
Abstract
Background:
Aging is a significant risk factor for the increased incidence of acute kidney injury and chronic kidney disease, posing significant challenges to global public health. The role of N6-methyladenosine (m6A) in the development of chronic kidney disease has been reported, but the regulatory mechanism of m6A in kidney aging remains unclear.
Results:
In this study, we identified a long noncoding RNA (lncRNA), called taurine up-regulated 1 (TUG1), which exhibited a significantly decreased level of m6A modification in human aged kidney through the m6A-lncRNA epitranscriptome microarray. Bioinformatics analysis and machine learning predicted that TUG1 had potentially strong interaction with PGC1-α. RNA immunoprecipitation and chromatin immunoprecipitation analysis showed that TUG1 promoted proliferator-activated receptor γ coactivator-1α (PGC1-α) expression by directly interacting with its TUG-1 binding element region, thereby impacting mitochondrial quality control (MQC), cellular senescence, and renal fibrosis. Silencing the RNA m6A methylase methyltransferase 14 (METTL14) or the reader protein insulin-like growth factor 2 mRNA-binding proteins (IGF2BP2) resulted in the weakened stability of lncRNA TUG1, contributing to an imbalance in MQC.
Conclusion:
Our study demonstrated that the m6A modification and stability of TUG1 were mediated by METTL14 in an IGF2BP2-dependent manner, and modulate the mitochondrial homeostasis in kidney aging by direct targeting PGC-1α. These findings provide a new perspective on potential therapeutic targets for kidney aging. Antioxid. Redox Signal. 41, 993–1013.
Innovation
We have discovered that the lncRNA TUG1 serves as a crucial molecular target implicated in mitochondrial quality control (MQC) and renal fibrosis during kidney aging. The dysregulation of m6A methylation triggers TUG1 RNA degradation and engagement in MQC processes by modulating expression of PGC1-α. This identified mechanism offers a plausible explanation for the observed cellular senescence and exacerbated renal fibrosis during aging. Our study offers a fresh perspective for unraveling the underlying mechanisms of renal aging.
Introduction
With the increasing population of elderly individuals, it is inevitable to shift attention toward addressing preventable factors contributing to tissue aging. The projections indicate that China’s population will reach a concerning 374 million by 2040, accounting for approximately 24.8% of the total population (Choudhury and Levi, 2011; O’Sullivan et al., 2017). The kidneys are highly metabolically active organs, subjected to significant oxidative stress and susceptible to the effects of aging. The presence of senescent cells may contribute to the progression of renal fibrosis following injury (Docherty et al., 2019). Renal tubules, which constitute more than 90% of the kidney, are primarily affected by renal aging. The expression of p16INK4A was found to be more prominent in the nuclei of distal tubules and collecting duct compared to other intrinsic kidney cells, as observed in a series of human kidney transplant biopsies (Sofue et al., 2018). In aged kidneys, there is a significant decline in the regenerative capacity of renal tubular cells, which promotes maladaptive repair and ultimately leads to accelerated chronic kidney disease progression and renal aging (Fang et al., 2020).
One important characteristic of the aging process is the occurrence of mitochondrial dysfunction. In aging tissues, there is a gradual decline in mitochondrial respiratory function, resulting in diminished efficiency in ATP production and an increased generation of reactive oxygen species (ROS) as byproducts of oxidative phosphorylation (Donate-Correa et al., 2023). This leads to the accumulation of oxidative damage in cells and acceleration of the aging process. By reshaping the morphology, quantity, and quality of mitochondria, a mechanism called mitochondrial quality control (MQC) evolves to repair damaged or dysfunctional mitochondria. This mechanism primarily involves processes such as mitochondrial fission, fusion, and mitophagy (mitochondrial autophagy) (Song et al., 2021; Pickles et al., 2018). Mitochondrial fission/fusion mechanisms interact with mitophagy (Pickles et al., 2018). On one hand, the timely elimination of aged or damaged mitochondria through mitophagy contributes to the maintenance of normal cellular quantity and function. In addition, increased biogenesis facilitates the generation of new mitochondria or partially repairs damaged ones to sustain optimal mitochondrial levels within cells (Ni et al., 2015; Su et al., 2023). The process of fission can promote mitophagy, while mitophagy itself also plays a critical role in regulating the processes of fission and fusion. Imbalances in these mechanisms and impairment of mitophagy can lead to cellular death (Bhatia et al., 2019). Therefore, maintaining MQC is crucial for preserving normal cellular functions and determining cell fate during kidney aging. However, research on changes in MQC during renal aging remains limited.
LncRNA TUG1 is a nonprotein coding RNA sequence that has been shown to be aberrantly expressed in various types of kidney diseases, and its dysregulation is closely associated with disease progression (Zhang et al., 2021). The emerging evidence suggests that TUG1 triggers the transcription of Ppargc1a mRNA in diabetic nephropathy, targeting the transcription factor PPARγ coactivator 1α (PGC-1α), which regulates mitochondrial function in podocytes (Li and Susztak, 2016; Long et al., 2016). It is demonstrated that the decreased proliferator-activated receptor γ coactivator-1α (PGC-1α) expression results in impaired mitochondrial function, leading to energy depletion, increased generation of ROS, and promotion of diabetic nephropathy development (Fontecha-Barriuso et al., 2020). However, whether TUG1 is involved in maintaining the balance of MQC during renal aging remains unclear.
The N6-methyladenosine (m6A) RNA methylation is a prevalent and conserved RNA modification in mammalian species (Sendinc and Shi, 2023). Researchers have confirmed that the core components of the RNA m6A methyltransferase complexes are methyltransferase 14 (METTL14), methyltransferase-like 3 (METTL3), and Wil1-associated protein. These components closely interact with each other, and play a crucial role in recognition, localization, and catalysis of m6a-modified RNAs. The RNA demethylase alkB homolog 5 and fat mass- and obesity-associated protein participate in dynamic regulation of m6A methylation by removing RNA m6A methylation marks. Transcriptionally modified RNA m6A modifications are recognized by m6A recognition proteins, which mediate RNA stability, nuclear export, transcription, and other cellular processes (Fu et al., 2014; Liu et al., 2014; Qu et al., 2022). The YTH domain protein (YTHDF) is currently the most extensively investigated and well-established m6A reader protein (Shi et al., 2017). There are also some modifier recognition proteins that do not contain the YTH domain, but only have common RNA binding domains, such as insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), which participate in m6A regulation by enhancing the stability and storage of their target RNAs (Cao et al., 2023). Notably, it is evidenced that m6A dysregulation leads to various kidney diseases (An and Duan, 2022; Jiang et al., 2022; Jiang et al., 2021). However, this is far from sufficient to explain the relationship between kidney aging and m6A modification.
