
Abstract
Significance:
The primary role of platelets is to generate a thrombus by platelet activation. Platelet activation relies on calcium mobilization from the endoplasmic reticulum (ER). ER resident proteins, which are externalized upon platelet activation, are essential for the function of platelet surface receptors and intercellular interactions.
Recent Advances:
The platelet ER is a conduit for changes in cellular function in response to the extracellular milieu. ER homeostasis is maintained by an appropriate redox balance, regulated calcium stores and normal protein folding. Alterations in ER function and ER stress results in ER proteins externalizing to the cell surface, including members of the protein disulfide isomerase family (PDIs) and chaperones.
Critical Issues:
The platelet ER is central to platelet function, but our understanding of its regulation is incomplete. Previous studies have focused on the function of PDIs in the extracellular space, and much less on their intracellular role. How platelets maintain ER homeostasis and how they direct ER chaperone proteins to facilitate intercellular signalling is unknown.
Future Directions:
An understanding of ER functions in the platelet is essential as these may determine critical platelet activities such as secretion and adhesion. Studies are necessary to understand the redox reactions of PDIs in the intracellular versus extracellular space, as these differentially affect platelet function. An unresolved question is how platelet ER proteins control calcium release. Regulation of protein folding in the platelet and downstream pathways of ER stress require further evaluation. Targeting the platelet ER may have therapeutic application in metabolic and neoplastic disease.
Introduction
Platelets are anucleate cells derived from megakaryocytes that are essential for clot formation. The platelet life span is ∼7–10 days in humans, and 100 billion platelets are produced daily in the steady state. Both circulating platelets and megakaryocytes (predominantly resident in the bone marrow) are exposed to extracellular signals that can affect their function and development. Platelets inherit cytoplasmic organelles, messenger RNA (mRNA), and proteins from the parent megakaryocyte. This appears to be an active process as megakaryocyte depletion of RNA binding (Heazlewood et al., 2022) and endoplasmic reticulum (ER) proteins (Jain et al., 2022; Lo et al., 2020; Passam et al., 2015; Zhao et al., 2019; Zhou et al., 2017; Zhou et al., 2014) lead to changes in the platelet mRNA and protein content. Platelets store procoagulant proteins and mediators in their granules (alpha and delta) and release them to the extracellular space upon activation.
As platelets circulate in the blood stream, they play multiple roles including: (i) interacting with other platelets and coagulation proteins in hemostasis (Klatt et al., 2018); (ii) with the endothelium to maintain vascular integrity (Coenen et al., 2017); and (iii) with immune cells, such as T cells, in immunomodulation and neutrophils or monocytes in inflammation (Ali et al., 2015).
Circulating platelets are prevented from adhering to intact blood vessels by the antiadhesive properties of the endothelium. In response to blood vessel injury, platelets first tether to extracellular matrix proteins that become exposed. Under high shear, platelet GPIb/V/IX interacts with von Willebrand factor, slowing down platelet movement to allow the interaction of platelet glycoprotein VI with collagen. Under low shear, glycoprotein (GP) VI can interact directly with collagen. Engagement of GPVI with collagen initiates platelet activation. Platelet activation increases the affinity of α2β1 for collagen, which induces firm platelet adhesion (Holtkötter et al., 2002; Nieswandt and Watson, 2003). Platelet activation results in platelet intracellular calcium mobilization, release of granule content, and activation of αIIbβ3 (Lecut et al., 2004; Nieswandt and Watson, 2003). Activated αIIbβ3 mediates platelet–platelet interactions via fibrinogen bridges to build a stable thrombus.
In addition to the release of granular content, platelets also mobilize normally ER-resident proteins to their surface (Holbrook et al., 2010). Many of these ER proteins belong to the protein disulfide isomerase (PDI) family of proteins, which catalyze the reduction, oxidation, and rearrangement of disulfide bonds. Since the first isolation of PDIA1 in the 1960s (Goldberger et al., 1963; Venetianer and Straub, 1963), more than 20 members of the PDI family have been identified (Galligan and Petersen, 2012). Several of these proteins have been described to play a role in regulating platelet activation and thrombosis (Fig. 1). The PDI family members share the thioredoxin fold and contain catalytic and non-catalytic sites. For example, the prototype member PDI (PDIA1) has two catalytic sites (a and a′) and two non-catalytic sites (b and b′). The catalytic site contains the CXXC sequence, which exists in dithiol, disulfide, and mixed disulfide states and is present in the majority of members. Most members have C-terminal ER localization motifs, for example, KDEL (Hatahet and Ruddock, 2007).
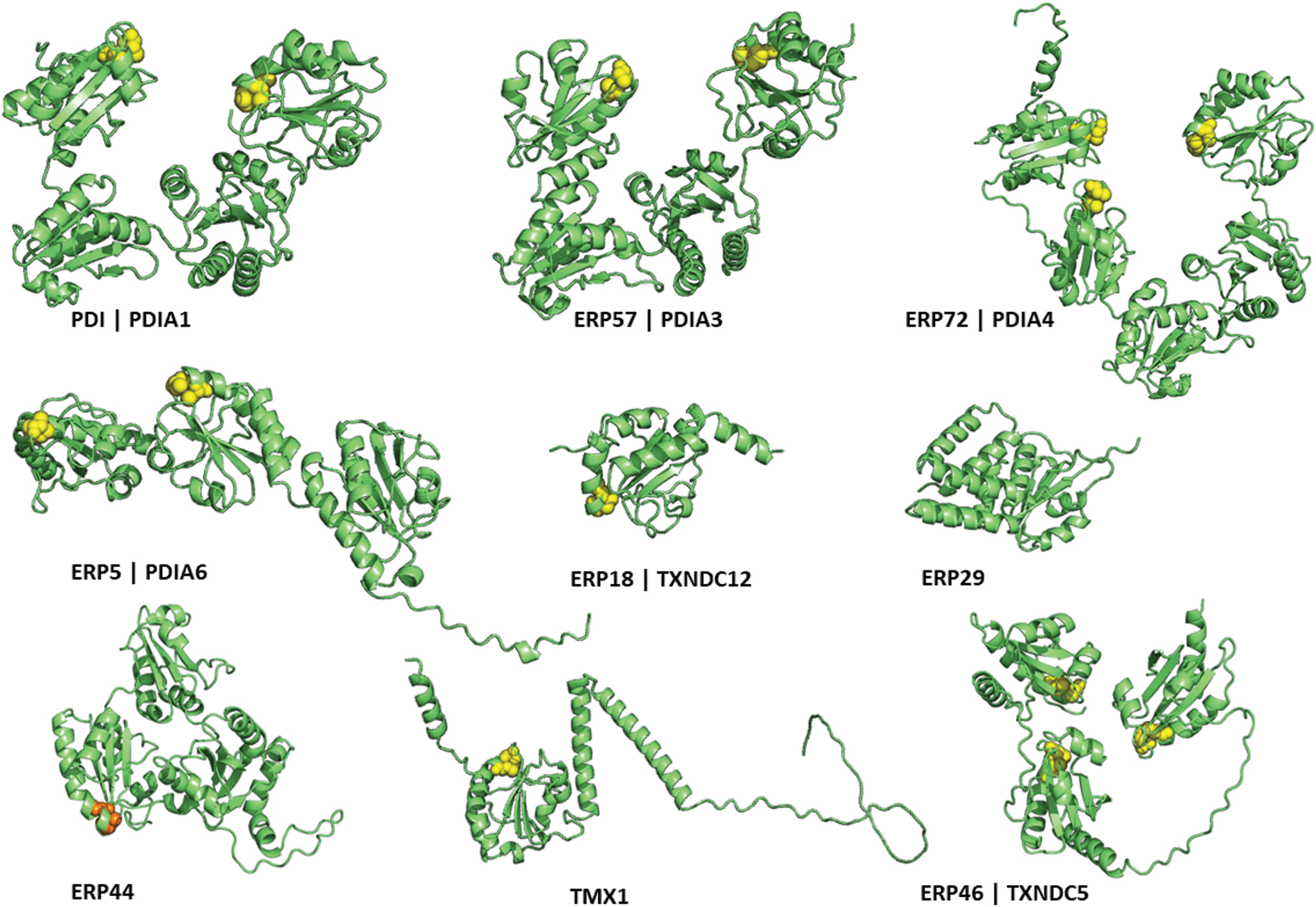
The structure of the PDIs influences their substrate specificity. The b′ domain of PDI (PDIA1) binds to delta-somatostatin, whereas the b′ domain of ERp57 (PDIA3) binds to the P-domains of calreticulin (CALR) and calnexin (Hatahet and Ruddock, 2007). ERp72 (PDIA4) associates with NOX1 via its N-terminus (Chen et al., 2008), whereas ERp5 (PDIA6) binds to the C-terminus of peroxiredoxin 4 (PRDX4) via its a0 domain (Sato et al., 2013). Identifying unique substrate partners for the PDI family members is challenging as they have broad specificities and multiple substrate-binding sites (Hatahet and Ruddock, 2007).
PDIs have been shown to be involved in retrotranslocation of misfolded proteins to the cytosol for ubiquitination and degradation by the proteasome. PDI (PDIA1) enabled cholera toxin's retrotranslocation, whereas ERp72 (PDIA4) mediated its retention in the ER (Forster et al., 2006). A complex of PDI (PDIA1), ERp5 (PDIA6), ERp72 (PDIA4), and glucose-regulated protein 78 (GRP78) occludes the cytosolic face of the Sec61 translocon, preventing the translocation of prion protein (Stockton et al., 2003). It is unknown if PDI “piggyback” proteins secreted to the extracellular space as family members are consistently mobilized to the surface and releasate of different cells, including platelets (Holbrook et al., 2010). The action of these proteins on the platelet surface predominantly affects the activation of platelet surface receptors. This directly facilitates platelet intercellular communications through ligand–receptor interactions.
