
Abstract
Aims:
Vascular smooth muscle cell (VSMC) ferroptosis is a pivotal event in the process of aortic dissection (AD), and a number of agents have a protective role against AD by inhibiting VSMC ferroptosis. While glycolysis is an ancient pathway related to almost all biological processes, its precise involvement in VSMC ferroptosis and AD remains unclear.
Results:
In this study, bioinformatics analysis revealed that glycolysis-related molecules and pathways were involved in VSMC ferroptosis and AD. We focused on the key enzyme of glycolysis, lactate dehydrogenase A (LDHA), and found that LDHA overexpression promoted ferroptosis and lipid peroxidation in cystine deprivation- or imidazole ketone erastin-treated VSMCs and vice versa. Clinical specimens showed a negative correlation between elevated LDHA levels in dissected aortae and ferroptosis-related molecules glutathione peroxidase 4 (GPX4), solute carrier family 7 member 11 (SLC7A11), and ferroptosis suppressor protein 1 (FSP1). In VSMC ferroptosis, LDHA overexpression led to the suppression of GPX4, SLC7A11, and FSP1. Furthermore, the interaction between LDHA and nuclear factor (erythroid-derived 2)-like 2 (NRF2) was identified, and the overexpression or agonist of NRF2 reversed the contribution of LDHA on VSMC ferroptosis and lipid peroxidation.
Innovation and Conclusion:
These results highlight a significant association between LDHA and VSMC ferroptosis in AD development mediated through NRF2. These findings present LDHA as a potential target for AD intervention by inhibiting its expression. Antioxid. Redox Signal. 42, 378–392.
Innovation
In summary, our study confirmed the link among glycolysis, VSMC ferroptosis, and AD and demonstrated that LDHA enhanced VSMC ferroptosis and lipid peroxidation via NRF2. Furthermore, our investigation revealed elevated LDHA expression in dissected human and mouse aortas, indicating that LDHA is a promising target for AD prevention and treatment.
Introduction
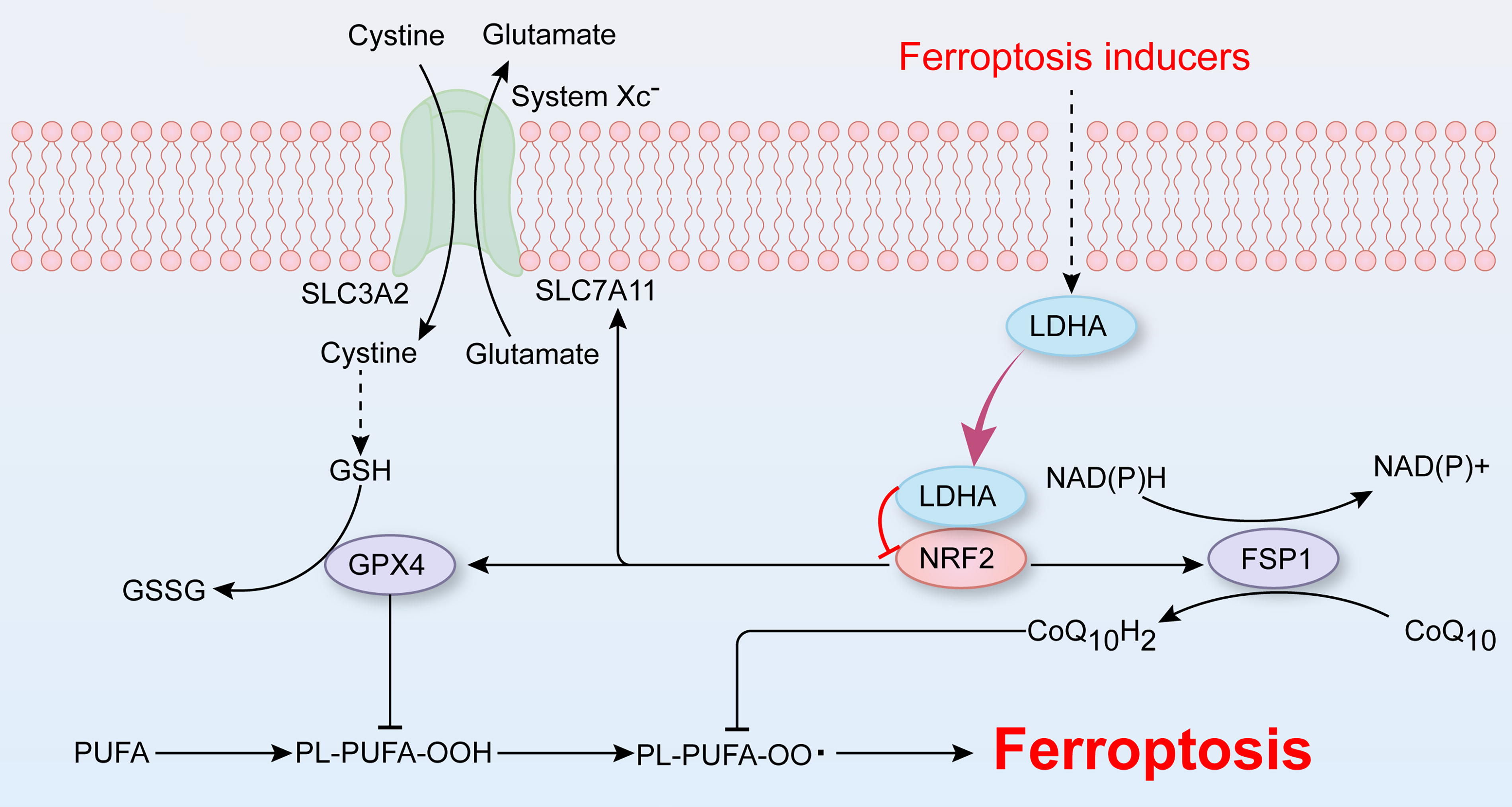
Aortic dissection (AD) is a life-threatening cardiovascular disease caused by disruption of the tunica intima, resulting in the separation of the layers of the aortic wall (Carrel et al., 2023). Our recent research revealed the participation of vascular smooth muscle cell (VSMC) ferroptosis in the development of AD (Chen et al., 2022; Li et al., 2022). Ferrous iron overload triggers the production of reactive oxygen species (ROS) via the Fenton reaction and leads to the accumulation of intracellular lipid peroxides, ultimately resulting in cell death, a process known as ferroptosis (Stockwell, 2022). In addition to iron overload, redox system imbalance serves as another catalyst for ferroptosis. The antioxidants glutathione and CoQ10H2 protect against ferroptosis by reducing lipid peroxidation and are synthesized through the system Xc−-glutathione peroxidase 4 (GPX4) and ferroptosis suppressor protein 1-coenzyme Q10 (FSP1-CoQ10) pathways, respectively (Jiang et al., 2021a, 2021b). Our recent investigations revealed the inhibition of these two ferroptosis-related pathways in the process of VSMC ferroptosis and AD (Chen et al., 2022). Treatment with the ferroptosis inhibitor BRD4770 has shown promise in mitigating aortic dilation and slowing disease progression by activating GPX4, SLC7A11, and FSP1 in a β-aminopropionitrile (BAPN)-induced mouse model of AD (Chen et al., 2022).
Previous research has demonstrated the critical involvement of glycolysis in both ferroptosis and AD (Ma et al., 2020; Sun et al., 2024). In Graves’ orbitopathy, the susceptibility of orbital fibroblasts to ferroptosis was increased by reducing glycolysis through pyruvate dehydrogenase kinase (PDK2) knockdown or treatment with dichloroacetic acid (Ma et al., 2022). Metabolic reprogramming in BAPN-treated macrophages leads to increased glycolytic metabolism and impaired mitochondrial oxidative phosphorylation, triggering vascular inflammation, extracellular matrix degradation, elastic plate breakage, and subsequent promotion of AD (Lian et al., 2019). As the core enzyme of glycolysis, lactate dehydrogenase A (LDHA) participates in cellular metabolism by converting pyruvate and nicotinamide adenine dinucleotide (NADH) to lactate and NAD+ (Feng et al., 2018). However, LDHA functions not only as a metabolism-associated enzyme but also as a noncanonical enzyme independent of lactate. Independent of its glycolytic activity, the activation of the small GTPase Rac1 by LDHA has been linked to breast cancer progression (Liu et al., 2022). In addition, the cooperation between LDHA and NADH has been shown to increase ROS production, activate the nuclear factor-kappa B (NF-κB) signaling pathway, and worsen chronic joint diseases such as osteoarthritis (Liu et al., 2022). Given its impact on oxidative stress, LDHA could affect glycolysis, VSMC ferroptosis, and AD, warranting further investigation.