This study has identified lncRNA TUG1 as a molecular target implicated in MQC and renal fibrosis during kidney aging, through screening m6A-lncRNA expression profiles. The dysregulation of m6A methylated TUG1 mediated the degradation of TUG1, which is involved in MQC by regulating PGC1-α expression. This mechanism may potentially contribute to the enhanced cell senescence and renal fibrosis observed during the aging process. This study provided novel insights into the underlying mechanisms of renal aging and offers a potential therapeutic target.
Results
The m6A methylation and expression levels of TUG1 decreased in human aged kidney
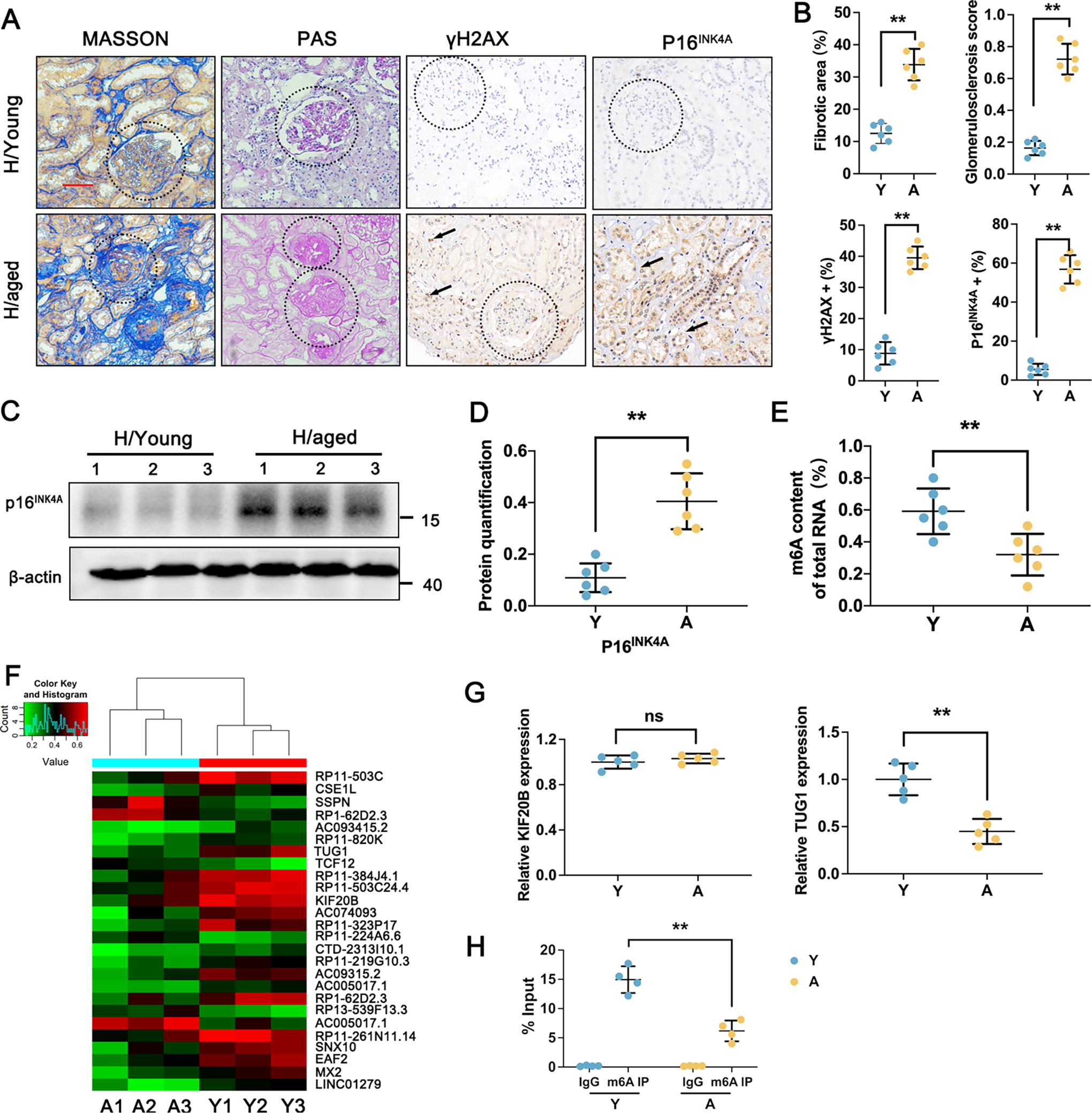
As recent evidence shows, m6A levels are crucial in the aging process and aging-associated diseases (Wu et al., 2020b; Jiang et al., 2021). To elucidate the important role of lncRNA m6A modification in aging kidneys, young and aged human kidney tissues were collected for the histopathology analysis. As shown in Figure 1A, B, both Masson and periodic acid-Schiff (PAS) staining indicated typical focal tubular atrophy, interstitial fibrosis, and glomerulosclerosis in the H/aged group. γH2AX is a novel biomarker for DNA double-strand breaks. Immunohistochemical staining (IHC) results showed that the level of γH2AX was increased in the H/aged group compared to the control, suggesting DNA damage during renal aging process (Fig. 1A, B). p16INK4A belongs to the INK4 gene family, which is a typical senescence-associated protein. As expected, IHC and Western blot analysis demonstrated a significant increase in p16INK4A expression level in H/aged group compared to the H/young group (Fig. 1B–D). In addition, we examined the methylation level in both H/control and H/aged groups to explore the involvement of m6A-associated lncRNA by colorimetric method. There was a remarkable downregulation of m6A methylation level in the H/aged group compared with the young group (Fig. 1E). To explore the important role of lncRNA m6A modification in aging kidneys, we conducted an m6A-lncRNA epitranscriptomic microarray analysis. Notably, we identified 63 hypermethylated m6A peaks and 297 hypomethylated m6A peaks in the H/aged group (Fig. 1F). Among the hypomethylated lncRNA, we identified RP11-503C, TUG1, and KIF20B as the most prominent ones; however, the most significant differential expression of lncRNAs is TUG1 (Fig. 1G). Thus, the lncRNA TUG1 has been speculated as a critical factor involved in aging kidney, with a significantly reduction in m6A modification observed in the H/aged group (Fig. 1H).