However, the precise mechanisms of how these ER-resident proteins migrate to the platelet surface are yet to be determined. Evidence from studies of other cell lines suggests that the mobilization of ER proteins may be linked to perturbations in the ER environment, including changes in ER redox homeostasis, ER calcium content, and proteostasis. In this review, we will examine the current understanding of the role of the platelet ER and its contributions to platelet activation, focusing on redox regulation, calcium homeostasis, and platelet protein synthesis. We will draw on evidence from other models to describe potential mechanisms of the escape of ER proteins to the platelet surface, and how this affects platelet intercellular interactions. Finally, we will discuss alterations in platelet ER in disease states and how the platelet ER can be manipulated for therapeutic benefit.
Platelet ER: Master Regulator of Platelet Response and Activation
Despite their anucleate structure, platelets have a developed ER and functional translational machinery including ribosomes and Golgi apparatus (Ts'ao et al., 1971; Yadav et al., 2017). The platelet ER consists mainly of smooth ER, which, in the platelet, is called the dense tubular system. Platelets also contain an elaborate network of intracellular membranes connected to the surface for protein transport inside and out of platelets (called the open canalicular system) (Fig. 2A). Approximately 100–1000 platelets can form from a megakaryocyte. Platelets are produced by proplatelet extensions formed by megakaryocytes and direct budding from the megakaryocyte body (Ellis et al., 2023; Patel et al., 2005; Potts et al., 2020). Megakaryocytes transfer mitochondria, granular content, and PDIs through the proplatelet shaft and into the terminal buds (Crescente et al., 2016; Italiano and Shivdasani, 2003). A study of mouse megakaryocytes and platelets identified glucose-6-phosphatase (an ER marker) in both, supporting the megakaryocyte origin of the platelet dense tubular system (Daimon and Gotoh, 1982).
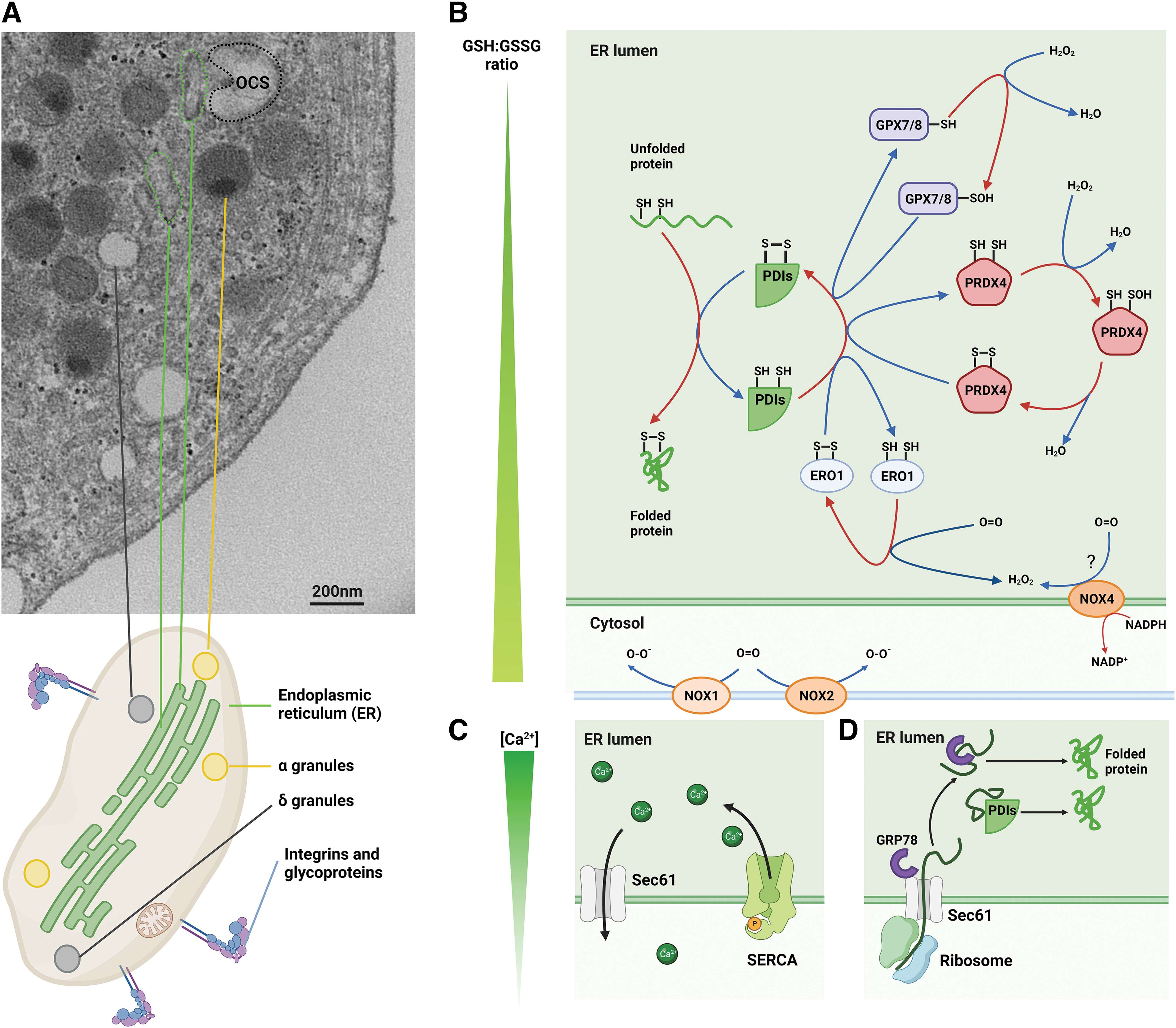
The platelet ER differs from nucleated mammalian cells. In nucleated cells, there is both rough ER and smooth ER. The rough ER is studded with ribosomes and has a primary function in protein folding of ∼7500 proteins (Hatahet and Ruddock, 2007). The smooth ER is the primary site of calcium storage and phospholipid synthesis, among other functions. The platelet contains predominantly smooth ER (initially identified by its peroxidase activity) (White, 1972) and only remnants of the rough ER (Gresele et al., 2017). However, the rough ER appears in nascent platelets following recovery from thrombocytopenia, further supporting its transfer from the parent megakaryocytes (Ts'ao, 1971).
The platelet dense tubular system is a dynamic structure that changes in morphology following platelet activation. In the resting platelet, electron microscopy studies show that the dense tubular system exists as thin and elongated structures, intertwined, but not continuous with the open canalicular system (Van Nispen Tot Pannerden et al., 2010). This positioning may be critical for regulating local release of calcium, membrane fusion, and shape change. A membrane complex has been described in platelets where the dense tubular system is coupled tightly to a portion of the open canalicular system. A functional Ca2+ nanodomain is contained within the membrane (Anand and Harper, 2020). The link of the open canalicular system to the extracellular space may serve to rapidly transfer extracellular signals for local release of calcium from the ER (Van Nispen Tot Pannerden et al., 2010).
In the activated platelet, the dense tubular system changes morphology within seconds to minutes after activation and becomes rounded and centralized in the platelet (Chung et al., 2021; Ebbeling et al., 1992). In the activated platelet, the dense tubular system remains closely associated with the open canalicular system (Chung et al., 2021), and this may facilitate the movement of PDI family proteins to the platelet surface after stimulation. In addition, the dense tubular system has also been proposed to contribute to the increased platelet surface area after activation, as the decrease in open canalicular system area does not appear sufficient to explain plasma membrane expansion based on scanning electron microscopy studies (Pokrovskaya et al., 2021).
One of the most important functions of the ER is the maintenance of an oxidizing environment to facilitate protein folding by the formation of disulfide bonds. This balance is maintained against the reducing environment of the cytoplasm by the function of oxidants including ER oxidoreductin 1 (ERO1), PRDX4, glutathione peroxidase 7 and 8 (GPX7, GPX8), and NADPH oxidase (NOX) members (Fig. 2B). PDI (PDIA1) is reduced as a consequence of catalyzing oxidative folding of polypeptides in the ER. ERO1 reoxidizes PDI (PDIA1) by transferring electrons from PDI (PDIA1), predominantly to oxygen, with hydrogen peroxide generated as a product of this reaction. Therefore, oxidative folding of proteins is linked to hydrogen peroxide generation.
Another major function of the ER is storage of calcium. Calcium is pumped from the cytoplasm into the ER by the sarcoendoplasmic reticulum calcium ATPase (SERCA). Calcium re-enters the cytoplasm through the inositol 1,4,5-triphosphate receptor (IP3R) and various “leak” channels including Sec61 (Fig. 2C). The increase in platelet cytosolic calcium is essential for platelet shape change (Paul et al., 1999), cytoskeletal reorganization (Ariyoshi and Salzman, 1996), and granule release (Flaumenhaft, 2003).
The platelet ER is a major site of lipid biosynthesis including many of the platelet-activating mediators. Thromboxane A2 is an important lipid mediator produced from arachidonic acid, which activates platelets via the thromboxane A2 receptor. The production of thromboxane A2 is controlled by thromboxane synthase in the ER and its synthesis is dependent on ER homeostasis (Spitler et al., 2013) and redox status (Lagarde et al., 2018). For example, treating platelets with the PDIA1 inhibitor bepristat 2a resulted in reduced platelet thromboxane A2 in response to collagen or thrombin stimulation (Przyborowski et al., 2022). Increased cytosolic calcium due to ER store release is also associated with thromboxane A2 production (Astarie-Dequeker et al., 1995). This suggests that multiple functions of the ER have an overlapping role in mediating biosynthetic regulation in platelets.