In this study, we first examined the differentially expressed genes (DEGs) involved in AD and VSMC ferroptosis to investigate the role of glycolysis in both processes. This study focused on the pivotal glycolytic enzyme LDHA and its involvement in VSMC ferroptosis and AD. To mimic the intricate ferroptosis processes, cystine deprivation (CD), which disrupts the system Xc−-GPX4 pathway, and the ferroptosis inducer imidazole ketone erastin (IKE), which targets SLC7A11 and VDAC 2/3, were used to induce VSMC ferroptosis. We examined the associations between LDHA and the system Xc−-GPX4 and FSP1-CoQ10 pathways using aortic samples. Finally, the regulatory mechanism of LDHA on VSMC ferroptosis was described in detail. Collectively, these findings shed light on the role and potential mechanisms of LDHA in VSMC ferroptosis, suggesting that LDHA is a promising target for AD treatment (Fig. 1).
Results
Glycolysis-related pathways participate in the process of AD and VSMC ferroptosis
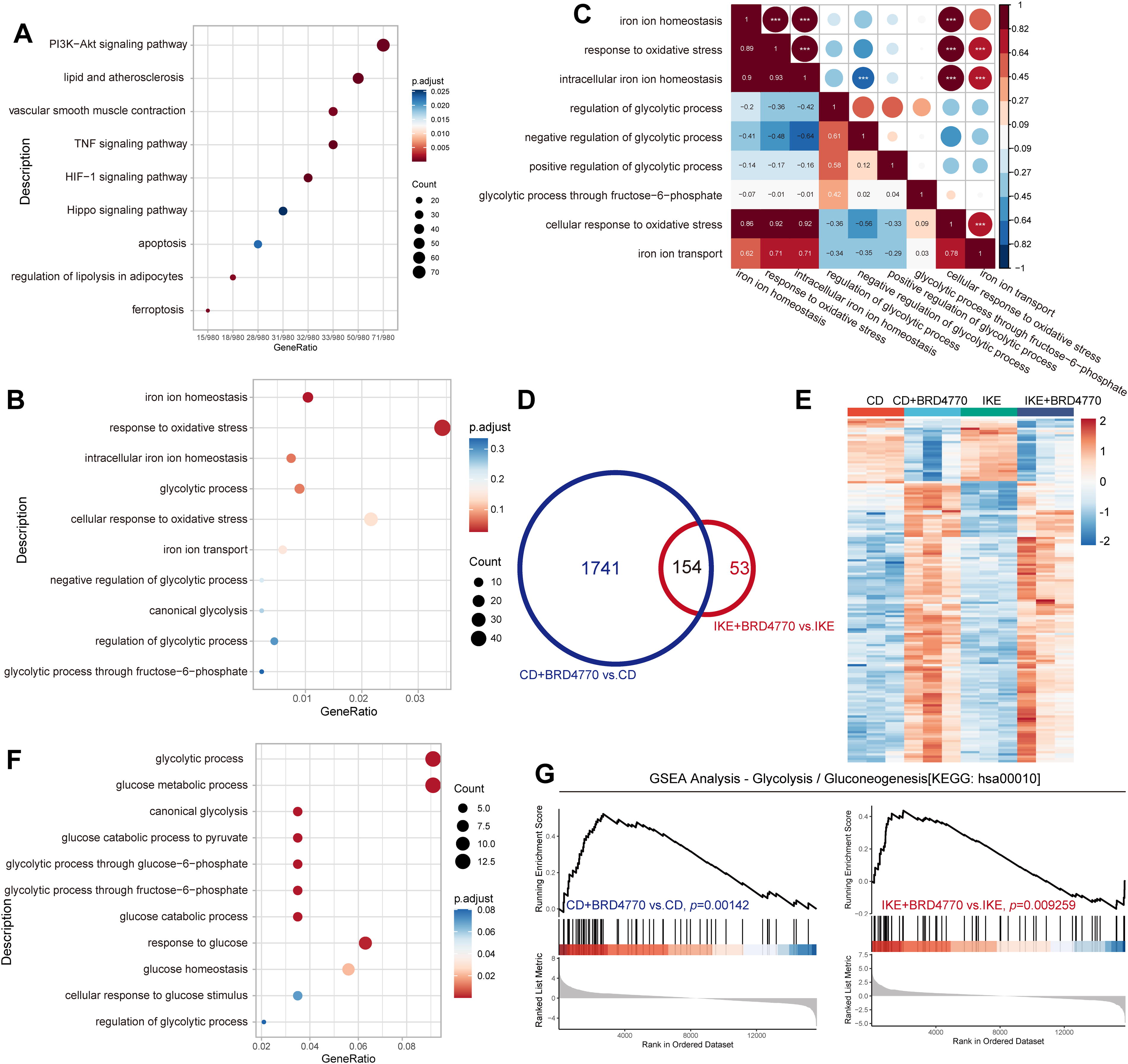
To identify genes implicated in AD, we reanalyzed the GSE153434 dataset, which comprises gene expression data of aortas from AD patients and controls obtained from the gene expression omnibus (GEO) database (Chen et al., 2021; Zhou et al., 2020). Bioinformatic analysis revealed 545 upregulated and 976 downregulated genes that were significantly differentially expressed between the AD aortas and normal aortas. As expected, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses highlighted ferroptosis and iron homeostasis, respectively, which is consistent with our prior findings (Fig. 2A and B). Within the biological process analysis of the DEGs, several molecules related to glycolysis and relevant pathways were enriched (Fig. 2B). Correlation analysis further revealed a negative relationship between the “intracellular iron ion homeostasis” and “negative regulation of glycolytic process” pathways, suggesting a potential link between ferroptosis and glycolysis (Fig. 2C). Building upon our earlier work demonstrating the protective effect of the histone methyltransferase inhibitor BRD4770 against CD- or IKE-induced ferroptosis in VSMCs, RNA sequencing was conducted to explore the mechanisms underlying the effects of BRD4770 on VSMC ferroptosis (Chen et al., 2022). This analysis revealed 1895 and 207 DEGs following BRD4770 treatment in CD- and IKE-induced ferroptosis, respectively (Fig. 2D). Additionally, 154 DEGs were coenriched upon BRD4770 treatment in both ferroptosis models (Fig. 2D and E). Multiple glycolysis-related pathways were highly enriched among these DEGs (Fig. 2F). Consistent with these findings, gene set enrichment analysis (GSEA) revealed significant differences between the indicated groups (Fig. 2G). According to the above datasets, glycolysis-related molecules and pathways are involved in VSMC ferroptosis and might play a role in the development of AD.