Renal fibrosis in aged mice model with the downregulation of TUG1
The aged kidneys show significantly increased fibrosis compared with young kidneys. The Western blot analysis revealed a significant increase in the expression levels of P16INK4A and γH2AX in kidneys of a 24-month-old mice group, which is consistent with the findings observed in human kidney sample (Fig. 2A, B). Masson and senescence-associated β-galactosidase (SA-β-gal) staining were conducted to evaluate the extent of renal fibrosis in mice. As shown in Figure 2C, D, the accumulation of SA-β-gal and extracellular matrix (ECM) were observed in kidney tissue from the 24-month-old group, but absent in 6-month-old mice. Consistent with the increased level of P16INK4A and γH2AX, the expression levels of collagen I (COL I), fibronectin (FN), and α-smooth muscle actin (α-SMA) were also upregulated, indicating age-related renal fibrosis occurrence in 24-month-old mice group (Fig. 2C, D, H). The expression of TUG1 decreased in the kidney tissue in 24-month-old mice (Fig. 2E). A decrease in fluorescence intensity of TUG1 and E-cadherin in 24-month-old mice or aged human kidney biopsy was detected using fluorescence in situ hybridization (FISH) and immunofluorescence (IF) analysis (Fig. 2F, G; Supplementary Fig. S1). This discovery has prompted further investigation into the association between TUG1 and age-related renal fibrosis.

TUG1 overexpression ameliorates renal fibrosis in accelerated aged model
To investigate the important role of TUG1 in age-related renal fibrosis, an accelerated aged mice model was constructed by continuous injection of D‐gal. Meanwhile, the TUG1-overexpressed aged mice model was established by TUG1-encoding virus transfection of D-gal-treated mice. Real-time quantitative PCR (RT-qPCR) analysis confirmed successful construction of the model (Fig. 3B). As shown in Figure 3C–E, IHC and Western blot analysis revealed that administration of D-gal resulted in renal fibrosis in the accelerated aged mice, as evidenced by positive staining for both β-gal and Masson, with increasing levels of p16INK4A, FN, COL I, and α-SMA. However, overexpression of TUG1 alleviated the process of renal fibrosis in the accelerated aged mice by suppressing β-gal accumulation, decreasing p16INK4A expression, and reducing ECM production (Fig. 3C–E).

TUG1 overexpression alleviates tubulointerstitial fibrosis in accelerated aged HK-2 cells
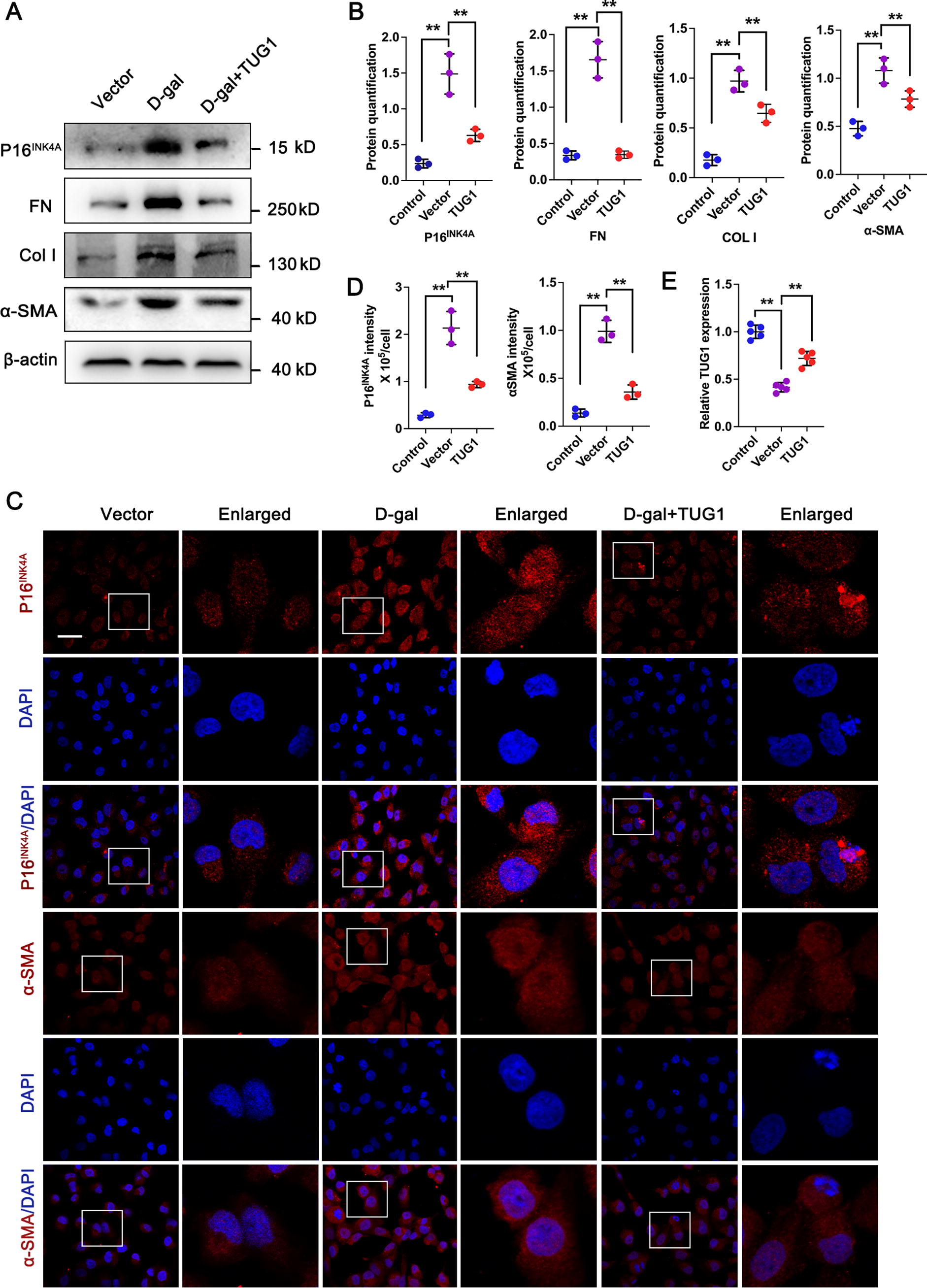
To further validate the protection effect of TUG1 on age-related renal interstitial fibrosis, renal tubular epithelial cells (HK-2) were treated with D-gal to successfully establish an accelerated aged cell model in vitro, as evidenced by the increased expression of p16INK4A (Fig. 4A, B). Consistent with the results in vivo, administration of D-gal resulted in renal fibrosis in the accelerated aged HK-2 cells, with higher expression of FN, COL I, and α-SMA compared with the control. Interestingly, transfection of HK-2 cells with an overexpressed TUG1 plasmid demonstrated a significant ability to counteract the upregulation of p16INK4A, as well as FN, COL I, and α-SMA induced by D-gal (Fig. 4A, B, E). In addition, IF staining results also confirmed that the overexpression of TUG1 could attenuate the elevated expression levels of p16INK4A and α-SMA in D-gal-treated HK-2 cells (Fig. 4C, D). The above results suggest that TUG1 may exhibit a protective effect against HK-2 cell aging and tubulointerstitial fibrosis in vitro.