Platelets utilize their ER for de novo protein synthesis in response to physiological and pathological changes, such as sepsis or platelet activation (Lindemann et al., 2001a; Middleton et al., 2019). Platelets are highly secretory cells, with proteins inherited from the parent megakaryocytes, taken up from the extracellular space and produced de novo in the circulating platelets. Surface-bound and secreted proteins in cells are typically translocated into the ER shortly after synthesis. There, the nascent peptides undergo folding, post-translational processing, and subsequent transport into secretory granules or to the cell surface. Platelet ribosomes have been found in the cytoplasm and associated with the small amounts of rough ER (White, 1972). Several proteins have been found to be synthesized de novo in platelets, which may contribute to thrombus formation and stability (Brogren et al., 2004; Panes et al., 2007; Savini et al., 2007).
The platelet ER is at the intersection between maintaining intracellular function and regulating platelet responses to external stimuli. In the following sections, we present the current understanding of the role of (i) the platelet ER redox environment (Fig. 2B); (ii) platelet ER calcium store (Fig. 2C); and (iii) protein synthesis in the ER (Fig. 2D), as mediators of platelet function.
The Platelet ER Redox Environment
The ER membrane provides spatial separation between the oxidizing environment within the ER lumen from the reducing environment in the cytosol. Glutathione is thought to be the main redox buffer in the endoplasmic reticulum, with the ratio of reduced (GSH) and oxidized (GSSG) glutathione contributing to the ER redox poise (Hatahet and Ruddock, 2009; Hwang et al., 1992). The oxidizing environment in the ER is maintained by the consumption of GSH as an electron donor and accumulation of GSSG, leading to a decreased GSH:GSSG ratio in the ER relative to the cytosol (Fig. 2B). GSSG accumulates as a consequence of multiple reactions in the ER, including: (i) PDI-mediated disulfide reduction or isomerization; (ii) unfolding of misfolded proteins in the ER for retrograde transport into the cytosol for degradation; and (iii) GPX-mediated clearance of hydrogen peroxide (Margittai et al., 2015).
The concentration of GSH in the ER is ∼10 mM (Hatahet and Ruddock, 2007). The oxidative status of the ER was first estimated in 1992 using a cell-permeable tetrapeptide, which estimated the GSH:GSSG ratio to be 1:1 to 3:1 in CRL 1606 hybridoma cells (Hwang et al., 1992). Estimates of the platelet ER oxidative status appear to be based on this original publication (Essex, 2009). Using newer fluorescent protein probes in cell lines (e.g., HeLa, fibroblasts), the ER redox potential was reported as −225 mV (Hudson et al., 2015).
Oxidative folding of proteins in the ER is performed by multiple ER chaperones that work cooperatively. These include PDI family members and non-disulfide isomerase chaperones such as CALR, calnexin, and GRP78 that are present in platelets. One human platelet expresses ∼32,000 PDI (PDIA1) molecules (Kim et al., 2013). Approximately 5%–10% of total PDI (PDIA1) is released from platelets after activation (Cho et al., 2012). Overexpression of PDI (PDIA1) results in threefold increase in basal hydrogen peroxide production in vascular smooth muscle cells in an NOX1-dependent mechanism (Fernandes et al., 2009). Inhibition of PDI (PDIA1) in platelets with bepristat 2a resulted in reduced reactive oxygen species (ROS) generation within the platelet (Bekendam et al., 2016; Przyborowski et al., 2022). This suggests that PDI (PDIA1) is essential to maintain the redox environment within the platelet.
Another redox-active intracellular protein in platelets is ERp5. ERp5 (PDIA6) interacts with GRP78 to assist in ER protein folding. In hyperoxidizing conditions, misfolded proteins become associated with GRP78, while the strength of the non-covalent ERp5 (PDIA6)-GRP78 interaction is decreased in reducing conditions. Therefore, ERp5 may play a role as a co-chaperone in assisting GRP78 in refolding misfolded proteins in hyperoxidizing conditions (Jessop et al., 2009). Depletion of intracellular ERp5 is associated with disruption of ER homeostasis and upregulation of other PDI members (Jordan et al., 2005; Lay et al., 2023).
The absolute quantity of PDIs may contribute to the redox environment in the ER. PDIs become reduced by catalyzing oxidative protein folding in the ER. They are reoxidized via several pathways, of which the most conserved is the ERO1 pathway.
ERO1 isoforms ERO1α and ERO1β are found in humans and are essential in maintaining redox homeostasis and regulating proteostasis in the ER. Both ERO1α and ERO1β have been identified in human platelets (Burkhart et al., 2012; Huang et al., 2021), with ERO1α function shown to promote platelet activation (Jha et al., 2023). ERO1 is a flavin adenine dinucleotide binding protein that uses molecular oxygen as an electron acceptor to catalyze oxidation of PDI (PDA1) and therefore lead to PDI (PDIA1)-mediated oxidative protein folding (Tu and Weissman, 2002) (Fig. 2B).
ERO1β has been reported to have greater in vitro enzymatic activity compared with ERO1α and is specific for the a′ domain of PDIA1 (Wang et al., 2011). Data for the role of ERO1α in oxidizing non-PDIA1 PDIs are variable. ERp57 (PDIA3) has been described to be a substrate for the Saccharomyces cerevisiae ERO1 analogue, ERO1p, in vitro (Kulp et al., 2006). Immunoprecipitation studies have identified that ERp57 (PDIA3), PDI (PDIA1), and ERp5 (PDIA6) associate with ERO1α in thrombin-activated human platelets (Jha et al., 2023). The reoxidation of other PDIs may be performed by PRDX4, GPX, and quiescin sulfhydryl oxidase (Bulleid and Ellgaard, 2011).
PRDX4 is detected in platelets (Wolny et al., 2023) and is reduced by PDI family members including PDI (PDIA1), ERp46 (TXNDC5), ERp5 (PDIA6), and ERp57 (PDIA3) (Tavender et al., 2010). Oxidation of PDI (PDIA) can be mediated by PRDX4 in a redox relay reaction from hydrogen peroxide (H2O2) (Tavender et al., 2010). However, knockdown of PRDX4 in a fibrosarcoma cell line did not result in changes to the redox status of PDI (PDIA1), ERp57 (PDIA3), ERp72 (PDIA4) (Tavender et al., 2008). GPX7 and GPX8 have been shown to increase the efficiency of PDI (PDIA1)-mediated oxidative protein folding, and in particular, GPX7 increases the rate of ERO1α-mediated PDIA1 oxidation (Kanemura et al., 2020).
NOXes are specialized enzymes for the production of ROS (Prieto-Bermejo and Hernández-Hernández, 2017), which play an important role in the ER redox. For example, of the seven NOXes, NOX4 is present in the ER (Helmcke et al., 2009), whereas NOX1 and NOX2 interact with PDIs and chaperones in the ER. PDI (PDIA1) binds and transfers p47phox to the membrane promoting the assembly of NOX2 (de A Paes et al., 2011). PDI (PDIA1) overexpression induces NOX1 and NOX4 overexpression in smooth muscle cells (Fernandes et al., 2021).
The above redox pathways are likely to operate in platelets and influence platelet function. Whereas the function of GPX7/8 has not been described in platelets, GPX3 deficiency promotes platelet-dependent thrombosis in vivo (Jin et al., 2011). Patients with diabetes have lower platelet GPX activity (Véricel et al., 2004). NOX1 and NOX2 are present in human platelets and regulate their function (Delaney et al., 2016). Platelet PDI (PDIA1) and NOX1 cooperatively contributed to GPVI signaling that involved the phosphorylation of p38 mitogen-activated protein kinase (MAPK), p47phox, protein kinase C (PKC), and Akt (Véricel et al., 2004). NOX1 increases platelet ROS production after stimulation with the G protein-coupled receptor-dependent agonists (Delaney et al., 2016). In addition, NOX4 mRNA is found in human platelets, and NOX4 protein has been identified in murine platelets (Delaney et al., 2016). However, a NOX4 knockout murine model did not demonstrate change in platelet aggregation in response to thrombin or collagen (Vara et al., 2021).
Redox reactions are rapid, whereas the downstream effect of their activation of transcription factors will take longer. The hydroxyl radical (the most powerful oxidant among the ROS) has a very short lifetime (few 10−6 s), whereas the effect of transcription factors may persist for minutes to hours (slow dynamics) (Swift and Coruzzi, 2017). Another time element for platelets is that ER redox responses to agonist stimulation are rapid (seconds), whereas ER redox imbalance in conditions of oxidative stress (i.e., the metabolic syndrome) is chronic. The oxidative burst in platelets occurs nearly instantaneously after stimulation with thrombin or arachidonic acid but occur after a lag of 25 s after stimulation with collagen with production of endoperoxides but not superoxide radicals (Bressler et al., 1979).
The ER redox status is subjected to local and systemic generation of oxidants. H2O2 is produced from Ero1α-PDI (PDIA1) protein folding and NOX. H2O2 reversibly oxidizes cysteine residues on enzymes and transcription factors controlling their activity (Lennicke and Cochemé, 2021). ROS produced from mitochondrial respiration localize to the redox contact “triangle” of ER, mitochondria, and peroxisomes (Chaube and Werstuck, 2016; Yoboue et al., 2018). Therefore, locally produced ROS can inhibit SERCA by S-oxidation of Cys674 (Tong et al., 2010b). ERO1α oxidizes stromal interaction molecule 1 (STIM1) and SERCA2, increasing cytosolic Ca2+ levels and thus promoting platelet activation and aggregation (Jha et al., 2023).