LDHA is upregulated in the aortas of patients and mice with AD
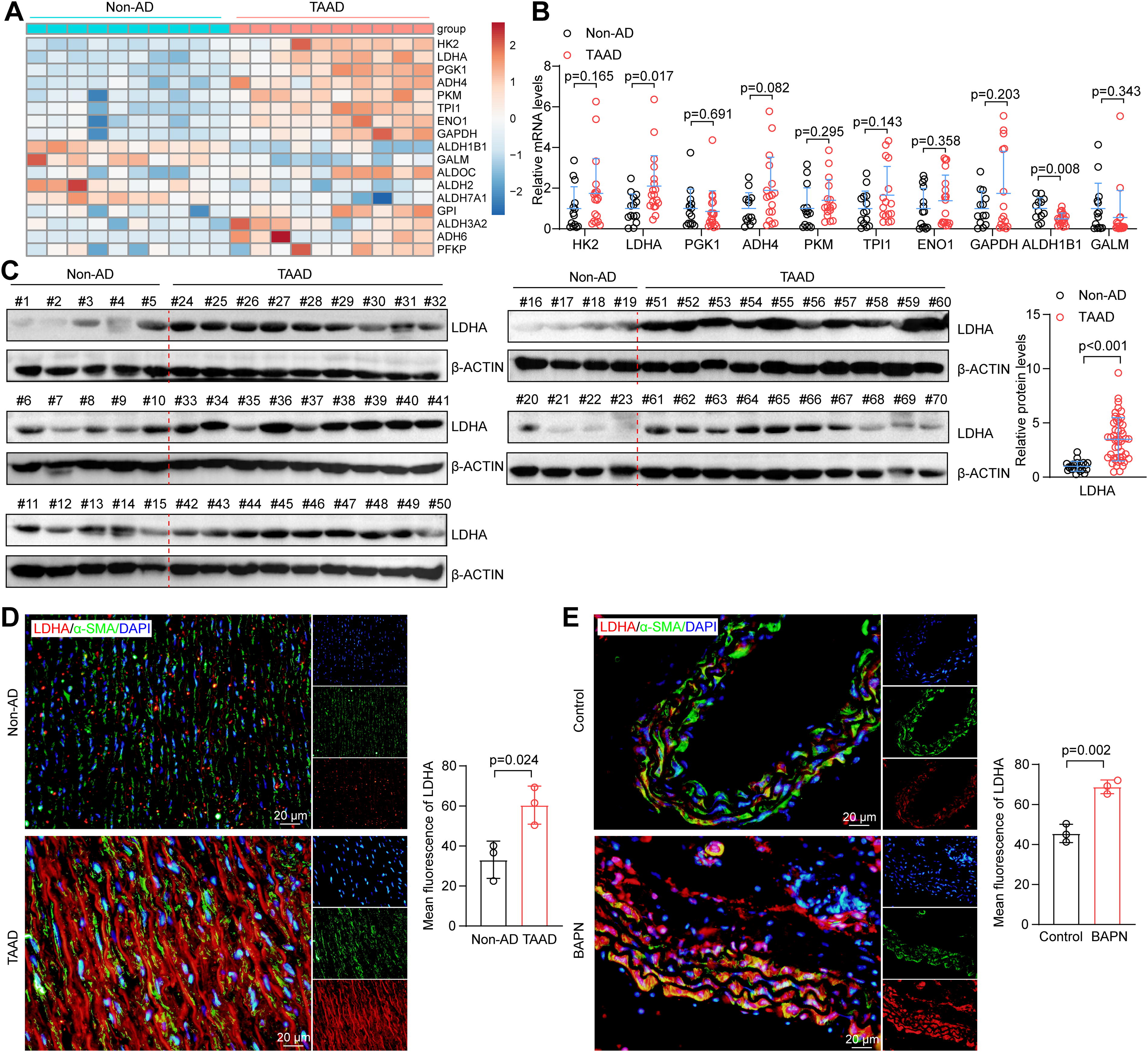
To elucidate the link among glycolysis, VSMC ferroptosis, and AD, the “glycolysis/gluconeogenesis” pathway of the GSE153434 dataset was reanalyzed, and 17 DEGs were identified (Fig. 3A). In the aortic tissues of non-AD and Stanford type A aortic dissection (TAAD) patients, the mRNA of the top 10 genes with significant changes was detected, and LDHA was the most prominently elevated (Fig. 3B). Therefore, we focused on LDHA, a pivotal member of glycolysis, for further research. Compared with those from non-AD aortas, dissected aortas from patients with TAAD exhibited dramatically increased LDHA protein expression (Fig. 3C). Immunofluorescence analysis revealed LDHA localization in both the cytoplasm and nucleus and greater LDHA expression in the TAAD aortas than in the non-AD samples (Fig. 3D). Similarly, in the BAPN-induced mouse model of AD, LDHA expression in the dissected aorta mirrored the clinical findings, showing a consistent upregulation of LDHA during AD development (Fig. 3E). This consistent increase in LDHA in dissected aortas from different species indicates a potential role for LDHA in AD development.
LDHA aggravates CD- or IKE-induced HASMC ferroptosis
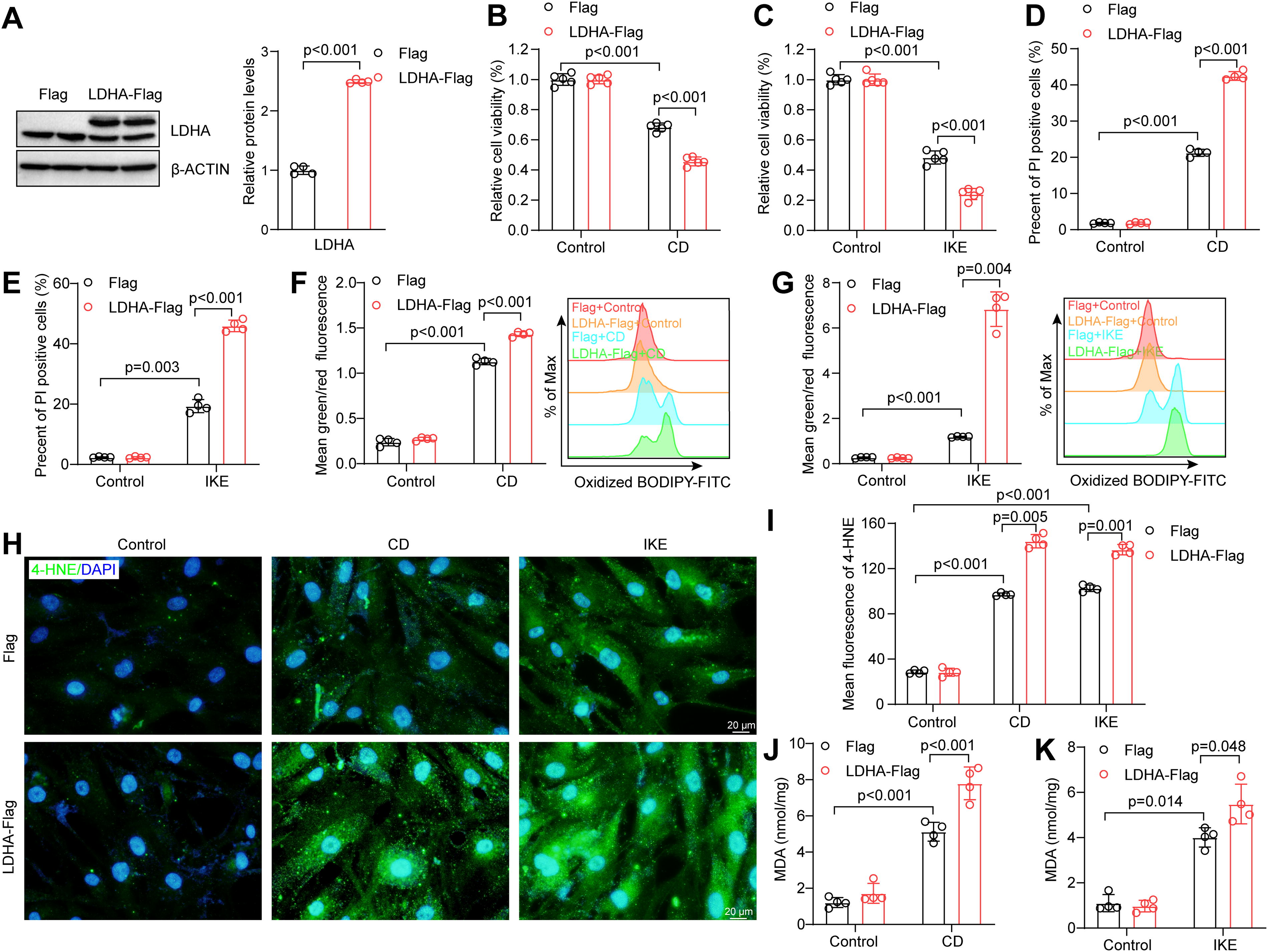
Based on the aforementioned results, we investigated the role of LDHA in ferroptosis by overexpressing it in human aortic smooth muscle cells (HASMCs) through lentivirus infection (Fig. 4A). As previously described, the medium with CD or IKE was used to induce ferroptosis in HASMCs (Chen et al., 2022). Overexpression of LDHA led to a significant decrease in cell counts compared with those of the controls in CD- or IKE-induced cell death (Fig. 4B and C). To validate these findings, propidium iodide (PI) staining was conducted and revealed an increase in the number of dead LDHA-overexpressing HASMCs upon CD or IKE treatment (Fig. 4D and E). Furthermore, multiple cell death-specific inhibitors, including emricasan for apoptosis, 3-methyladenine for autophagy, necrosulfonamide for necroptosis, and ferrostatin-1 for ferroptosis, were used to intervene the function of LDHA. As expected, only ferroptosis-specific inhibitor ferrostatin-1 reversed the CD- or IKE-induced cell death, whereas the other inhibitors had no effect on the process of cell death (Supplementary Fig. S1A and B), suggesting the cell death promoted by LDHA upon CD or IKE treatment is indeed ferroptosis. The accumulation of lipid peroxide is a hallmark of ferroptosis, as confirmed in HASMCs (Jiang et al., 2021a, 2021b). BODIPY 581/591 C11 is a sensor of lipid oxidation, and the ratio of 581–591 was elevated in CD- or IKE-induced HASMC ferroptosis. Overexpression of LDHA further elevated the ratio of oxidized C11-BODIPY during HASMC ferroptosis (Fig. 4F and G). Additionally, the levels of two lipid peroxides, 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), were consistently greater in the LDHA-overexpressing group than in the control group after treatment with CD or IKE (Fig. 4H–K). Overall, LDHA overexpression promotes VSMC ferroptosis and exacerbates lipid peroxidation.