TUG1 formed potentially strong interaction with PGC1-α predicted by bioinformatics analysis and machine learning
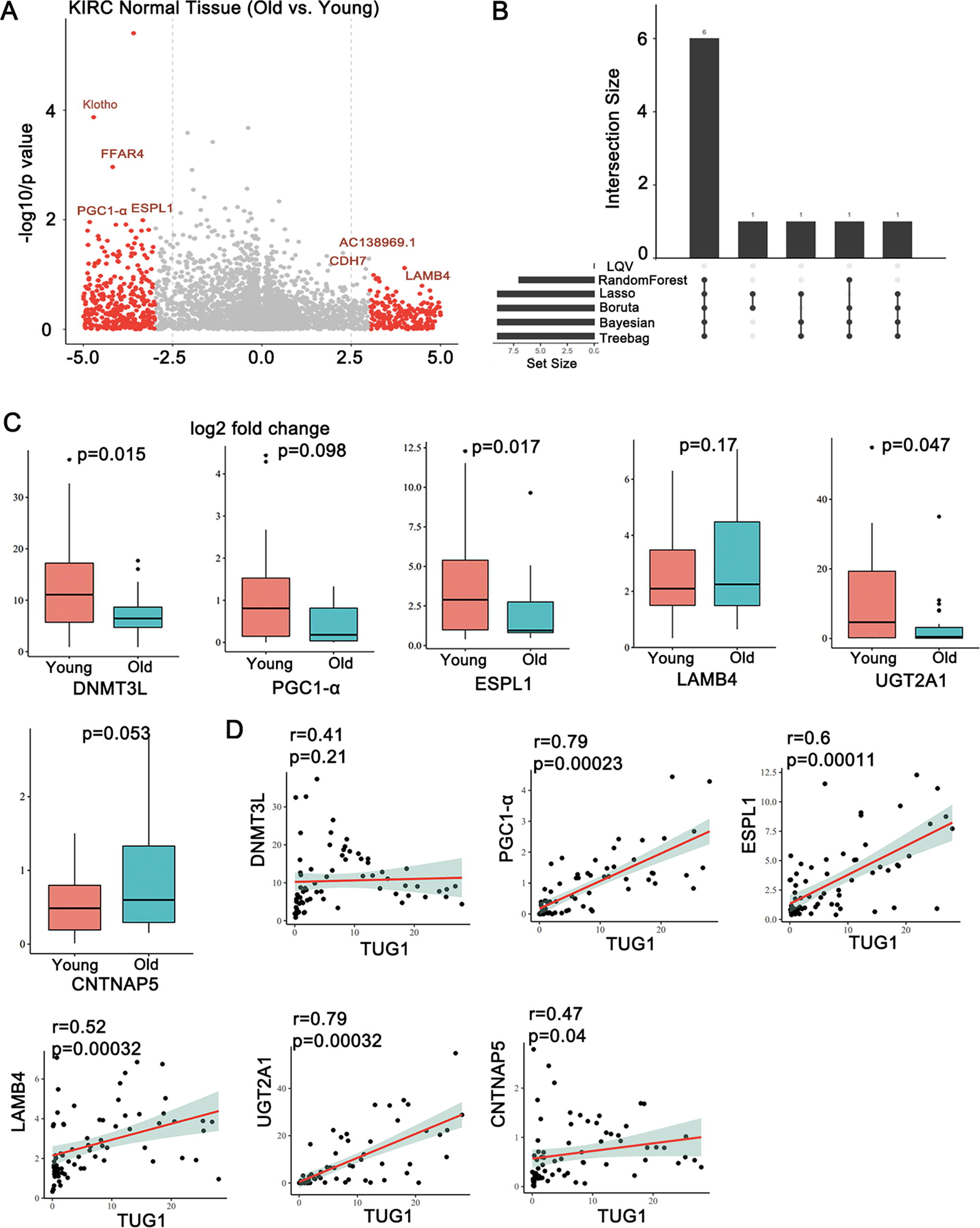
To investigate the targeted mRNA associated with TUG1-mediated age-related renal fibrosis, we chose normal kidney tissue samples from The Cancer Genome Atlas (TCGA)-KIRC dataset for further research, with age information included. We divided the samples into two groups based on a cutoff value of 65 years: the Young group (<65 years) and the Old group (>65 years), followed by differentially expressed gene analysis on these two datasets. The results showed that there were 1010 genes differentially expressed between the young group and the old group based on the criteria of p < 0.05 and fold change >3 | fold change <−3. Among that, 498 genes were upregulated and 512 genes were downregulated (Fig. 5A). For the outcome of Old and Young groups, we conducted visualization diagram using six common machine learning (ML) algorithms, including learning vector quantization (LVQ), random forest (RF), LASSO, Boruta, Bayesian, and Treebag, screening for relevant mRNA associated with age. The mRNAs that appeared in five or more of these algorithms were selected as Hub mRNA. Hub mRNA refers to: aging-related feature genes selected through various ML algorithms, to some extent avoiding algorithm bias. As a result, six mRNAs were identified by five algorithms, which included PGC1-α, DNMT3L, ESPL1, LAMB4, UGT2A1, and CNTNAP5 (Fig. 5B). As shown in Figure 5C, the expressions of PGC1-α (p = 0.0098), DNMT3L (p = 0.015), ESPL1 (p = 0.017), and UGT2A1 (p = 0.047) were significantly decreased in Old group compared with Young group from TCGA-KIRC normal tissue, whereas the levels of LAMB4 (p = 0.17) and CNTNAP5 (p = 0.053) between these two groups did not exhibit a significant difference. Next, we conducted a correlation analysis between Hub mRNAs and TUG1. The coplot indicated a robust correlation between PGC1-α (r = 0.79), UGT1A1 (r = 0.79), ESPL1 (r = 0.6), and lncRNA TUG1. The Hub mRNA PGC1-α was collectively chosen as the targeted mRNA of lncRNA TUG1, based on its positive correlation (r >0.4) and statistical significance (p < 0.01). PGC1-α, as a key regulator, regulates mitochondrial DNA transcription through the modulation of mitochondrial transcription factor A (TFAM) activity, ultimately inducing mitochondrial biosynthesis, playing a key role in the development of kidney disease (Ito et al., 2024). Besides, a previous study has also found that PGC1-α is a target gene of TUG1; thus, we selected PGC1-α as the target gene for further study.