The ER is also subjected to non-endogenous production of oxidants, which when produced in excess are referred to as “oxidative stress.” These can reach the platelet ER from other sources such as endothelial cells or circulating lipids. Excessive vascular superoxide production has been demonstrated in hypercholesterolemia (Mugge et al., 1991). Superoxide promotes lipid peroxidation leading to the generation of lipid alkoxyl and peroxyl radicals. Systemic inflammation in cardiovascular disease can be an additional source of exogenous oxidants for platelets. Soluble CD40L mediates stimulation-induced platelet release of ROS through activation of Akt and p38 MAPK signaling pathways (Freedman, 2008). ROS produced by neutrophil oxidative burst are released extracellularly potentially affecting neighboring platelets (Silvestre-Roig et al., 2020).
It is clear that there are multiple players involved in the ER redox status in platelets. Many of these players have been found to modulate the activity of the main regulators of intracellular calcium flux.
The ER Calcium Store
Calcium is maintained at ∼10,000-fold greater concentration in the ER than the cytosol. Multiple members of the PDI family and ER chaperone proteins act as buffering molecules for calcium and maintain the ER calcium homeostasis. CALR and GRP78 are thought to contribute to ∼50% and 25% of the calcium buffering capacity of the ER, respectively (Fig. 3A) (Prins and Michalak, 2011). The calcium gradient across the ER membrane is maintained by a mixture of active release from the ER stores by IP3R, reuptake by SERCA, and “leak” of calcium from the ER via channels, including the Sec61 translocon complex (Fig. 3B).
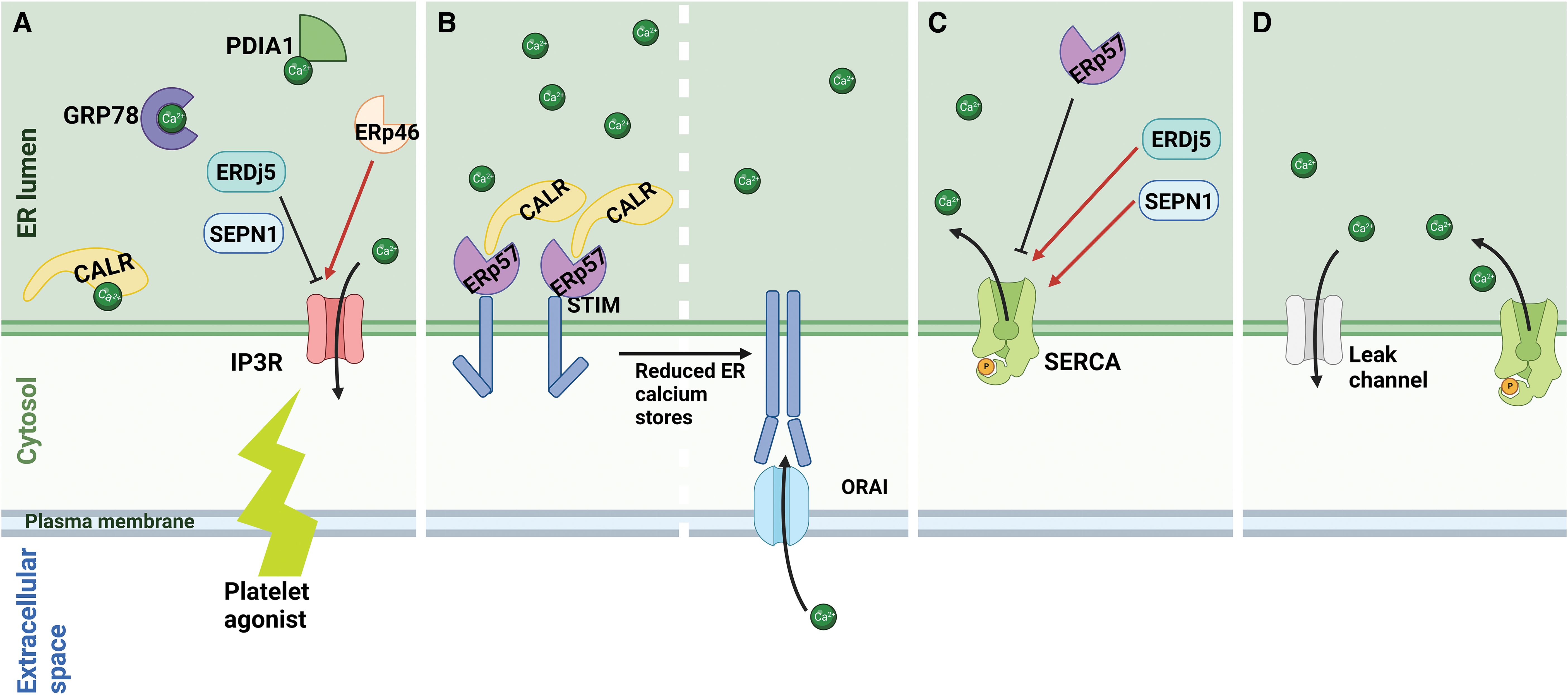
The IP3R is one of the key channels involved in calcium mobilization from the ER in platelets. Upon platelet activation, IP3R is activated downstream of phospholipase C, and calcium is released from the ER into the cytosol. Activity of the IIP3R is regulated by ER redox proteins: ERp46 (TXNDC5), which oxidizes and activates; whereas ER disulfide reductase 5 (ERDj5) reduces and inhibits IP3R-mediated calcium release (Fig. 3A). This occurs via oxidation or reduction of the conserved Cys2505 and Cys2513 residues of IP3R, near the channel pore on the ER luminal surface (Fujii et al., 2023).
Decrease in intracellular calcium stores, such as after platelet activation, results in calcium uptake from the extracellular space in a process known as store-operated calcium entry (SOCE). This process occurs due to dissociation of calcium from the EF-hand domain of STIM1 in the calcium-deplete ER, leading to STIM1 activation. The activated STIM1 undergoes oligomerization and closely associates with the ORAI channel to form the activated STIM1-ORAI complex. Calcium taken up through this complex is then pumped into the ER by SERCA to replenish calcium stores.
STIM1 contains five conserved cysteine residues that can undergo redox modification to regulate its activity. The cysteine residues are located within the signal peptide (Cys5), upstream of the EF-hand domain (Cys49 and Cys56), in the transmembrane domain (Cys227) and in the cytosolic domain (Cys437) (Bhardwaj et al., 2016). ERp57 (PDIA3) via its interaction with CALR inhibits SOCE in a process dependent on its interaction with the STIM1 ER luminal Cys49 and Cys56 residues (Fig. 3B) (Prins et al., 2011). Cys437 in STIM1 is within the STIM1-ORAI-activating region and is S-acylated following ER calcium depletion. Cys437 acylation facilitates SOCE by assisting in the colocalization of STIM1 with ORAI (Kodakandla et al., 2022). STIM1-deficient murine platelets have reduced calcium mobilization from intracellular stores after agonist stimulation and form smaller and less stable thrombi (Varga-Szabo et al., 2008).
The SERCA pump is the only active calcium transporter of the ER and is encoded by three genes, SERCA1, SERCA2, and SERCA3, with alternative slicing generating more than 10 isoforms (Periasamy and Kalyanasundaram, 2007). Of these isoforms, SERCA2b and SERCA3 have been shown to function in platelets (Enouf et al., 1992; Wuytack et al., 1994). The activity of SERCA2b in high ER calcium situations is inhibited by the formation of a disulfide bond at the intraluminal loop 4 of SERCA2b mediated by ERp57 (PDIA3) (Li and Camacho, 2004). In situations of low ER calcium, ERp57 (PDIA3) dissociates from SERCA2b, allowing the pump to activate.
The activity of the pump is increased by the reduction of the intraluminal disulfide bond by ERDj5 and selenoprotein N (Fig. 3C) (Chernorudskiy et al., 2020; Ushioda et al., 2016). In contrast, SERCA3, although also found to colocalize with ERp57 (Crescente et al., 2016), has been shown to be more resistant to oxidative inactivation compared with SERCA2b (Grover et al., 1997). Recently, ERO1α has been shown to regulate platelet cytosolic calcium by directly interacting with STIM1 and SERCA2 (Jha et al., 2023), thus providing a link between the redox environment and calcium homeostasis in the platelet ER.
The activity of SERCA is balanced by “leak” from the ER through various channels, including the Sec61 translocon, leucine-rich repeat-containing protein 8B (LRRC8B), mistsugumin 23 (MG23), presenilins, and pannexins. The Sec61 translocon (Fig. 3D) has been proposed to be one of the main sources of passive calcium leak from the ER (Parys et al., 2022). While many of the ER leak channels have been identified at the protein or mRNA level in platelets (Huang et al., 2021; Wright et al., 2016), their effect on platelet function is unknown.
Protein Synthesis in the Platelet ER
Despite being anucleate in humans, platelets contain the requisite components for protein synthesis, including mRNAs, ribosomes, and regulatory factors necessary for translation initiation and control. The first evidence for de novo platelet protein synthesis arose from the observation that radiolabeled amino acids were incorporated into proteins in intact platelets in the 1960s (Booyse and Rafelson, 1967; Warshaw et al., 1967). This incorporation of labeled amino acids was inhibited by puromycin, a translation inhibitor, but not actinomycin D, a transcription inhibitor. Both radiolabeled amino acids and radiolabeled carbohydrates are incorporated into newly synthesized glycoproteins in platelets, thus necessitating the presence of a functional endoplasmic reticulum for glycoprotein synthesis (Zimmerman and Weyrich, 2008).
Exposure of platelets to agonists such as thrombin results in production of proteins including interleukin 1β and B cell chronic lymphoma factor 3 (bcl-3), which have roles in inflammation and clot retraction, respectively (Lindemann et al., 2001a; Zimmerman and Weyrich, 2008). Platelet activation regulates the intracellular distribution of mRNAs and the mRNA-binding protein eukaryotic initiation factor 4E, thus offering a mechanism for signal-dependent protein synthesis in platelets (Lindemann et al., 2001b).
