role of CaMKII-δ in DOX-induced assembly of the CBM complex and MALT1-dependent k48-linked ubiquitination of Nrf2, cells were transfected with CaMKII-δ siRNAs (RiboBio Co., Ltd., 50 nM) at 24 h before DOX treatment. The efficiency of gene knockdown was evaluated by Western blot. Scrambled siRNA was used as a negative control. To confirm that inhibition of CaMKII-δ inhibits Nrf2 ubiquitination through MALT1 inhibition, cells were transfected with adenovirus (AD) carrying rat MALT1 (AD-MALT1, Genechem, Shanghai, China) or a negative control (AD-NC) at 24 h before DOX and hesperadin (0.5 μM) treatment. In the end, the cells and culture medium were collected for morphological, biochemical, and molecular analysis.
Echocardiography
Transthoracic echocardiography was performed on anesthetized mice using a FUJIFILM Visual Sonics Vevo2100 Imaging System with a 30-MHz high-frequency transducer. Left ventricular diastolic and systolic anterior or posterior wall thickness and left ventricular end-diastolic and end-systolic internal dimensions were measured from two-dimensional short-axis view under M-mode tracings at the level of the papillary muscle. Then, left ventricular volume, EF, and FS were calculated using a software provided by the supplier.
Measurement of activities of CK and LDH
CK activity was detected following the instruction provided by the supplier (Biosino Bio-Technology). Briefly, 4 μL serum and 200 μL work solution were mixed in a 96-well plate, then the plate was incubated for 2 min at 37°C and measured on microplate reader at kinetic mode to acquire ΔA at 340 nm, and the CK activity was calculated as described in the protocol.
For detection of LDH activity, 20 μL serum, 25 μL matrix buffer, and 5 μL coenzyme I were mixed in a 96-well plate and incubated for 15 min at 37°C, 25 μL dinitrophenylhydrazine was added into the plate and incubated for 15 min at 37°C, and then 250 μL sodium hydroxide solution (0.4 M) was added into the plate and incubated for 5 min at room temperature. The absorbance was recorded at 450 nm, and LDH activity was calculated with a formula provided by the supplier (Jiancheng Bioengineering Institute, Nanjing, China).
Detection of oxidative stress
ROS production was evaluated by DHE staining in vivo and DCFH-DA staining in vitro. Briefly, the cryosections of fresh heart samples or H9c2 cells were incubated with DHE (Beyotime, Shanghai, China, 5 μM) or DCFH-DA (Beyotime, 10 μM) at 37°C for 30 min and protected from light. Images were obtained using a fluorescent microscope (Carl Zeiss, Germany), and then quantification was performed by using ImageJ.
To further assess oxidative stress in vivo and in vitro, the levels of H2O2, MDA, and GSH and the activities of SOD and GSH-Px in the myocardium or H9c2 cells were measured with the commercially available kits (MDA, GSH, SOD, and GSH-Px [Jiancheng Bioengineering Institute] and H2O2 [Beyotime]).
Transmission electron microscopy
Samples of myocardium were quickly removed from the left ventricle and immediately fixed with 5% glutaraldehyde for 2 h, postfixed in 1% osmic acid for 2 h, dehydrated in graded ethanol, embedded in epon at 37°C for 12 h, and solidified at 60°C for 24 h. The embedding blocks were sliced into 70-nm-thick ultrathin sections on a microtome (Leica, Germany). The ultrathin sections were sequentially stained with both 3% uranyl acetate and lead citrate. The images were captured and analyzed with the transmission electron microscope (FEI Tecnai G2 Spirit, USA).
Analysis of mitochondrial function
The ΔΨm of the myocardium or H9c2 cells was evaluated by using the fluorescent dye JC-1 (Beyotime). A shift in fluorescence from red (JC-1 aggregates) to green (JC-1 monomers) indicates a loss of ΔΨm. Briefly, cryosections of fresh heart samples or H9c2 cells were incubated with the JC-1 working solution at 37°C for 20 min in the dark. Images were obtained using a fluorescent microscope (Carl Zeiss).
The ATP level of heart tissue or H9c2 cells was measured by using an ATP detection kit (Beyotime) according to the manufacturer’s instructions.
Analysis of apoptosis and necrosis
The apoptotic cells in paraffin-embedded left ventricular tissue were analyzed by a terminal deoxynucleotidyl TUNEL assay. The assay was performed according to the manufacturer’s instructions (Roche, Nutley, NJ, USA). The tissue slices were examined microscopically and photographed by a high-resolution digital camera (Nikon Eclipse 80i, Tokyo, Japan). Then, the images were quantified using ImageJ.