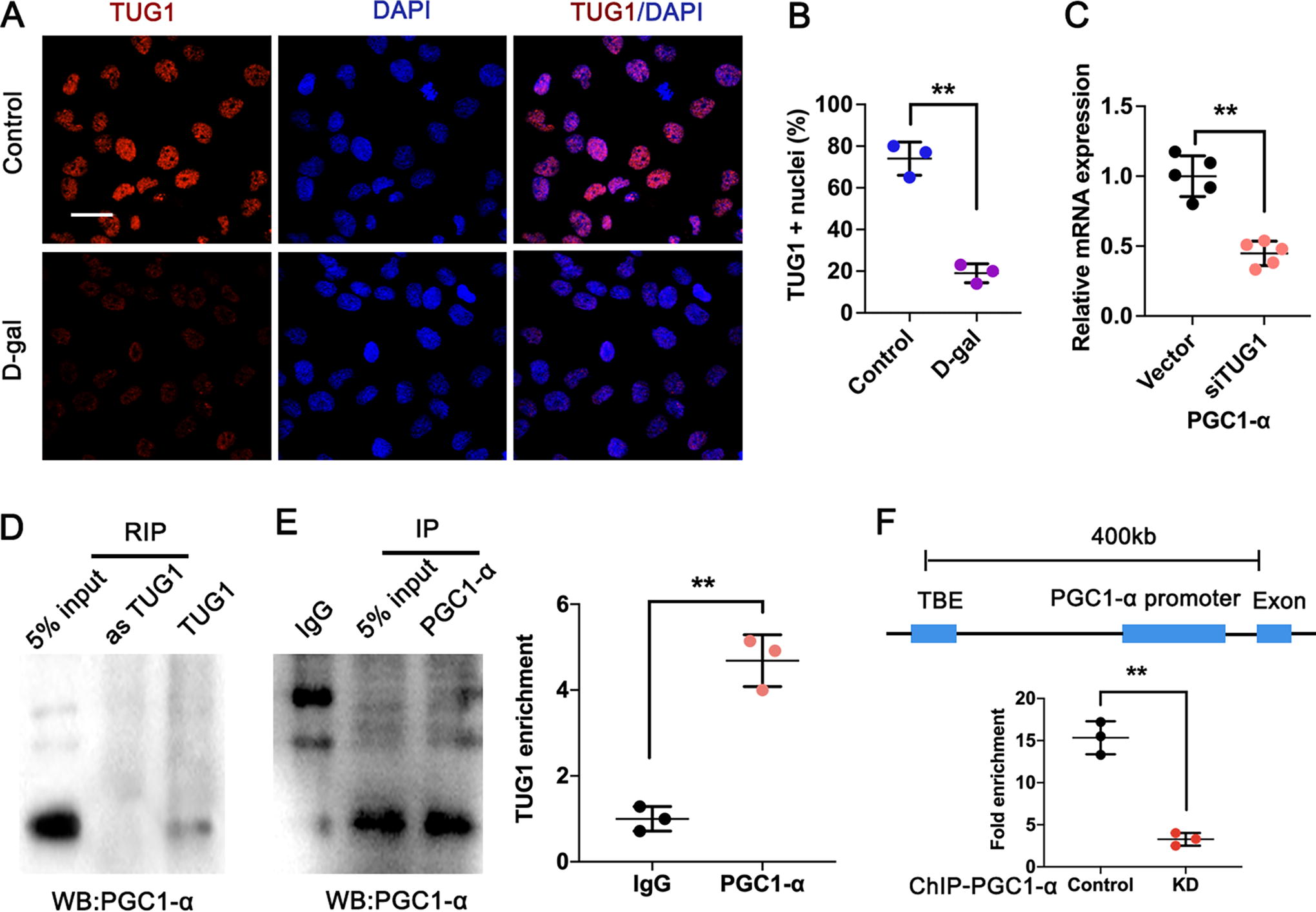
TUG1 enhanced PGC1-α expression by directly interacting with TBE region of PGC1-α
To elucidate the underlying mechanism through which TUG1 modulates the expression of PGC1-α, FISH and subcellular fraction analysis were performed to detect its distribution in accelerated aged HK-2 cells using a specific cy3-labeled TUG1 probe. The result showed that TUG1 predominantly localized within the nucleus and exhibited downregulation in accelerated aged HK-2 cells, suggesting its potential role as a transcriptional regulator in the nucleus (Fig. 6A, B). Then we detected the expression of PGC1-α in TUG1-silenced HK-2 cells using RT-qPCR. As depicted in Figure 6C and Supplementary Figure S2, knockdown (KD) TUG1 significantly suppressed the expression of PGC1-α in HK-2 cells. The RIP assay was subsequently conducted to validate the interaction between TUG1 and PGC1-α by employing biotinylated sense and antisense TUG1 RNA mixed with nuclear extracts from cultured HK-2 cells. Immunoblot analysis targeting PGC1-α demonstrated that only the sense TUG1, not the antisense counterpart, interacted with PGC1-α (Fig. 6D). We confirmed this result through the implementation of reciprocal IP using a biotin-conjugated PGC1-α antibody. Furthermore, RNA transcript levels of TUG1 were measured in the IP complex. The results showed a great abundance of TUG1 in the PGC1-α-pulldown RNA complex compared with IgG controls (Fig. 6E). ChIP analysis of the PGC-1α, followed by qPCR validation, demonstrated a significant enrichment in the TCF/LEF binding (TBE) element of PGC1-α in TUG1 WT HK-2 cells. However, this enrichment was significantly weakened in TUG1-KD HK-2 cells (Fig. 6F). These findings suggested that TUG1 might enhance PGC1-α expression by directly interacting with the TBE elements in the promoter region of PGC1-α.