The knockdown of LDHA alleviated VSMC ferroptosis
Subsequently, LDHA knockdown plasmids were constructed using the PLKO.1 vector, and LDHA-sh2 and LDHA-sh3 were selected based on their knockdown efficiency for further investigations into the role of LDHA in VSMC ferroptosis (Fig. 5A). As expected, compared with that in the PLKO.1 group, the cell count in the LDHA knockdown group was greater after CD- or IKE-induced ferroptosis (Fig. 5B and C). Moreover, LDHA knockdown reduced cell death following treatment with CD or IKE (Fig. 5D and E). Knockdown of LDHA affected lipid peroxidation and reduced oxidized C11-BODIPY during HASMC ferroptosis (Fig. 5F and G). Additionally, the levels of two lipid peroxides, 4-HNE and MDA, were notably lower in the LDHA knockdown groups than in the PLKO.1 group upon ferroptosis induction, suggesting that LDHA alleviated CD- or IKE-mediated lipid peroxidation (Fig. 5H–K). Therefore, LDHA knockdown has an antiferroptotic effect and protects against lipid peroxidation.

LDHA participates in the regulation of ferroptosis-related molecules
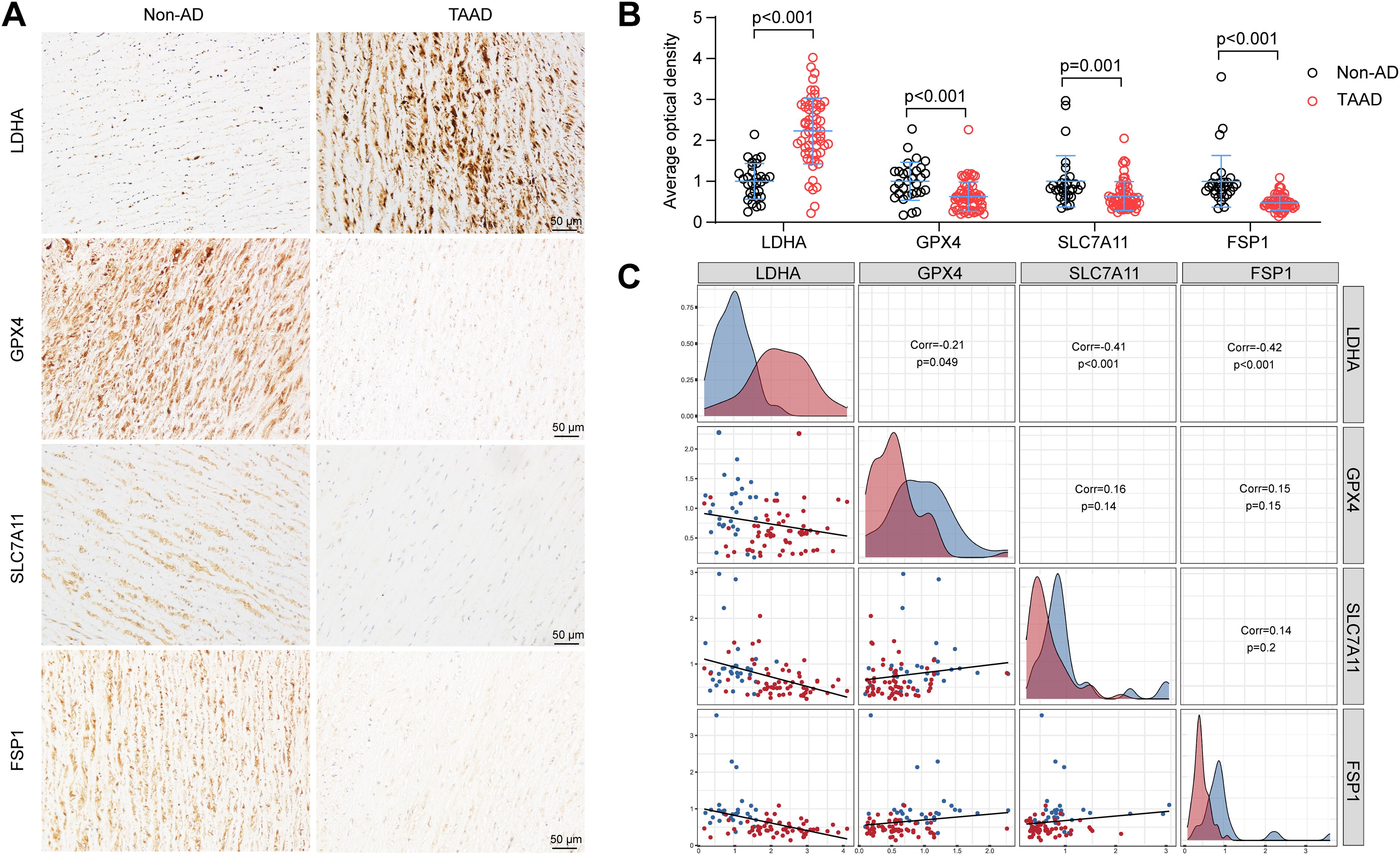
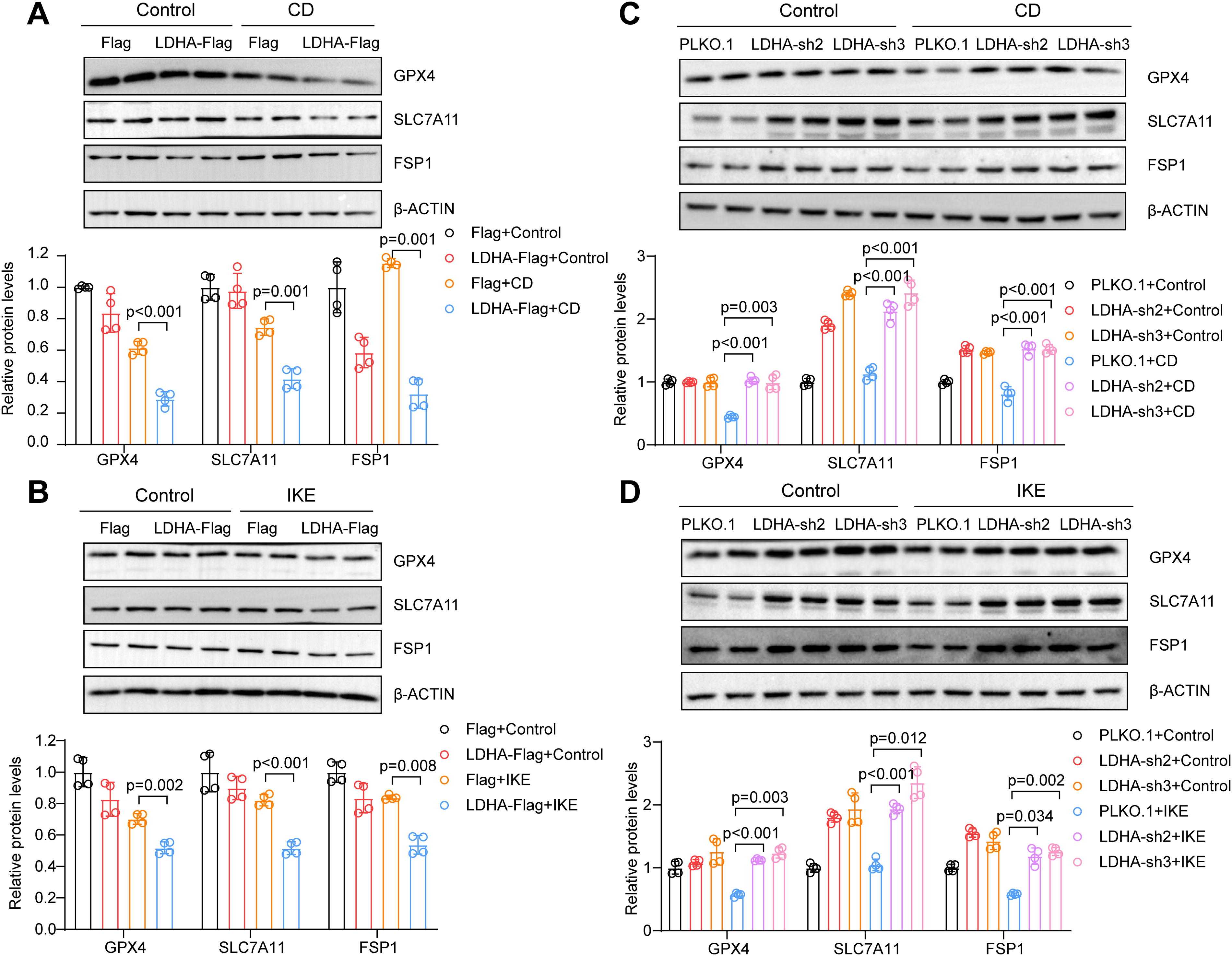
As previously mentioned, GPX4, SLC7A11, and FSP1 are the core regulatory molecules that control lipid peroxidation during ferroptosis (Stockwell, 2022). To investigate the associations among LDHA, GPX4, SLC7A11, and FSP1, tissue chips containing the aortas of 30 non-AD patients and 60 TAAD patients were used. Immunohistochemical staining revealed increased LDHA expression and decreased GPX4, SLC7A11, and FSP1 expression in the aortas of AD patients, as expected (Fig. 6A and B). Next, the correlation analysis among LDHA, GPX4, SLC7A11, and FSP1 was conducted, and the LDHA expression was negatively correlated with GPX4, SLC7A11, and FSP1 in the aortic tissue chips (Fig. 6C). Next, the