Nascent peptides that are destined for the cell surface or secretion are translocated into the endoplasmic reticulum for folding and post-translational processing (Fig. 2D). This process is dependent on the Sec61 translocon, a heterotrimeric complex that associates with polyribosomes and delivers the newly synthesized polypeptide into the ER. The Sec61 translocon has also been proposed to act as a calcium leak channel (Figs. 2C and 3D) (Lang et al., 2017). This may provide an additional mechanistic link between the calcium homeostasis in the ER and its protein folding capacity. However, there have been no studies to date describing the functional significance of the Sec61 translocon in platelets.
Although PDI family members play an important role in catalyzing oxidative protein folding in the ER, whether changes in their content lead to alterations in total platelet protein synthesis is unknown. Depletion in specific platelet PDIs can variably lead to upregulation of other family members in some models (Kim et al., 2013; Lay et al., 2023) but not others (Zhou et al., 2022). This could potentially be explained by overlap in the client peptides for disulfide isomerases. Further studies are needed to ascertain the effect of altered PDI content on de novo platelet protein synthesis as a result to external stimuli.
Perturbation of Platelet ER Homeostasis and ER Stress
Platelets maintain ER homeostasis by a tight regulation of redox balance, calcium, and protein folding. ER redox, calcium homeostasis, and ER protein folding function are intrinsically linked. For example, ER proteins that play an essential role in protein synthesis, such as GRP78, PDI (PDIA1), CALR, and glucose-regulated protein 94 (GRP94), also act as reservoir and buffering molecules for calcium to maintain ER calcium homeostasis (Fig. 3A). While calcium affects the chaperone activity of GRP78, CALR, calnexin, and PDI (PDIA1); calcium does not affect PDI (PDIA1) reductase/oxidase activity (Primm et al., 1996). However, alterations in calcium may indirectly affect PDI (PDIA1) reductase/oxidase activity: calcium depletion sequesters PDI (PDIA1) due to association with CALR and therefore reduces the available PDI (PDIA1) for protein folding (Avezov et al., 2015). ER calcium depletion is associated with reduced ER folding capacity and ER stress (Lebeau et al., 2021; Preissler et al., 2020).
Given platelets can respond to the extracellular environment and stimuli via de novo protein synthesis, there is a variable load of nascent polypeptides that require folding in the ER. The ER can adapt to changes in protein folding load; however, physiological insults can cause the ER capacity to be overwhelmed and lead to “ER stress” (Jain et al., 2022). Platelet ER stress is a recently described mechanism of platelet activation (Jain et al., 2022; Lay et al., 2023). ER stress-induced oxidation of IP3R sensitizes it to calcium release and therefore ER calcium depletion. In platelets, this may lead to increased activation and granule secretion (Paul et al., 1999).
The primary role of ER stress is to address excessive protein folding and reduce the unfolded protein burden in the ER. This is achieved by the unfolded protein response (UPR) that decreases the synthesis of proteins globally, but specifically increases the synthesis of selected proteins, such as chaperones and transcription factors, that restore ER homeostasis. In addition, abnormally folded proteins are degraded by translocation from the ER into the cytosol, and nascent proteins may undergo co-translational degradation to further reduce the protein folding load (Plumb et al., 2015).
There are three branches of the UPR mediated by the ER sensors inositol-requiring enzyme 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Fig. 4). An increase in unfolded proteins in the ER results in dissociation of GRP78 from the ER-luminal domains of IRE1, PERK, and ATF6. This frees GRP78 and allows it to participate in protein folding, but additionally relieves the inhibitory effect of GRP78 on UPR signaling (Wang et al., 2009).

Although UPR has been more completely studied in nucleated cells, anucleate platelets contain proteins involved in UPR pathways. Given platelet phenotype is altered with UPR despite mammalian platelets lacking a nucleus (Jain et al., 2022; Lay et al., 2023), this suggests that platelet activation by ER stress is likely due to changes in platelet ER redox balance, calcium stores, or proteostasis.
IRE1 homodimerizes upon UPR activation and undergoes autophosphorylation, which results in the activation of its kinase and RNAse activities. The IRE1 RNAse cleaves unspliced X-box binding protein 1 (XBP1) mRNA to its spliced form. XBP1s mRNA is subsequently translated to a potent transcription factor that enhances the production of various ER proteins aimed at relieving the UPR, including GRP78 (Acosta-Alvear et al., 2007; Gebert et al., 2021; Pramanik et al., 2018). In addition, the IRE1 RNAse is thought to degrade other mRNAs and microRNAs to reduce the translational burden, in a process called IRE1-dependent decay (RIDD).
PDI family members modulate the IRE1 pathway activation and its sequelae (Fig. 4A). In addition to its role as a co-chaperone for GRP78, ERp5 (PDIA6) also modulates the ER stress response by attenuating the RNase activity of IRE1, thus inhibiting RIDD (Eletto et al., 2016; Eletto et al., 2014). Phosphorylation of PDI (PDIA1) at Ser357 appears to be an early event in ER stress, with phosphorylated PDI (PDIA1) interacting directly with IRE1 to suppress sustained IRE1 signaling (Yu et al., 2020).
PERK activation results in phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (eIF2α), which attenuates translation globally and therefore reduces the unfolded protein burden. Phosphorylated-eIF2α also enhances the translation of a select subset of mRNAs, including activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) (Palam et al., 2011). ATF4 and CHOP exert their function by altering gene transcription in nucleated cells, but their functions in anucleate platelets are unknown. PDI family members also affect PERK activation, in addition to modulating the IRE1 pathway (Fig. 4B). PDI (PDIA1) and ERp57 (PDIA3) appear to play opposing roles on PERK; ERp57 maintains PDIA1 in the reduced state, and depletion of ERp57 results in increased PDI (PDIA1) oxidation and PERK signaling (Yu et al., 2020). Loss of ERp5 (PDIA6) has also been shown to augment PERK activation (Eletto et al., 2014).
ATF6 exists as two isoforms, ATF6α and ATF6β, predominantly located in the ER. ATF6 signaling after UPR activation occurs via trafficking and subsequent cleavage in the Golgi to a soluble 50 kDa fragment (ATF6-f) that acts as a transcription factor (Fig. 4C) (Oka et al., 2022). In nucleated cells, ATF6-f induces the expression of ER stress response proteins, including GRP78 and XBP1 (Haze et al., 1999; Yoshida et al., 2001). Oxidative stress induction in platelets in vitro results in increased ATF6-f (Jain et al., 2022), although further studies are needed to determine the role of ATF6-f in the anucleate platelet. ERp18 (TXNDC12) has been shown to interact with ATF6 only during ER stress, to specifically reduce an interchain Cys467-Cys467 disulfide in the ATF6 dimer to regulate ATF6 trafficking to the Golgi, where it can be further cleaved into the active ATF6 (Oka et al., 2022; Oka et al., 2019). mRNA transcripts for ERp18 (TXNDC12) are highly expressed in megakaryocytes (Holbrook et al., 2010), although a specific role of ERp18 in platelets has not yet been identified.
Misfolded proteins driving ER stress are transported from the cytosol into the ER for degradation (Fig. 4D), in a process known as ER-associated degradation (ERAD). This is dependent on ERp46 (TXNDC5) triggering the mannose trimming activity of ER Degradation Enhancing Alpha-Mannosidase Like Protein 3 (EDEM3) through covalent interaction at its redox site in situations of ER stress (Yu et al., 2018). Whether ERp46 (TXNDC5) depletion in platelets alters activity because of reduced capacity to activate ERAD, particularly in pathological states that trigger platelet ER stress, remains unknown.
Secretory cells may be more prone to ER stress due to the increased burden of protein synthesis to generate secreted proteins. Until recently, there has been little data regarding the contribution of ER stress to platelet activity, and most of the evidence for ER stress in blood cells comes from studies of lymphocytes. Activation of the IRE1 pathway promotes type-2 helper cell activation-induced proliferation and cytokine secretion (Pramanik et al., 2018). B cell differentiation into plasma cells, which are highly secretory cells producing large quantities of immunoglobulins, appears to be dependent on the activation of the IRE1 pathway and XBP1 splicing (Brewer and Hendershot, 2004).
SEC61A1, the pore-forming subunit that is essential for the function of the Sec61 translocon, is a transcriptional target of XBP1s and strongly induced from B cell differentiation into plasma cells (Schubert et al., 2018). In addition to inducing Sec61 translocon expression, ER stress in differentiating B cells also lead to ER expansion. This increase in ER size appears to alleviate ER stress in a mechanism dependent on ER lipid biosynthesis but independent of ER chaperone expression (Schuck et al., 2009). In B cells, this expanded ER is accompanied by increased ER calcium mobilization mediated by the Sec61 translocon complex (Pick et al., 2021).
How Does ER Stress Regulate Platelet Function?
Like plasma cells, platelets are also highly secretory cells with protein cargo inherited from the parent megakaryocyte, as well as produced in vivo in response to physiological stimuli. The role of specific UPR pathways in platelet function has only recently been studied in mouse megakaryocyte-specific knockout models. PERK and XBP1 (downstream of IRE1) knockout in megakaryocytes resulted in increased platelet apoptosis, platelet protein aggregates, and activation compared with wild type. In contrast, ATF6 knockout resulted in increased platelet activation and protein aggregation but did not increase the proportion of apoptotic platelets compared with wild type (Jain et al., 2022). ER proteins including GRP78 and GRP94 were also found to be upregulated in response to UPR derangements due to the knockout of select UPR pathways. These findings suggest that the different branches of the UPR play different roles in platelet biogenesis and platelet activation.