TUG1 improved age-related MQC in accelerated aging model
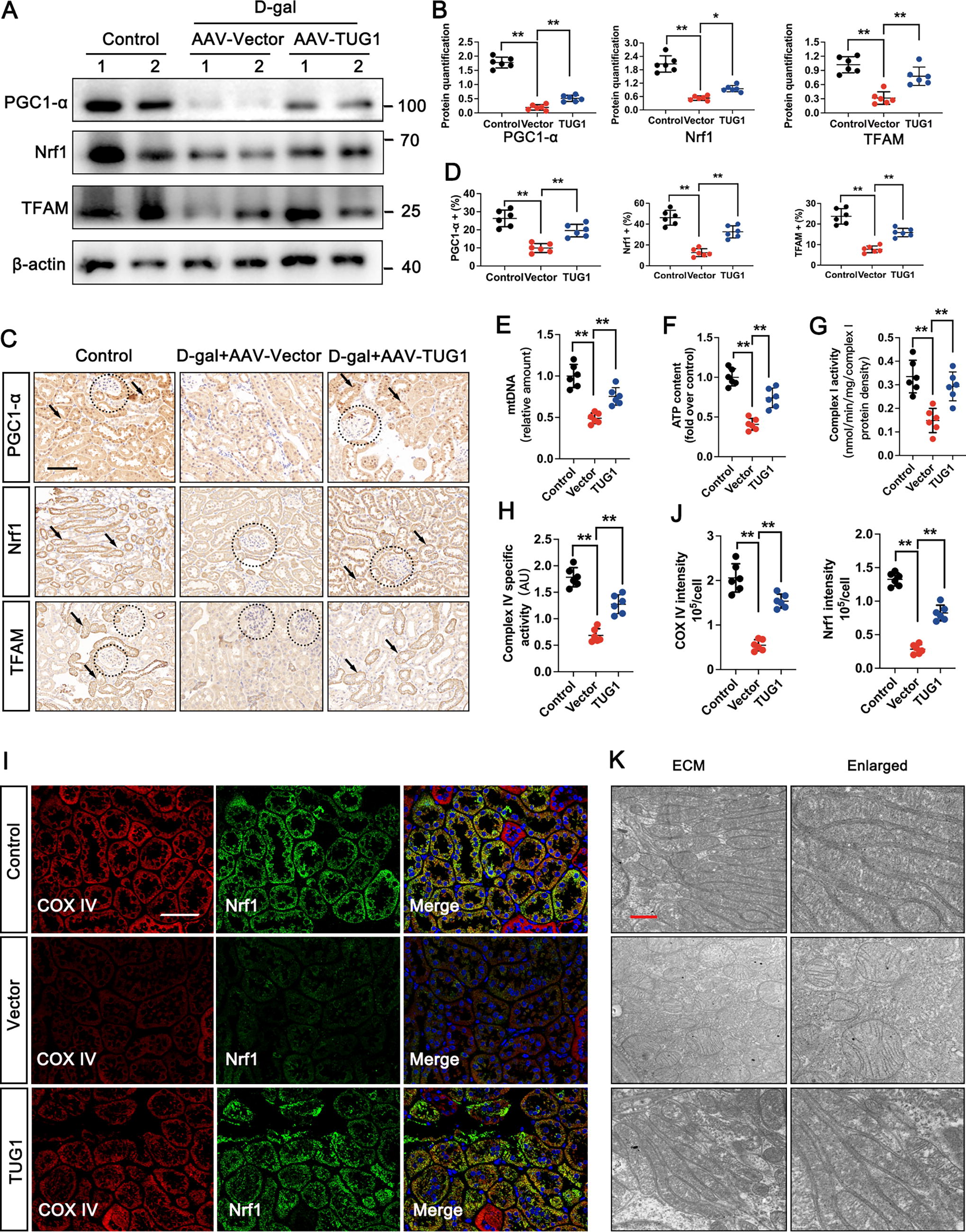
Given the observed direct interaction between TUG1 and PGC1-α in aged kidneys, it was deemed important to investigate whether TUG1-driven kidney aging involved MQC. We established a mouse model of accelerated aging with TUG1 overexpressed by administering AAV-TUG1. Both Western blot and IHC assays demonstrated that overexpression of AAV-TUG1 effectively restored the decreased expressions of PGC1-α, nuclear respiratory factor 1 (Nrf1), and TFAM in the D-gal-treated group (Fig. 7A–D). The results depicted in Figure 7E-H also revealed a reduction in mitochondrial DNA (mtDNA) and ATP content, as well as decreased expressions of COX I and COX IV in the D-gal-treated group. However, these effects were significantly reversed upon overexpression of TUG1. IF staining of the colocalization between COX IV and Nrf1 revealed that TUG1 overexpression in the D-gal-treated group maintained normal mitochondrial redox homeostasis by enhancing the expression of COX IV and Nrf1 (Fig. 7I, J). In addition, the D-gal-treated group exhibited mitochondrial swelling and disorganized cristae, while TUG1 overexpression preserved structurally intact mitochondria in accelerated aging model (Fig. 7K).
Downregulated expression of METTL14 resulted in reduced TUG1 m6A modification
Based on the m6A-lncRNA epitranscriptomic microarray analysis, we identified TUG1 as a critical player involved in aging kidney with significantly reduced m6A modification observed in the H/aged group. METTL14, the most significant RNA m6A methylase, was further investigated for the potential correlation with TUG1. Therefore, the expression and cellular localization of METTL14 were determined. Western blot showed that the protein expression of METTL14 was lower in the 24-month-old group (Fig. 8A). Both IHC and IF staining confirmed METTL14 was mainly localized in the cell nucleus. Importantly, the results indicated that the level of METTL14 was significantly decreased in the 24-month-old group when compared with the 6-month-old group (Fig. 8B–E).

Next, HK-2 cells transfected with siRNA of METTL14 (siMETTL14) were cultured. The level of TUG1 was significantly lower in the presence of siMETTL14 (Fig. 8F). Furthermore, RIP-PCR and RIP-qPCR assay confirmed that the direct interaction between TUG1 and METTL14, and the binding ability of TUG1 to METTL14 are reduced in 24-month-old mice (Fig. 8H–J). Interestingly, m6A level of TUG1 was also significantly decreased in the presence of siMETTL14 (Fig. 8K). According to the enriched and specific m6A peaks of TUG1 predicted with the sequence-based RNA adenosine methylation site predictor (SRAMP) tool, two putative m6A sites of TUG1 were mutated from A to G, referring to TUG1-Mut1, TUG1-Mut2 (one potential m6A site), and TUG1-Mut3 (two potential m6A sites) (Fig. 8L, left panel). The levels of TUG1 m6A modification in HK-2 cells with siMETTL14 and/or TUG1-mutant genes were investigated by using MeRIP-qPCR. As we hypothesized, downregulation of METTL14 reduced the m6A modifications of TUG1-WT, TUG1-Mut1, and TUG1-Mut2 in HK-2 cells when compared to the control group. Surprisingly, the m6A modification of TUG1-Mut3, which contained mutation at two m6A sites, did not exhibit significant changes with siMETTL14 (Fig. 8L, right panel). It was manifested that the m6A site mutation resulted in a reduced binding efficacy between lncRNA TUG1 and METTL14. Thus, we demonstrated that the dysregulation of m6A modification of TUG1 was attributed to the abnormal expression of METTL14 during kidney aging.