expression of GPX4, SLC7A11, and FSP1 in LDHA-overexpressing cells was detected, and in CD- or IKE-induced HASMC ferroptosis, the expression of these proteins was consistently downregulated (Fig. 7A and B). Conversely, LDHA knockdown increased the protein levels of GPX4, SLC7A11, and FSP1 during ferroptosis in HASMCs (Fig. 7C and D). Together, LDHA might participate in ferroptosis and AD by regulating GPX4, SLC7A11, and FSP1.
System Xc−-GPX4 and FSP1-CoQ10 pathways were involved in the regulatory effect of LDHA on VSMC ferroptosis
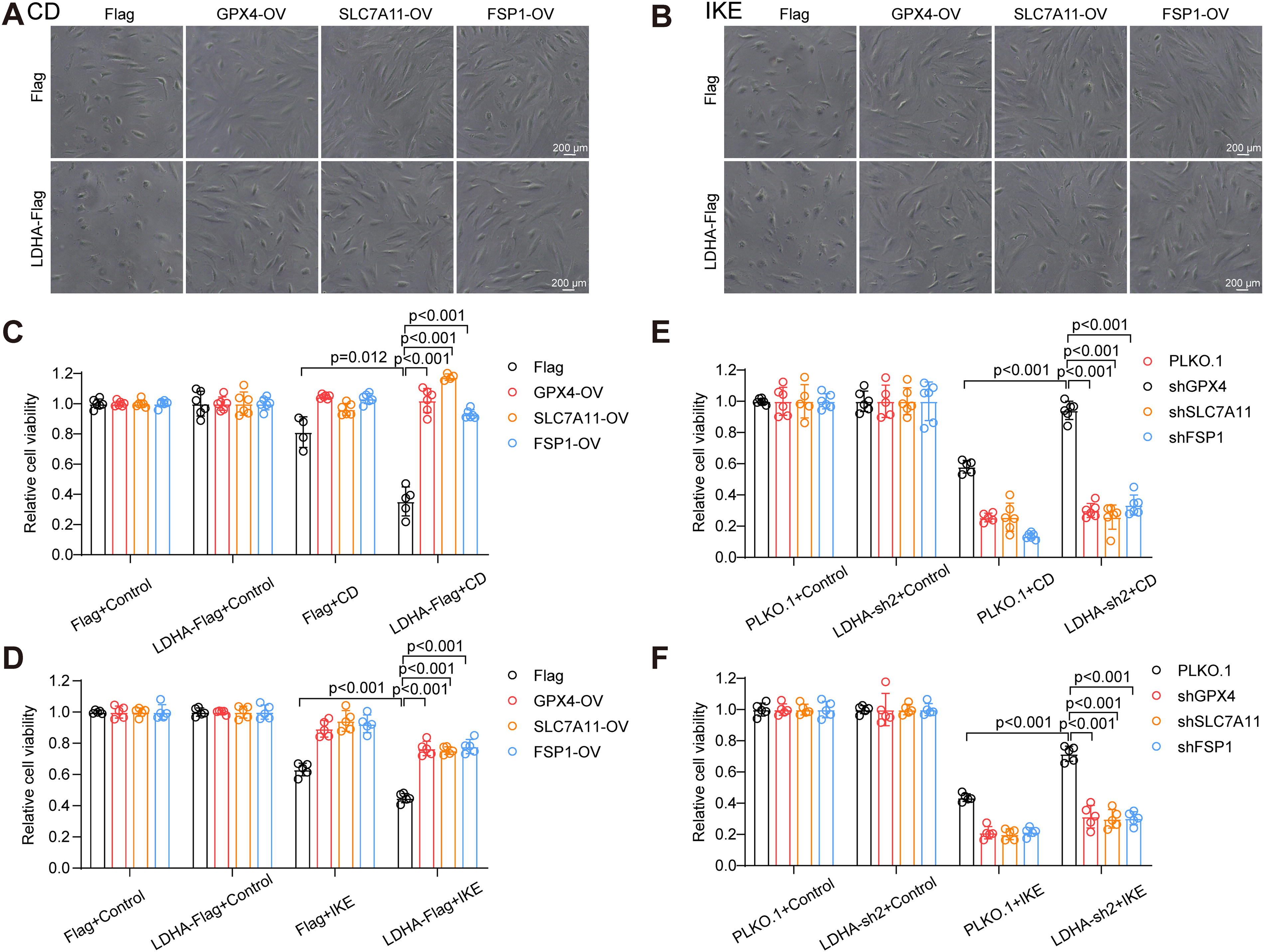
To further elucidate the regulatory mechanism of LDHA on VSMC ferroptosis, lentivirus infection was utilized to overexpress GPX4, SLC7A11, or FSP1 in LDHA-overexpressing HASMCs (Fig. S2A–C). As expected, the overexpression of GPX4, SLC7A11, or FSP1 mitigated CD- or IKE-induced cell death in LDHA-overexpressing HASMCs and counteracted the ability of LDHA to promote ferroptosis (Fig. 8A–D). Similarly, knockdown of GPX4, SLC7A11, or FSP1 in LDHA knockdown HASMCs attenuated the protective effect of LDHA knockdown against CD or IKE treatment (Fig. 8E and F, S2D–2F). Furthermore, the impact of GPX4, SLC7A11, and FSP1 on the pro-oxidative function of LDHA was investigated. Overexpression of these proteins reduced the ratio of oxidized C11-BODIPY in LDHA-overexpressing HASMCs treated with CD or IKE (Fig. 9A and B). Moreover, the overexpression of GPX4, SLC7A11, or FSP1 reduced the excessive accumulation of MDA in LDHA-overexpressing HASMCs during CD or IKE treatment (Fig. 9E and F). Conversely, knockdown of GPX4, SLC7A11, or FSP1 weakened the protective effect of LDHA knockdown on lipid peroxidation during VSMC ferroptosis (Fig. 9C, D, G, and H). In conclusion, system Xc−-GPX4 and FSP1-CoQ10 pathways were indeed involved in the influence of LDHA on VSMC ferroptosis.