Although ER chaperone proteins and PDIs are typically increased in response to UPR activation, alterations to intracellular ER protein content or function may also affect UPR activation (Fig. 4). Disruption of the platelet ER by deletion of PDIs is accompanied by upregulation of other ER redox and chaperone proteins in a likely compensatory mechanism. We have identified that the deletion of ERp5 (PDIA6) in platelets was accompanied by upregulation of ERp57 (PDIA3), PDI (PDIA1), and the activation of IRE1. GRP78 depletion is associated with ER stress (Borok et al., 2020), which is unsurprising given its role in associating with ER stress sensors and preventing inappropriate ER stress activation. This suggests that ER-resident proteins, at least both GRP78 and ERp5, act as a “brake” for inappropriate ER signaling.
ER stress results in the selective upregulation of ER chaperone proteins that help to reduce the unfolded protein load. In platelets, these ER-resident proteins are present in the ER but also migrate to the cell surface upon platelet activation. These extracellular ER chaperones and PDIs have essential roles in platelet activation, thrombus formation, and intercellular communication. While many of the platelet PDIs are involved in prothrombotic mechanisms, there is a family member, thioredoxin related transmembrane protein 1 (TMX1), which localizes to the ER and the cell membrane and is associated with an antithrombotic effect on the platelet surface (Table 1) (Zhao et al., 2019).
Endoplasmic Reticulum Chaperone Proteins Have Multiple Roles in the Intracellular Space, Including Redox Control in Protein Folding, Maintaining Endoplasmic Reticulum Calcium Homeostasis, and Modulating Endoplasmic Reticulum Stress Responses. However, Data on the Specific Intracellular Roles of These Proteins in Platelets are Limited
4-PBA, 4-phenylbutyrate; ATF6, activating transcription factor 6; CALR, calreticulin; EDEM3, ER Degradation Enhancing Alpha-Mannosidase Like Protein 3; eIF2α, eukaryotic translation initiation factor 2 subunit alpha; ER, endoplasmic reticulum; ERAD, ER-associated degradation; ERO1, endoplasmic reticulum oxidoreductin 1; ERp5, endoplasmic reticulum protein 5; ERp29, endoplasmic reticulum protein 29; ERp46, endoplasmic reticulum protein 46; ERp57, endoplasmic reticulum protein 57; GRP78, glucose-regulated protein 78; GRP94, glucose-regulated protein 94; GSH, glutathione; Ig, immunoglobulin; IP3R, inositol 1,4,5 triphosphate receptor; IRE1, inositol-requiring enzyme 1; PDI, protein disulfide isomerase; PDIA1, protein disulfide isomerase A1; PDIA3, protein disulfide isomerase A3; PDIA4, protein disulfide isomerase A4; PDIA6, protein disulfide isomerase A6; PERK, PKR-like ER kinase; RIDD, IRE1-dependent decay; SERCA, sarcoendoplasmic reticulum calcium ATPase; SOCE, store-operated calcium entry; TMX1, thioredoxin related transmembrane protein 1; TXNDC5, thioredoxin domain-containing protein 5.
NOXes and PDIA1 share parts of the same signaling pathways in ER stress responses. NOX4 and NOX2 are signaling elements in the UPR during ER stress, with NOX4 playing a prosurvival or proapoptotic role, whereas NOX2 enhances proapoptotic signaling (Laurindo et al., 2014). PDI (PDIA1) supports growth factor-dependent NOX1 activation, and PDI (PDIA1) overexpression induces acute spontaneous NOX activation (Fernandes et al., 2021). Upregulation of NOX4 inhibits SERCA (Tong et al., 2010a). Are NOXes increased in ER stress? There is evidence showing that under stress conditions such as ER stress, ROS production is increased via NOX2 and NOX4 (Santos et al., 2014). ER stress upregulates the expression of NOX4 and NOX1 in vascular smooth muscle cells of spontaneous hypertensive rats (Camargo et al., 2018).
Further evidence from perturbation of ER stress affecting platelet function has been seen with in vitro treatment of platelets with chemical ER stress inducers and ER chaperones. Platelets exposed to the SERCA inhibitor thapsigargin mobilize calcium to the cytosol (Lebeau et al., 2021), develop ER stress (Lay et al., 2023) and aggregate. Platelet treatment with the N-glycosylation inhibitor tunicamycin is also associated with ER stress and markers of activation such as elevated P-selectin (Jain et al., 2022). It is not unexpected that in vitro induction of ER stress with thapsigargin, which inhibits reuptake of calcium into the ER, results in elevated cytosolic calcium.
However, other ER stress inducers, such as tunicamycin that does not have a direct role in disturbing ER calcium homeostasis, have also been shown to cause reduced ER calcium (Lebeau et al., 2021). ER calcium depletion causes destabilization of GRP78 interactions with its substrates and therefore disturbs proteostasis (Preissler et al., 2020). In contrast, chemical chaperones such as 4-phenylbutyrate (4-PBA) and tauroursodeoxycholic acid have been shown to alleviate ER stress and reduce thapsigargin- and tunicamycin-mediated ER calcium loss (Lebeau et al., 2021). In vitro treatment of platelets with 4-PBA has been associated with reduced agonist-induced platelet aggregation (Jain et al., 2022).
In summary, platelet ER redox environment, ER calcium homeostasis, and platelet ER stress are interdependent on each other, due to common proteins involved in these pathways. A rise in platelet cytosolic calcium, either as a direct or indirect result of these three mechanisms, leads to activation of platelet surface receptors and release of mediators for intercellular communication. In addition, ER chaperone proteins and PDIs are also externalized to the platelet surface or released as a result of platelet activation, which also play a role in the platelet interaction with its environment.
ER Proteins on the Platelet Surface: Escape from a Stressed Cell or Orderly Release?
Many ER-resident proteins, including PDI (PDIA1), ERp5 (PDIA6), ERp29, ERp46 (TXNDC5), ERp57 (PDIA3), ERp72 (PDIA4), and TMX1, are mobilized to the platelet surface after agonist stimulation. These proteins participate in the redox control of platelet surface integrin function, most importantly integrin αIIbβ3 (Fig. 5A).
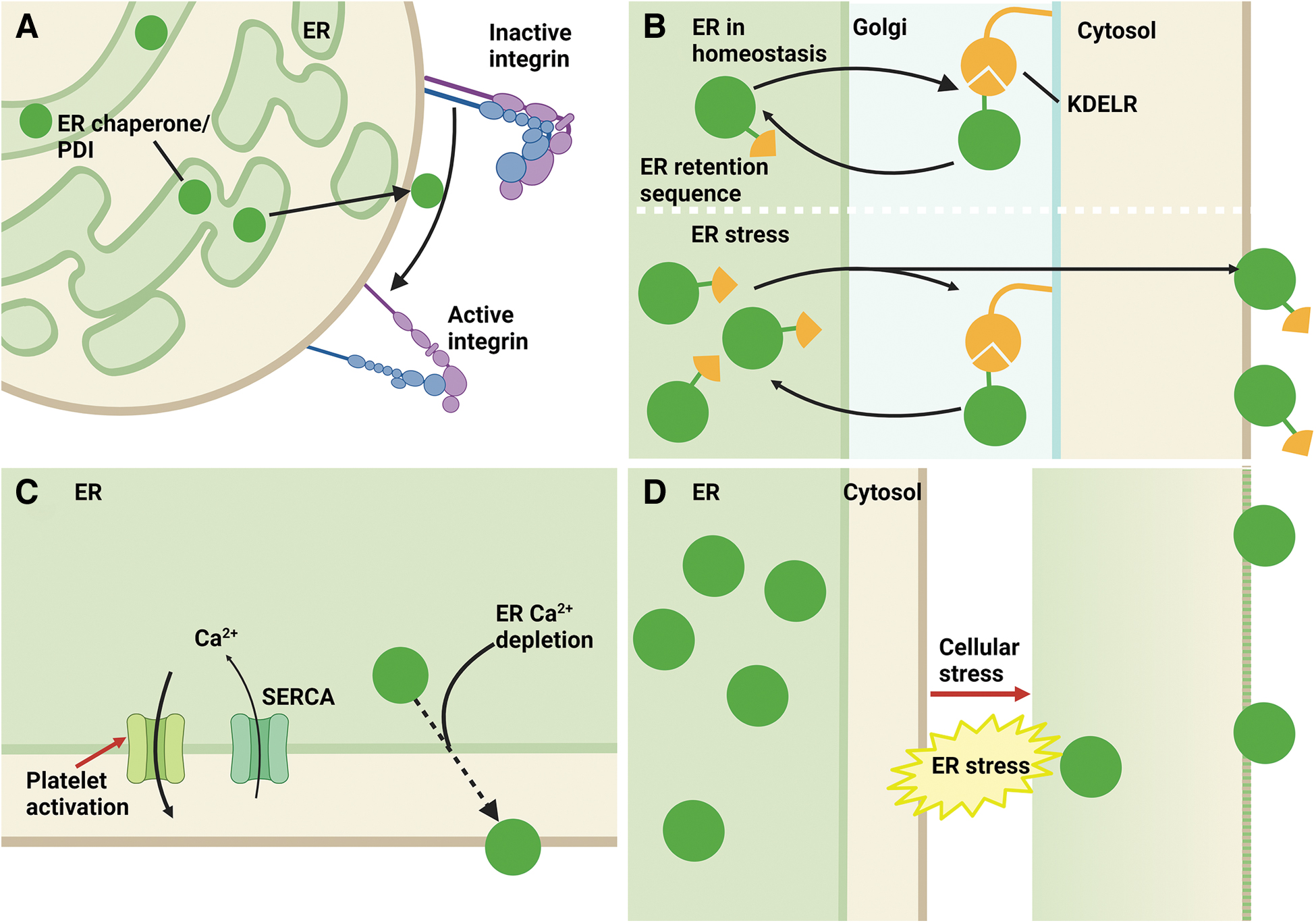
Whether platelet ER PDIs migrate to the cell surface via the same mechanism as seen in other cell lines is unknown. Many of the ER proteins have a KDEL or related ER retention sequence, which allows retrieval of these proteins from the Golgi to the ER, as there is greater affinity for the KDEL sequence to its receptor (KDELR) in the acidic environment of the Golgi. One proposed mechanism for the cellular escape of ER PDIs is that upregulation of these may overwhelm the retrieval capacity of KDELRs and thus lead to cell surface expression (Fig. 5B). ER stress may also play a role in this pathway, as two of the three isoforms of KDELR (KDELR2 and KDELR3) appear to be targets of XBP1s, the transcription factor that is produced downstream of IRE1 pathway activation. Silencing RNA knockdown of KDEL2 significantly reduced the secretion of an ER-resident protein, mesencephalic astrocyte-derived neurotrophic factor (Trychta et al., 2018).