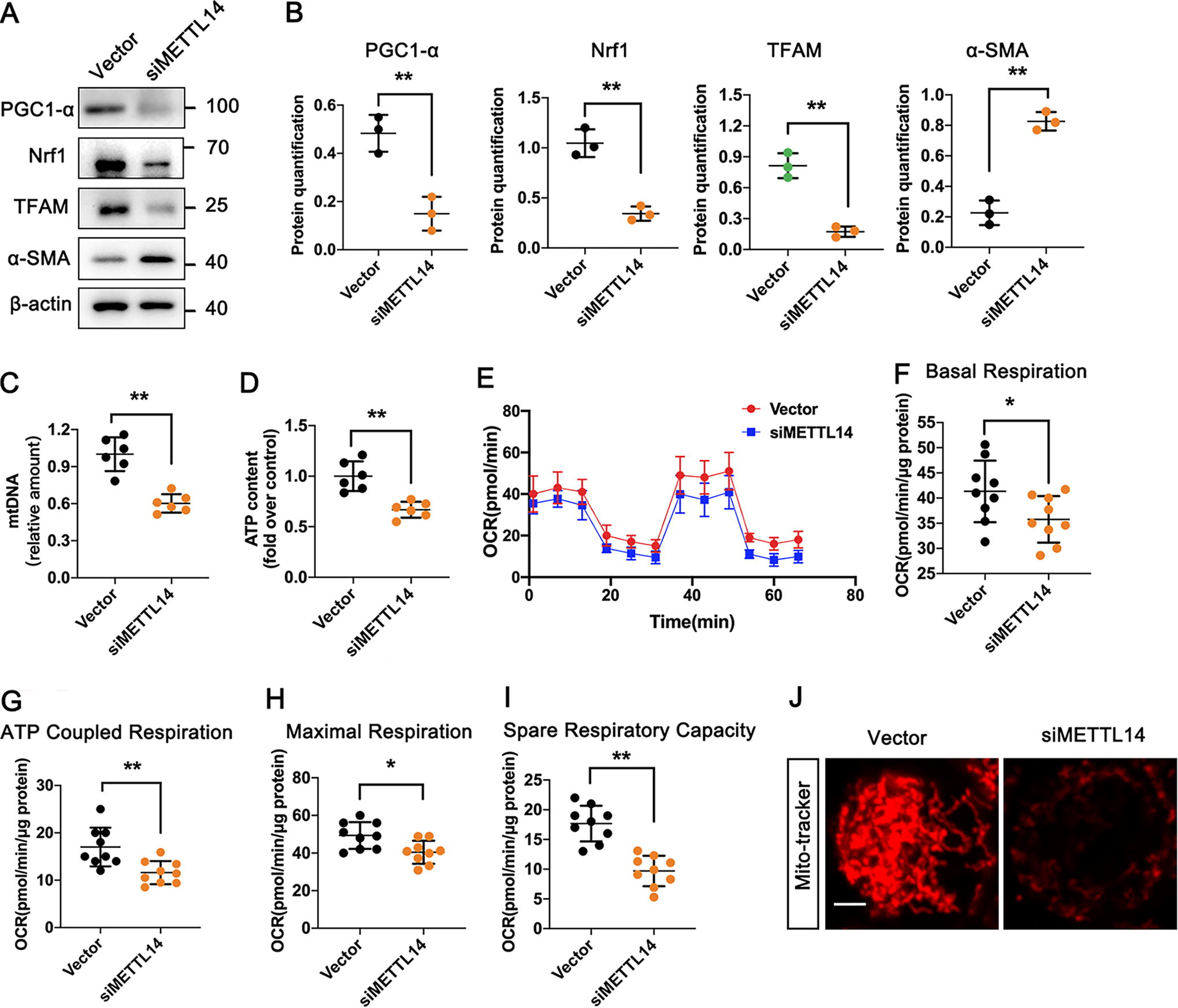
METTL14 regulated the homeostasis of MQC in kidney aging
As we have already confirmed that TUG1 could regulate the homeostasis of MQC during kidney aging, it was reasonable to presume that inhibition of METTL14 may affect mitochondrial functions through TUG1 in senescent HK-2 cells. After transfecting with siMETTL14, the expression of PGC1-α, Nrf1, and TFAM was downregulated, while α-SMA was upregulated (Fig. 9A, B). Moreover, HK-2 cells with siMETTL14 exhibited lower mtDNA and ATP contents (Fig. 9C, D), accompanied by the reduced mitochondrial respiration activities, including basal respiration, ATP-coupled respiration, maximal respiration, and spare respiration capacity by oxygen consumption rate analysis (Fig. 9E-1). Consistently, the mitochondrial morphology exhibited normal filamentous shape in control group (vector only); however, it transformed into short rod shape with siMETTL14 (Fig. 9J).