Targeting NRF2 reversed the promoting effect of LDHA on VSMC ferroptosis
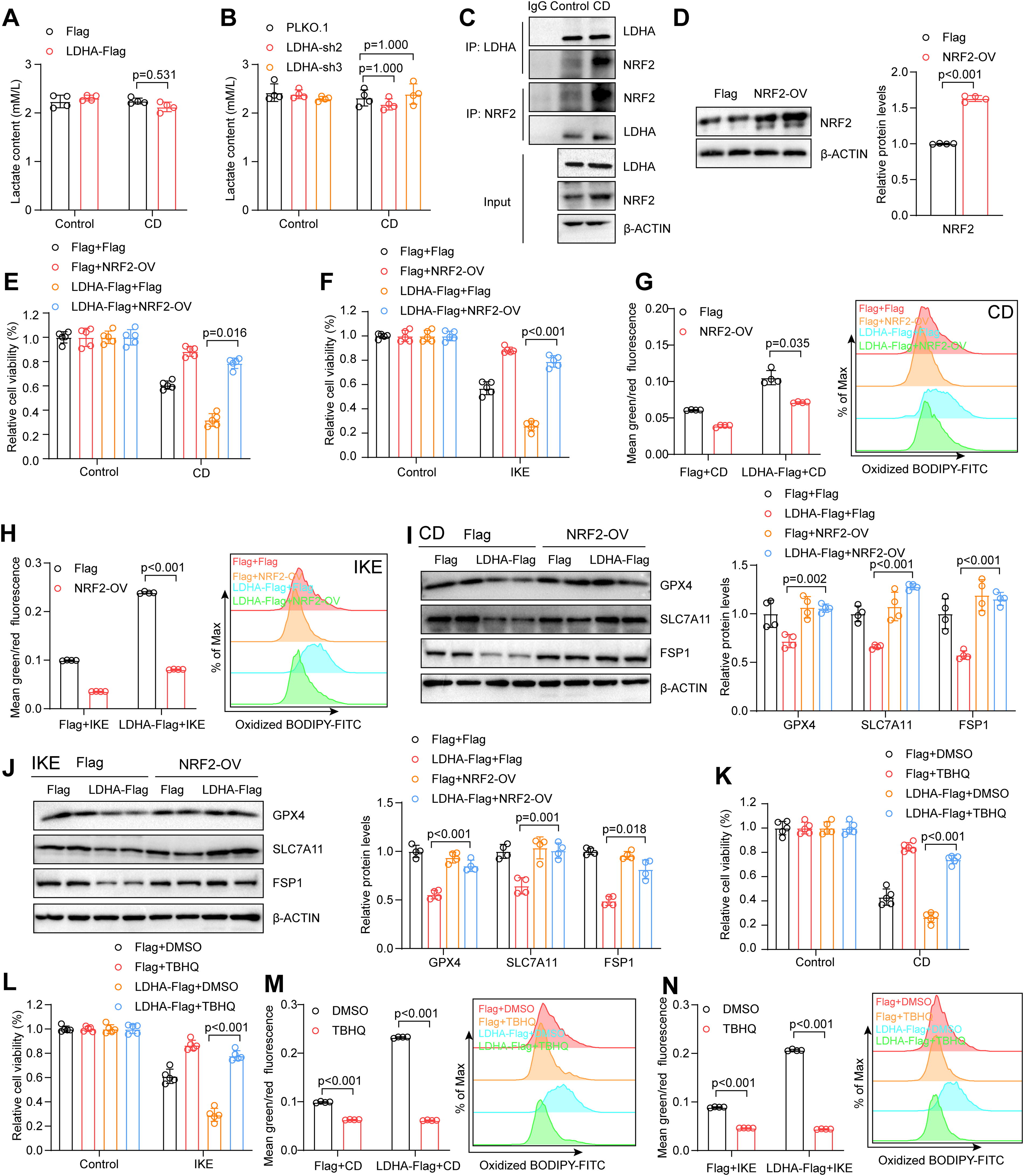
Given the role of LDHA in glycolysis, lactate levels were assessed; however, manipulating LDHA expression did not affect lactate concentration following CD treatment, suggesting that LDHA might promote VSMC ferroptosis via a nonenzymatic pathway (Fig. 10A and B). Previous studies have reported that the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2) is involved in regulating the expression of GPX4, SLC7A11, and FSP1 and protects against ferroptosis through the activation of the System Xc−-GPX4 and FSP1-CoQ10 pathways (Hu et al., 2024; Yang et al., 2024; Zhao et al., 2024). Thus, it is plausible that LDHA may regulate VSMC ferroptosis via NRF2. Immunoprecipitation assays revealed that LDHA and NRF2 indeed interacted in HASMCs, and this interaction was enhanced upon CD treatment (Fig. 10C). Furthermore, the overexpression of NRF2 was performed in HASMCs and effectively mitigated the impact of LDHA on ferroptosis and lipid peroxidation in response to CD or IKE treatment (Fig. 10D–H). Consistent with these findings, NRF2 upregulated the expression of GPX4, SLC7A11, and FSP1 in LDHA-overexpressing HASMCs upon CD or IKE treatment (Fig. 10I–J). Additionally, the antioxidant tert-butylhydroquinone, a known NRF2 agonist, reversed the effects of LDHA on VSMC ferroptosis, showing its potential application in the treatment of AD progression (Fig. 10K–N).
Discussion
VSMC ferroptosis plays a crucial role in AD, as revealed in our prior research in which inhibiting VSMC ferroptosis effectively delayed disease progression in vivo (Chen et al., 2022; Li et al., 2022). This current investigation revealed that abnormal glycolysis is a contributing element to VSMC ferroptosis and AD, which was confirmed through extensive bioinformatics analysis. Specifically, the key glycolytic enzyme LDHA exacerbated cell death and lipid peroxide accumulation in CD- or IKE-treated HASMCs. The downregulation of the ferroptosis-associated molecules GPX4, SLC7A11, and FSP1 displayed an inverse relationship with LDHA levels in both AD and non-AD patient aortas, and their suppression was evident in LDHA-overexpressing HASMCs. Notably, the overexpression of these molecules counteracted the impact of LDHA on ferroptosis in HASMCs. Moreover, the interaction between LDHA and the transcription factor NRF2 was found to be strengthened following CD treatment. The impact of LDHA on VSMC ferroptosis was effectively reversed by the overexpression or agonist of NRF2, elucidating the regulatory mechanism of LDHA in VSMC ferroptosis. Overall, LDHA serves as the connecting factor among glycolysis, VSMC ferroptosis, and AD, highlighting its potential as a promising target for AD therapy (Fig. 1).