The pattern of externalization of these ER-resident proteins onto the platelet surface differs. ERp29, ERp44, ERp57 (PDIA3), and ERp72 (PDIA4) are mobilized to the platelet surface after thrombin activation at 1 U/mL, but there is a reduction in platelet surface ERp29, ERp44, and ERp57 (PDIA3) at high thrombin concentration of 5 U/mL, consistent with either shedding of these proteins or internalization at higher agonist concentrations (Holbrook et al., 2010). This is in contrast with the different pattern of ERp5 (PDIA6) exposure on the platelet. ERp5 (PDIA6) increases in a concentration-dependent manner to convulxin, collagen, and thrombin stimulation. Although convulxin and thrombin results in a time-dependent increase in platelet surface ERp5 (PDIA6), collagen stimulation results in a peak and subsequent decline in platelet surface ERp5 (Jordan et al., 2005). TMX1, an antithrombotic member of the PDI family of proteins, is increased on the platelet surface after thrombin 1 U/mL (Zhao et al., 2019), but whether the increase is agonist- or time dependent is unknown.
Given agonist stimulation of platelets converges on increased cytosolic calcium as a mediator of downstream signaling, it is plausible that externalization of these ER proteins occurs in a calcium-dependent process (Fig. 5C). This may also be supported by the colocalization of ER proteins, such as PDI (PDIA1) and ERp57 (PDIA3), with SERCA-dependent calcium stores (Crescente et al., 2016). It is challenging to compare the effect of platelet calcium flux on externalization of PDIs between reports, as agonist doses and experimental conditions vary. In addition, the calcium flux after different agonists, for example, thrombin compared with convulxin, differs in terms of absolute peak, time to peak, and duration of the “plateau” phase in cytosolic calcium concentration (Nagy et al., 2018).
We have shown that ERp5-deficient (PDIA6) platelets have increased platelet ER stress and increased calcium mobilization after platelet stimulation with the thromboxane A2 receptor agonist U46619 and increased secretion of PDI family proteins constitutively and after agonist stimulation (Lay et al., 2023). In other cell lines, ER calcium depletion results in externalization of ER-resident proteins in a process known as “exodosis” (Trychta et al., 2018). Further studies are warranted to determine if different ER-resident proteins mobilize to the platelet surface at different cytosolic calcium concentrations, as this may be a potential mechanism to regulate the presence and ratio of different PDIs on the platelet surface.
Some ER proteins such as CALR and ERp57 (PDIA3) have been described as damage-associated molecular patterns, particularly in the setting of tumor cells, as a signal for immunogenic cell death (Wiersma et al., 2015). Cellular stress, for example, induced by cytotoxic therapy, results in CALR and ERp57 (PDIA3) exposure on the cell surface in an ER stress-dependent manner (Panaretakis et al., 2008). Lipotoxicity has been shown to be associated with ER stress in multiple tissues (Han and Kaufman, 2016). In vitro induction of lipotoxicity in Chinese hamster ovary cells resulted in dilated ER morphology with GRP78 and PDIA1 escape into the cytosol, potentially due to impaired ER membrane integrity (Fig. 5D) (Borradaile et al., 2006). Evidence against indiscriminate membrane disruption as the cause of ER protein externalization includes the lack of membrane continuity between open canalicular system and the dense tubular system seen in resting platelets by electron tomography (Van Nispen Tot Pannerden et al., 2010), and the centralization of the dense tubular system within the activated platelet by 2D STORM imaging (Chung et al., 2021).
ER stress may affect other cellular homeostasis mechanisms, such as response to oxidative stress, that normally contribute to maintaining the intracellular location of ER chaperones. For example, pretreatment of a murine colorectal cell line (CT 26) with antioxidants N-acetyl cysteine or GSH, introduction of mutant eif2α that is unable to be phosphorylated and therefore activated, or depletion of PERK resulted in reduction of surface CALR and ERp57 (PDIA3) after mitoxantrone (a cytotoxic anthracycline) treatment (Panaretakis et al., 2009). This increase in surface CALR and ERp57 (PDIA3) appears to be independent of cellular membrane derangement due to oxidative or ER stress, as other proteins such as GRP78, PDI (PDIA1), and GRP94 were not increased.
In other cell lines, induction of ER stress upregulates ER proteins and results in the externalization of a subset of the upregulated proteins. In vitro ER stress induction with tunicamycin and thapsigargin resulted in increased intracellular and surface GRP78 in multiple cell lines; however, GRP78 was not detected in the releasate from these cells (Dorners et al., 1990; Zhang et al., 2010). In contrast, exogenous overexpression of ER-resident proteins such as GRP78 and ERp72 (PDIA4) (Dorners et al., 1990) is not associated with ER stress but is associated with the specific increased release of the overexpressed protein, but not other ER-resident proteins. These two findings suggest that mobilization of ER-resident proteins to the cell surface can occur via various mechanisms, including ER stress and overproduction of the protein.
ERp5 (PDIA6) knockout platelets had constitutively increased secretion of PDI (PDIA1), ERp57 (PDIA3), and ERp72 (PDIA4), with similar resting cytosolic calcium compared with wild-type platelets (Lay et al., 2023). It is possible that the increased production of ERp57 (PDIA3), ERp72 (PDIA4), and GRP78 in these megakaryocytes and platelets in response to ER stress overwhelmed the ER retention capability of the KDELR and thus resulted in increased secreted ER proteins in this setting (Fig. 5B).
The affinity of the ER retrieval sequence for the KDELR may also play a role. For example, ERp29 contains a C-terminal KEEL motif, which has a lower affinity for the KDELR and therefore ERp29 is more likely to be exposed on the extracellular surface or secreted (Bikard et al., 2019). Cell-based assays and computational modeling have identified amino acids near the four C-terminal amino acids as potentially predictive of whether the protein will escape to the cell surface or be secreted secondary to ER calcium depletion (Trychta et al., 2021). Platelet surface ERp29 and ERp46 (TXNDC5) (KEEL and RDEL retrieval sequences, respectively) are less than that of ERp57 (PDIA3) and ERp72 (PDIA4) (QEDL and KEEL retrieval sequences, respectively) after thrombin stimulation (Holbrook et al., 2010), and this suggests that there are mechanisms beyond affinity of the ER retention signal and saturation of the KDELR in ER protein escape.
Further studies are required to answer the question of how externalization of ER proteins on the platelet surface occurs, and the regulatory mechanisms that contribute. In addition, knockout of one ER PDIs may lead to multiple compensatory changes, including alterations in ER stress, calcium homeostasis, and upregulation of other ER proteins. Therefore, the interpretation of the mechanisms leading to their release requires careful interpretation.
Released ER Proteins: Mediators of Platelet Interactions?
Platelet surface ERp5 (PDIA6), ERp46 (TXNDC5), ERp57 (PDIA3), ERp72 (PDIA4), and PDI (PDIA1) interact with integrin αIIbβ3 to facilitate its action and modulate its binding to ligands such as fibrinogen. All three thioredoxin sites in ERp46 (TXNDC5) appear to contribute to αIIbβ3 activation and ATP release. Perhaps akin to the intracellular role of ERp46 (TXNDC5) in rapidly introducing disulfide bonds in immature peptides before PDI (PDIA1) “editing,” ERp46 (TXNDC5) introduces disulfides into αIIbβ3 before fibrinogen binding, and this is followed by fibrinogen-dependent action of PDI (PDIA1) on the β3 integrin (Zhou et al., 2022). ERp57 (PDIA3) is postulated to target different sites on αIIbβ3 compared with PDI (PDIA1), although the precise targets are unknown.
The second active site of ERp57 (PDIA3) is essential for the interaction between ERp57 (PDIA3) and β3 integrin on the platelet surface to facilitate platelet aggregation and accumulation in a thrombus (Wang et al., 2013). ERp72 (PDIA4) interacts with disulfide bonds in both αIIb and β3 integrins via its a and a′ active sites, with ERp72-deficient (PDIA4) platelets having reduced response to agonist induced aggregation, and reduced alpha and dense granule release (Zhou et al., 2017). Unlike ERp46 (TXNDC5), ERp57 (PDIA3), ERP72 (PDIA4), and PDI (PDIA1), which act upon and activate αIIbβ3, ERp5 (PDIA6) appears to have a regulatory role by reducing the affinity of αIIbβ3 to fibrinogen and fibrin.
Other substrates for the PDI family members have not been systematically identified. The most studied of these is PDIA1, which has been shown to interact with platelet surface GP1bα (Li et al., 2019), αvβ3 (Ponamarczuk et al., 2018; Swiatkowska et al., 2008) on endothelium and tissue factor to support platelet agglutination, thrombosis, and thromboinflammation. Substrates for ERp46 (TXNDC5) beyond αIIbβ3 have not yet been identified, although this has been postulated to include the α2β1 integrin as well as regulators of P-selectin and ATP secretion by platelets (Zhou et al., 2022). Both ERp57 (PDIA3) and ERp72 (PDIA4) play a role in directly activating the coagulation cascade, as although ERp57 (PDIA3) deficiency results in reduced platelet and fibrin deposition, mutant ERp57 (PDIA3) with an active second catalytic site has intact fibrin deposition (Zhou et al., 2014), and recombinant ERp72 (PDIA4) in a β3 null mouse cremasteric arteriole laser injury model results in increased fibrin but not platelet deposition (Zhou et al., 2017).