The Annexin V-FITC/PI detection kit (Yeasen Biotechnology, Shanghai, China) was used to assess cell apoptosis (Annexin V+/PI− [early apoptosis] + Annexin V+/PI+ [late apoptosis]) and necrosis (Annexin V+/PI+) in H9c2 cells. Briefly, H9c2 cells were digested with trypsin and collected after centrifugation. Then, the cells were incubated with Annexin V-FITC and PI staining solution at room temperature for 15 min protecting from light. The stained cells were detected by flow cytometry, and the results were analyzed by the NovoExpress software.
To further assess cell apoptosis in vivo and in vitro, the caspase 3 activity of cardiac tissue or H9c2 cells was measured with a commercial kit (Beyotime). Briefly, the sample was lysed in ice-cold lysis buffer and then centrifuged to collect the supernatant. Detection buffer (40 μL), 50 μL of supernatant, and 10 μL of Ac-DEVD-pNA (2 mM) were mixed in a 96-well plate and incubated for 120 min at 37°C. The absorbance was recorded at 405 nm.
EBD was used to evaluate myocardial necrosis. EBD was dissolved in saline (10 mg/mL) and was injected into the tail vein of mice (100 µg/g body weight). After injection of EBD for 30 min, mice were sacrificed and the ventricular myocardium was harvested in liquid nitrogen. The cryosections were imaged using a fluorescent microscope (Carl Zeiss).
The LDH release was analyzed to assess cell necrosis in H9c2 cells. In brief, 120 µL culture medium and 60 μL LDH work solution were mixed in a 96-well plate and incubated for 30 min at room temperature. The absorbance was recorded at 490 nm. The percentage of LDH release was calculated by a formula as described in the protocol (Beyotime).
Western blot and coimmunoprecipitation
Cardiac tissue or H9c2 cells were lysed in ice-cold lysis buffer and then centrifuged to collect the supernatant. The protein concentration was determined by a BCA protein assay kit (Beyotime). A total of 30–40 μg of protein samples was separated by 8% SDS-PAGE, transferred to the PVDF membrane, blocked with 5% nonfat milk at room temperature for 2 h, then incubated with the primary antibodies against MALT1 (Proteintech, Wuhan, China), Nrf2 (ABclonal, Wuhan, China), p-CaMKII-δ (CST, Boston, MA, USA), t-CaMKII-δ (Proteintech), p-CARD11 (CST), CARD11 (Affinity Biosciences, Jiangsu, China), BCL10 (Boster, Wuhan, China), Cul3 (Proteintech), Keap1 (Proteintech), k48-ubiquitin (CST), or GAPDH (Proteintech) overnight at 4°C, and then washed with TBST and incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (Beyotime) at room temperature for 1 h. The protein signals were detected by Luminata Crescendo Western HRP substrate via Molecular Imager ChemiDoc XRS System (Bio-Rad, Philadelphia, PA, USA) and quantified by Image Lab Software.
Co-immunoprecipitation (Co-IP) was used to detect the interactions between CARD11, BCL10, and MALT1, the interaction between Nrf2 and MALT1, or the k48-linked ubiquitination of Nrf2. The Co-IP assay was performed using a commercial kit (Absin, Shanghai, China). Briefly, the protein lysates of cardiac tissue or H9c2 cells were incubated with the primary antibody against MALT1 or Nrf2, or rabbit immunoglobulin G (Beyotime) on a rotating incubator overnight at 4°C, and then incubated with protein A/G agarose beads at 4°C for 4 h. After centrifugation, the collected precipitates were washed with a washing buffer. The immunopurified proteins were used for Western blot.
Immunofluorescent staining
The H9c2 cells were fixed with 4% paraformaldehyde for 15 min, followed by permeabilization for 20 min, blocked with 5% bovine serum albumin for 1 h, then incubated with the primary antibodies against Nrf2 overnight at 4°C, then washed with PBST, and incubated with secondary fluorescent antibodies for 1 h. The cell nuclei were stained with DAPI for 5 min. The confocal microscope (Leica) was used to take the pictures.
Statistical analysis
Prism GraphPad 8.0 was used for statistical analysis. Data were presented as mean ± standard error of mean. Differences among multiple groups were analyzed by one-way analysis of variance followed by Tukey’s test. Differences were considered statistically significant when the p value is <0.05.
Footnotes
Authors’ Contributions
L.-Q.L.: Conceptualization, formal analysis, funding acquisition, investigation, methodology, and writing—original draft. M.-R.L., X.-Y.L., D.P., and H.-R.L.: Formal analysis and investigation. X.-J.Z.: Project administration. X.-J.L.: Funding acquisition and project administration. J.P.: Funding acquisition, project administration, and writing—reviewing and editing.
Data Availability
Data will be made available on request.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
This work was supported by the National Natural Science Foundation of China (No. 82173815 and 81872873 to J.P., No. 82073849 to X.-J.L.), the Natural Science Foundation of Hunan Province, China (No. 2020JJ4770 to J.P., No. 2021JJ30032 to X.-J.L.), and the Fundamental Research Funds for the Central Universities of Central South University, China (No. 1053320215455 to L.-Q.L.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.