Glucose metabolic reprogramming is a hallmark of AD and contributes to VSMC death, degradation of the extracellular matrix, endothelial dysfunction, and phenotypic transition of VSMCs (Li et al., 2023). Increased glycolytic activity has been shown to drive aortic remodeling by regulating endothelial-to-mesenchymal transition (Wang et al., 2023). In this study, dysfunction of glycolysis signaling pathways in AD was identified through bioinformatic analysis, and the expression of glycolysis-related molecules in the dissected aorta significantly differed from that in normal aortas. Furthermore, the connection between glycolysis and ferroptosis, a novel mechanism of AD pathogenesis, was elucidated. Previous research has demonstrated that taurine upregulated 1 (TUG1) promotes PDK4-mediated glycolysis, protecting against ferroptosis in hepatic stellate cells and driving the progression of liver fibrosis (Zhang et al., 2023a, 2023b). In HASMCs, LDHA, a key glycolytic enzyme, enhanced its interaction with NRF2 during ferroptosis and led to the inhibition of the system Xc−-GPX4 and FSP1-CoQ10 pathways, thereby exacerbating CD- or IKE-induced cell death and lipid peroxidation. Additionally, an inverse relationship between LDHA and GPX4, SLC7A11, or FSP1 was confirmed in aortic samples from both normal subjects and AD patients. These findings suggest that LDHA may serve as a critical link between glycolysis and VSMC ferroptosis, providing insights into the pathogenesis of AD.
As a glycolytic enzyme, LDHA is mainly involved in lactate production (Feng et al., 2018). However, ongoing research has revealed nonenzymatic roles for LDHA. In osteoarthritis, the LDHA–NADH interaction promoted ROS production and enhanced IκB-ζ stabilization, worsening inflammation and disease progression (Arra et al., 2020). Moreover, a cell-permeable derivative of itaconate, 4-octyl itaconate, inhibited LDHA-mediated ROS generation and prevented pancreatic beta cell death under oxidative stress (Wu et al., 2022a, 2022b). Similarly, our study revealed LDHA contributed to CD- or IKE-induced lipid peroxidation and exacerbated HASMC ferroptosis. Furthermore, LDHA cooperated with NRF2 during VSMC ferroptosis and inhibited the transcriptional promotion of GPX4, SLC7A11, and FSP1 by NRF2, which expanded the nonenzymatic role of LDHA in cell death. These findings underscore the multifunctional nature beyond the metabolism of LDHA, warranting further exploration. Additionally, increased LDHA expression in HASMCs was observed in aortic samples from AD patients, consistent with prior research. Wu et al. demonstrated that LDHA facilitated VSMC transformation and MMP2/9 expression via the NDRG3-ERK1/2 pathway, and the pharmacological inhibition of LDHA attenuated the progression of AD (Wu et al., 2022a, 2022b). In summary, LDHA not only modulates the biological function of surviving VSMCs but also influences cell death processes. Nevertheless, in vivo experiments are essential to further elucidate the role of LDHA in VSMC ferroptosis and AD.
Compared with that in normal aortas, LDHA expression in VSMCs was greater in dissected human and mouse aortas in VSMCs; however, the potential regulatory mechanism involved remains unknown. Previous studies have provided some insights into the regulation of LDHA expression in AD. Histone modification, a classical regulatory mechanism, has been implicated in AD pathogenesis (Guo et al., 2019). The histone demethylase KDM6B was reported to mediate H3K27me3 demethylation, leading to increased LDHA expression in osteosarcoma cells (Jiang et al., 2021a, 2021b). Notably, in abdominal aortic aneurysms, an increase in KDM6B expression was found to mediate inflammatory cytokine release by reducing H3K27me3 levels (Davis et al., 2021). Thus, upregulated LDHA in AD may be attributed to enhanced KDM6B-mediated H3K27me3 demethylation. In addition to transcriptional regulation, protein degradation also plays a role in regulating protein levels. In gastric cancer, CPT1A functions as a lysine succinyltransferase, succinylating LDHA at K222 and inhibiting its binding to SQSTM1, thus reducing lysosomal degradation (Li et al., 2020). Remarkably, increased lysine succinylation has been observed in aortic aneurysm and dissection pathogenesis, and protein–protein interaction network analysis revealed increased lysine succinylation in LDHA (Zhang et al., 2023a, 2023b). The above results indicated that lysine succinylation might account for LDHA expression in AD. Overall, the elevation of LDHA in AD is likely a comprehensive result of multiple factors, and further experimental validation is needed to confirm these hypotheses.
The present study has certain limitations. These studies have focused mainly on in vitro research and elucidated the role and underlying mechanisms of LDHA in VSMC ferroptosis and AD. The potentially similar function of LDHA in vivo requires further exploration. Additionally, this study primarily emphasized the nonenzymatic role of LDHA, and investigation of the enzymatic function of LDHA in VSMCs is necessary to better understand the impact of glycolysis on ferroptosis and AD.
Methods
HASMC culture and treatment
HASMC primary culture was obtained using the primary culture of aortas obtained from pruned donor tissues during heart transplantation, as previously described (Chen et al., 2022), and this process was approved by the Human Research Ethics Committees of Tongji Hospital, Tongji Medical College, and Huazhong University of Science and Technology. Briefly, the aortas were cleaned of inner and adventitial tissues under microscopic observation, and the medial tissues were then finely minced into 1 × 1 mm fragments using sterile scissors within culture flasks. The minced tissues were carefully positioned in the flasks and incubated for 30 min to ensure a close fit with the bottom of the flask. Subsequently, Dulbecco's modified eagle medium (DMEM)/F12 supplemented with 10% fetal bovine serum (SH30084.03; HyClone) and 1% penicillin–streptomycin (15140-122; ThermoFisher Scientific) was added to the flasks. The tissues were cultured at 37°C in a humidified atmosphere containing 5% CO2. Approximately 7 days later, the HASMCs began to migrate from the aortic tissues and were subsequently passaged 3–7 times for further experimentation. To induce ferroptosis, HASMCs were subjected to CD or IKE at a concentration of 2.5 μM (S8877, Selleck) for 16 h, as previously described.
Western blotting
Western blotting was conducted in accordance with the established methods (Chen et al., 2022). A mixture of radio immunoprecipitation assay (RIPA) lysis buffer containing protease and phosphatase inhibitor cocktails was used to lyse the aortic tissues and HASMCs, followed by quantification of total protein using the bicinchoninic acid (BCA) protein assay. Approximately 5 μg of proteins was separated through 10%–12% acrylamide gels using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVH00010, Millipore) membrane. The membranes were initially blocked with 5% nonfat milk for 1 h at room temperature and then were incubated overnight at 4°C with specific primary antibodies. This step was followed by the addition of horseradish peroxidase-conjugated antirabbit or antimouse secondary antibodies for 1 h at room temperature. The protein bands were then incubated with Clarity Western ECL substrate and visualized using a ChemiDoc XRS+ system (Bio-Rad). β-Actin served as the loading control, and the results were quantified using Image Lab 6.1 software. The antibodies used in this study were LDHA (19987-1-AP, Proteintech), SLC7A11 (Proteintech, 26864-1-AP), GPX4 (ab125066, Abcam), FSP1 (HPA042309, ATLAS), NRF2 (380773, Zen-Bioscience), and β-Actin (AC026, ABclonal).
Cell viability assay
Cell viability was assessed using a Cell Counting Kit-8 (CCK-8, CK04, Dojindo) and following the manufacturer’s instructions. HASMCs were seeded at a density of 8000 cells per well in 96-well plates and subsequently treated with CD or IKE to induce ferroptosis. Following treatment, 10 μL of CCK-8 reagent was added to each well, and the cells were incubated for 2 h at 37°C. The number of living cells in each well was analyzed by detecting the absorbance at 450 nm using a multifunctional microplate reader (ELx808, BioTek). Cell viability was calculated using the following formula: (Abs of experimental group-Abs of blank group)/(Abs of control group-Abs of blank group) × 100%.
PI staining
HASMCs were plated in 6-well plates and treated with CD or IKE for 16 h. After treatment, the supernatant from each well was collected, and the cells were digested using trypsin. Next, the mixture containing the supernatant and cells was centrifuged at 1000 rpm to collect all cells. PI dyes (5 μg/mL, P4170, Sigma-Aldrich) were added to cell suspension and incubated in the dark for 15 min. The fluorescence intensity was measured using flow cytometry, and the results were analyzed using FlowJo 10 software.