Alterations in Platelet ER Redox, Calcium, and Protein Homeostasis in Disease
There is evidence of ER dysregulation resulting in alterations of platelet function. Glycosylation is a conserved process that is essential for correct protein function and cellular interactions, and inhibition of glycosylation is associated with ER stress (Denzel et al., 2014). Patients with congenital disorders of glycosylation have been described to have variable hematological deficits, including abnormalities in platelet count or function, increased risk of bleeding, and thrombosis (Chang et al., 2018). Pathological states such as malignancy and diabetes mellitus (DM) have also been shown to result in defective N-glycosylation (Mammadova-Bach et al., 2020), and this may contribute to the increased thrombotic risk in these conditions.
In pathological states, such as cardiovascular disease, hyperlipidemia, and DM, platelets may be more prone to activate than normal, resulting in inappropriate thrombus formation and potentially life-threatening thrombotic complications. Potential mechanisms contributing to platelet hyperactivity include alterations in ER redox, platelet cytosolic calcium concentration, alterations in PKC signaling and platelet ER stress (Kaur et al., 2018).
Oxidant modifications of the PDI family may affect their intracellular and extracellular function (Fig. 6A). S-sulfenylation appears to be an intermediate step in the formation of disulfide PDI (PDIA1) in oxidative stress (Yang et al., 2023). S-nitrosylation inhibits platelet surface PDI (PDIA1) activity (Bekendam et al., 2018). In addition, SNO-PDI (PDIA1), SNO-ERp5 (PDIA6), and SNO-ERp57 (PDIA3) added exogenously to platelets inhibited platelet aggregation. Intracellularly, S-nitrosylation of PDI (PDIA1) has been linked to inhibition of its function; S-nitrosylation of PDI (PDIA1) inhibits its enzymatic activity, leads to the accumulation of polyubiquitinated proteins, and activates the UPR (Uehara et al., 2006).

Platelets from patients with DM have increased activation of the IRE1 pathway and evidence of protein aggregates, which are seen in ER stress (Jain et al., 2022). Whether platelet ER stress in DM occurs due to increased or dysregulated protein synthesis is unknown, and it is possible that platelet ER stress could be a downstream effect of dysregulated platelet ER calcium homeostasis or oxidative stress leading to disturbed ER redox balance.
DM is well known to induce oxidative stress in many tissues resulting in end-organ complications, and platelet oxidative stress is one proposed mechanism leading to platelet hyperactivity (Ferroni et al., 2004; Zhang et al., 2020). In a diabetic rat model, the expression of GRP78 was reduced, and PDI (PDIA1) was increased in platelets. This was associated with an increased in vivo thrombosis tendency and increased activation of the extrinsic coagulation cascade (Hernández Vera et al., 2012).
Although many studies investigating role of specific PDIs in platelet-knockout models have evaluated compensatory changes in other PDIs, this may be insufficient to determine the downstream effect on ER redox status. This is because the quantity of PDIs does not necessarily correlate with the redox status of these proteins. For example, while total PDI (PDIA1) is unchanged in cardiomyocytes from a streptozotocin-induced diabetic mouse model, the proportion of oxidized PDI (PDIA1) was significantly reduced, leading to a disturbed redox balance (Panaretakis et al., 2008). A similar mechanism, although not yet explored, could potentially lead to disturbed redox homeostasis in diabetic platelets.
Basal cytosolic calcium is increased in platelets from patients with type 2 DM (Randriamboavonjy et al., 2012). Platelets from these patients have reduced SERCA2 function via nitration of tyrosine 294 and 295 and alterations in calcium signaling (Randriamboavonjy et al., 2012). It is possible that the mobilization of platelet ER proteins to the surface is also altered and may be an additional mechanism of increased thrombosis in this condition (Fig. 6B).
Myeloproliferative neoplasms (MPNs) are malignant disorders of blood cells that are associated with increased risk of thrombosis. MPNs are associated with several genetic mutations, and ∼20% of patients carry mutations in CALR. Mutations in these cases result in a change to the C-terminus of the protein and loss of the KDEL sequence (How et al., 2019). Despite the CALR-ERp57 (PDIA3) interaction being mediated by the P domain rather than C-terminus of CALR, mutant CALR loses its association with ERp57 (PDIA3) and STIM1, leading to excessive SOCE in the megakaryocytes from the affected patients. This is thought to drive megakaryocyte proliferation seen in this disease (Di Buduo et al., 2020). However, the finding of dysregulated calcium homeostasis is not limited only to CALR-mutated MPNs. Transcriptomic analysis of platelets from patients with MPN with all mutational signatures has identified candidates involved in proteostasis, ER stress response, and calcium homeostasis (Mosale Seetharam et al., 2023; Shen et al., 2021).
Circulating PDIs are thought to be released by activated platelets and endothelial cells and appear to predominantly circulate in the reduced form (Oliveira et al., 2019). Given MPNs are associated with evidence of ER dysregulation, it is possible that changes on platelet surface PDI could contribute to the increased thrombosis seen in this condition. Thus far, platelet-surface PDIs in MPNs have not been specifically assessed. Plasma PDI (PDIA1) was elevated in patients with MPNs (Sharda et al., 2021). Endothelial cells carrying the JAK2 V617 mutation, present in 50%–95% of MPNs, have increased (PDI) PDIA1 release (Sharda et al., 2021). How plasma PDIA1 quantity relates to platelet surface PDIA1 is unknown. Healthy donors with low plasma PDIA1 did not have a difference in platelet aggregation compared with healthy donors with high plasma PDIA1 (Oliveira et al., 2019). It is therefore possible that platelet-surface PDI (PDIA1) is more relevant to platelet aggregation than circulating PDI, as demonstrated in platelet-specific PDIA1-deficient mice (Kim et al., 2013). Different effects are also possible due to the redox status of released PDI (PDIA1).
In both DM and MPNs, platelets show evidence of ER dysfunction and increased tendency to activate. Whether ER stress causes mobilization of ER chaperones and PDIs to the platelet surface has not yet been defined but may present an additional explanation for the increased thrombosis seen in these conditions.
Whereas the ER stress response is an attempt to restore homeostasis, this may not always be successful. ER stress failure has been identified as an ineffective response in metabolic diseases, such as diabetes and obesity, conditions associated with platelet hyperactivity (Fig. 6C). For example, excessive ROS and/or reactive nitrogen species induce sulfonation (SO3H) or S-nitrosylation of IRE1, which can decrease IRE1 ribonuclease activity. This thereby inhibits the production of XBP1s with subsequent failure to upregulate chaperones in response to the oxidative/nitrosative stress (Uehara et al., 2006). It is plausible that ER stress failure also affects platelet function in metabolic disease.
Future Directions
The platelet ER has multiple functions including redox homeostasis, calcium storage, and protein synthesis. Derangements in these aspects of ER function can result in ER stress and platelet activation. When platelets are activated, multiple processes occur including shape change, degranulation, and migration of ER proteins to the platelet surface to activate platelet receptors. Activated platelet surface receptors facilitate platelet intercellular interactions, leading to clot formation and modulation of inflammatory and immune responses.
How different pathological conditions initiate and sustain platelet ER stress need to be further clarified. Dysfunctional ER stress responses have been seen in chronic diseases including diabetes, obesity, and steatohepatitis, whereby downstream targets of the ER stress response pathways fail to be activated or are only partially activated. This ER stress response failure appears to contribute to end-organ dysfunction (Sasako et al., 2019). Whether ER stress response failure occurs in platelets, for example, in DM, leading to platelet dysfunction remains to be investigated. In addition, platelets may be more prone to ER stress failure than other cells, given the lack of a nucleus as a signaling target in the UPR. Further studies on this topic will require investigation of multiple downstream markers of each ER stress pathway.
Some of the key mediators in maintaining platelet ER homeostasis are members of the PDI family of proteins. Studies of these proteins predominantly focus on the impact of their extracellular depletion and the resultant effect on thrombosis, with the role of these molecules on regulating αIIbβ3 activation being a frequent theme. However, contribution of intracellular PDIs on platelet function and megakaryocyte development should also be considered, as many of these proteins play a role in cellular calcium homeostasis, proteostasis, and regulation of ER stress signaling. In addition, intracellular depletion of a PDI may lead to derangements in ER redox and calcium, which may cause downstream changes in platelet function independent of the extracellular function of these proteins.
The exact mechanisms by which PDI family members regulate platelet interactions via αIIbβ3 and other platelet surface molecules remain to be elucidated. ERp5 (PDIA6) has been shown to cleave the Cys177-Cys184 disulfide bond in β3 enabling fibrinogen detachment, whereas ERp46 (TXNDC5) cleaves the Cys473-Cys503 disulfide bond in β3; however, information on other members is required. In addition, data on the contribution of PDIs via non-αIIbβ3 substrates remain incomplete.
Targeting PDIs may provide novel antithrombotics with a safer bleeding profile compared with traditional agents. Elevated released or circulating PDIs have been found in various thrombotic disorders. In addition, in vitro and in vivo induction of ER stress is accompanied by externalization or release of PDIs. Therefore, in addition to targeting extracellular PDIs that mediate thrombosis, another therapeutic strategy could be to address signaling pathways that result in the externalization and release of these proteins. This may be increasingly pertinent as ER stress has recently been found to contribute to platelet hyperactivation, and chemical chaperones aimed at relieving ER stress may prove to be additional antithrombotic agents.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.