MDA assay
The MDA content in HASMCs was assessed using an MDA assay kit (MAK085, Sigma-Aldrich) according to the manufacturer’s instructions (Chen et al., 2022). Following treatment, the cells were collected and lysed in RIPA lysis buffer on ice. The lysate was sonicated to ensure complete cell lysis, and the protein concentration of each sample was measured using a BCA protein assay. The MDA working solution, which contains thiobarbituric acid and the antioxidant butylated hydroxytoluene (BHT), was added to the lysate, and the samples were incubated at 95°C for 15 min. After cooling to room temperature, 200 μL of the reaction mixture was transferred to a 96-well plate, and the absorbance at 532 nm was measured using a multifunctional microplate reader. The MDA content of each sample was calculated according to a standard curve and normalized to the protein concentration.
Lipid ROS assay
Cellular lipid ROS accumulation was assessed using the C11 BODIPY 581/591 molecular probe (D3861, Invitrogen). The HASMCs were treated with CD or IKE for 16 h and then incubated with a 5 μM Lipid Peroxidation Sensor in the dark for 30 min. After washing with phosphate buffer solution (PBS) three times, the cells were trypsinized and resuspended in PBS. The fluorescence intensity was detected using flow cytometry. The shift in the fluorescence emission peak from 590 nm to 510 nm reflects proportionality to the generation of lipid ROS.
Plasmid constructs and lentivirus infection
The full-length sequence of human LDHA and NRF2 was cloned and inserted into the Phage-Flag lentiviral vector, respectively. The shRNA sequences targeting LDHA were inserted into the PLKO.1 lentiviral vector. To avoid off-target effects, three LDHA knockdown plasmids were designed with the following sequences (5′–3′): LDHA-sh1, GGCTATACTTACACCCAAACG; LDHA-sh2, GGACTTGGCAGATGAACTTGC; LDHA-sh3, GCCCGATTCCGTTACCTAATG. Lentivirus generation was performed as described previously. The packaging plasmids psPAX2 (12260, Addgene) and pMD2.G (12259, Addgene) and LDHA overexpression/knockdown plasmids were transfected into HEK293T cells using the transfection reagent polyethyleneimine. After 8 h, the medium was changed to remove the transfection reagent, and the lentivirus-containing supernatant was harvested at 32 and 56 h posttransfection. The supernatant was filtered through a 0.45 µm filter (SLGP033RB, Millipore) to eliminate impurities before HASMCs were infected with 10 µg/mL hexadimethrine bromide (H9268, Sigma-Aldrich). After a 24-h infection period, the supernatant was discarded, and the infected cells were utilized for subsequent experiments. Prior to application, the LDHA protein level in the infected cells was assessed to verify the infection efficiency. The overexpression and knockdown plasmids for GPX4, SLC7A11, and FSP1 have been described in a previous study (Chen et al., 2022). The target sequences of the knockdown plasmids (5′–3′): GCTCACAGCAATTCTGATAAT for SLC7A11, GTGGATGAAGATCCAACCCAA for GPX4, and CAACATCGTCAACTCTGTGAA for FSP1.
Bioinformatic analysis
Bioinformatic analysis of VSMC ferroptosis was performed based on the protective function of BRD4770 on ferroptosis in HASMCs (GSE272018). Four groups were collected and analyzed, including CD, CD with BRD4770 treatment (CD+BRD4770), IKE, and IKE with BRD4770 treatment (IKE+BRD4770) groups, as reported in a previous study (Chen et al., 2022). DEGs were identified using the R (version 4.0.2) package DEseq2, and p-values were adjusted using the Benjamini and Hochberg approach. Adjusted p value <0.05 and a |logFC| >1 were defined as the thresholds for DEG selection. The clusterProfiler (v3.16.1) R package was used for GO, KEGG and GSEA analyses. The dataset “GSE153434,” which includes human aortas with or without AD, was acquired from the GEO database, and the detailed analysis has been described in a previous study (Chen et al., 2021; Zhou et al., 2020).
Immunostaining
Thirty non-AD and 60 TAAD aortic tissues were fixed, embedded, and sliced to create a tissue chip. The detailed clinical information is available in a previous study (Li et al., 2022). The sections were deparaffinized with xylene, and antigen retrieval was performed using EDTA solution (pH 9.0, MVS-0099, MXB biotechnologies) for 20 min. After being blocked with bovine serum albumin (BSA, FA016-100 G, Genview), the resections were treated with 3% hydrogen peroxide for 30 min to eliminate endogenous peroxidase. Following overnight incubation with primary antibodies, the sections were incubated with the corresponding secondary antibodies for 1 h at room temperature. The reaction products were detected using a 3,3’-diaminobenzidine (DAB) horseradish peroxidase color development kit (ZLI-9017, ZSGB-BIO). The average optical density of each sample was analyzed using ImageJ software. For immunofluorescence staining, sections were incubated with Alexa Fluor-conjugated goat antimouse/rat serum secondary antibodies after treatment with primary antibodies, and nuclei counterstaining was performed with 4’,6-diamidino-2-phenylindole (DAPI).
For 4-HNE fluorescence staining, HASMCs were fixed in a 4% paraformaldehyde solution at room temperature for 30 min in a 24-well plate and rinsed with PBS three times in a shaker. Next, 0.2% Triton X-100 was added to each well for 15 min to rupture the cell membranes, and the cells were blocked with 5% BSA to avoid nonspecific antigens. After washing three times, the HASMCs were incubated with primary antibodies overnight at 4°C, followed by incubation with Alexa Fluor-conjugated secondary antibodies for 1 h at room temperature. Finally, the cells were incubated with DAPI for 5 min to stain the nucleus and observed under a fluorescence microscope (Olympus). Fluorescence images were acquired, and the fluorescence intensity was calculated using ImageJ software.
Coimmunoprecipitation assays
The HASMCs were harvested in prelysis buffer supplemented with proteinase inhibitors and then sonicated for complete lysis. After centrifugation, 100 μL of the supernatant was set aside as the input, whereas the remaining supernatant was combined with 1 μg of NRF2 or LDHA antibody (each sample) for the immunoprecipitation assay. To ensure optimal antigen-antibody binding, the samples were rotated overnight at 4°C. Subsequently, magnetic beads were introduced to the samples and incubated for an additional 3 h at 4°C, followed by three washes with washing buffer to remove nonspecific binding. The coprecipitation product was eluted using SDS loading buffer and heated at 95°C for 15 min prior to being loaded onto an SDS-PAGE gel for western blotting.
Real-time PCR
Total RNA was extracted using TRIzol reagent (A33251, Invitrogen) following standard protocols. Approximately 5 μg of the extracted RNA was reverse transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (11141ES60, Yeason). The resulting reverse transcription products were then diluted 10-fold in RNase-free water before proceeding to quantitative polymerase chain reaction (PCR). The relative mRNA levels of the target genes were measured using the SYBR Green PCR Master Mix (11201ES08, Yeasen) as previously described (Chen et al., 2022). Data normalization was performed relative to 18S expression. Primer sequences are listed in Supplementary Table S1.
Lactate concentration measurement
The lactate concentration was determined using a lactate assay kit (A019-2-1, Nanjing Jian Cheng Institute) in accordance with the manufacturer’s guidelines. After treatment, HASMCs were harvested and lysed to extract the supernatant. The enzyme working solution and color developer were added to the lysate, followed by a 10-min incubation at 37°C. Upon addition of stop solution, the absorbance at 530 nm was measured using a multimode microplate reader. The lactate levels in each sample were determined based on a standard curve and then normalized to the protein concentration.
Statistical analysis
All data were presented as mean ± standard deviation. Student t test and one-way analysis were used to compare between two groups and multiple groups, respectively. Pearson’s correlation analysis was used for correlation analyses. Statistical analysis was performed using SPSS 23.0 software (IBM), and differences with a two-tailed probability <0.05 were considered statistically significant.
Footnotes
Data availability
The raw data for this study can be obtained from the corresponding author upon a reasonable request. Electronic laboratory notebook was not used in this study.
Authors’ Contributions
X.F. and X.Y. collected clinical specimens and wrote the original draft. B.H. and H.L. conducted the experiments and provided bioinformatics analysis support. J.C. performed statistical analysis. X.G. cultured the primary HASMCs and constructed the plasmids. Z.-M.F., F.-H.G., and X.W. participated in project discussions. D.-S.J. and Y.C. designed the experiments and revised the article. All authors read and approved the final article.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This work was supported by grants from the
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Tables S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.