IGF2BP2-mediated regulation of lncRNA TUG1 stability in mice kidney aging
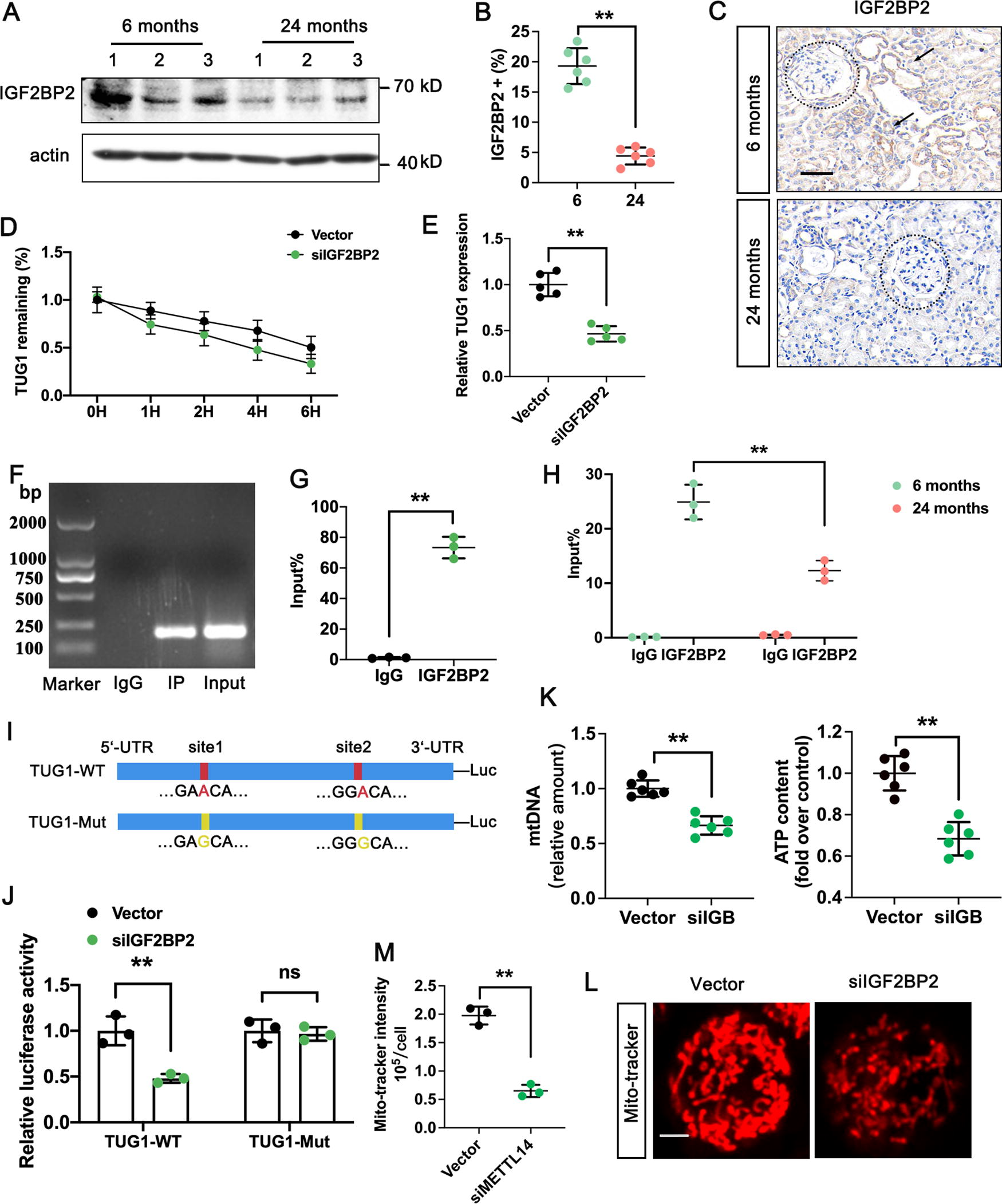
The m6A-modified RNA is identified by “reader” proteins that affect downstream. Recently, IGF2BP2 was identified as one of these “reader” proteins (Cao et al., 2023). In our study, we observed that IGF2BP2 was weakly expressed in the 24-month-old group and mainly localized in the renal tubules (Fig. 10A–C). To explore the function and regulatory mechanism of IGF2BP2 on TUG1 in kidney aging, we used siRNAs to silence IGF2BP2 in HG-induced HK-2 cells and test the stability of lncRNA TUG1 by RNA decay assay. The results showed that the stability of lncRNA TUG1 decreased when IGF2BP2 was depleted. In addition, the RNA level of TUG1 was significantly lower in the presence of siIGF2BP2 (Fig. 10D, E). We confirmed the specific interaction between IGF2BP2 and lncRNA TUG1 through the RIP assay, and the binding ability of TUG1 to IGF2BP2 was reduced in 24-month-old mice (Fig. 10F–H). The luciferase reporter assay was performed to further identify whether IGF2BP2 could directly bind to the m6A modification sites of lncRNA TUG1. The results showed that the luciferase activity of the reporter carrying the wild-type 3′ UTR fragment of TUG1 was significantly reduced in the siIGF2BP2 group, while the activity in the TUG1-Mut group did not change significantly (Fig. 10I, J). Moreover, silence of siIGF2BP2 in HK-2 cells exhibited lower mtDNA and ATP contents (Fig. 10K), along with abnormal mitochondrial morphology compared with vector group (Fig. 10L). Our research demonstrated that the methylated lncRNA TUG1 was directly identified by IGF2BP2, maintaining the stability of methylated lncRNA TUG1 in HK-2 cells through an m6A-IGF2BP2-dependent mechanism.
Discussion
The kidney is one of the organs that undergoes rapid aging (Choudhury and Levi, 2011, Docherty et al., 2019). With advanced age, the kidney is subjected to morphological changes such as tubular atrophy, interstitial fibrosis, and glomerulosclerosis, and contributes to a decline of kidney function (Docherty et al., 2019). Aging is also an independent risk factor for chronic kidney disease, which is more common among elderly individuals. Various cellular processes and molecular pathways are involved in the complex process of kidney aging, but the underlying mechanisms remain unclear (Fang et al., 2020). Current research has shown a direct correlation between mitochondrial dysfunction and renal aging (Miao et al., 2019). In this study, we demonstrated that lncRNA TUG1 acts as a protective factor to maintain mitochondrial homeostasis by directly targeting PGC1-α, thereby alleviating cellular senescence and kidney aging.
Mitochondria is the center of cellular energy metabolism, maintaining dynamic balance through continuous fusion and division to adapt to the energy needs of cells under different environmental conditions (Pickles et al., 2018). Dysfunction in mitochondrial function leads to a reduction in ATP synthesis, oxidative stress, and inflammatory reactions that collectively contribute to the development of various diseases (Doke and Susztak, 2022; Mito et al., 2022; Wang et al., 2022). The proximal tubule and thick ascending limb of Henle’s loop are the most densely populated regions for mitochondria in the kidney; therefore, stable mitochondrial structure and function are critical factors for maintaining renal tubular epithelial cell function (Yao et al., 2022). Increasing evidences suggest that abnormal mitochondrial dynamics are associated with various pathological processes, including renal aging (Gallage and Gil, 2016; Wang et al., 2022; Xu et al., 2023). Several studies have shown that aged mice exhibit multiple changes in their renal tubules’ mitochondria, including decreased mitochondrial numbers, loss of cristae structure, hypertrophy, and membrane rupture (Rosa et al., 2023). Similarly, our research has observed damaged mitochondrial structures such as swelling and fragmentation along with disrupted cristae, while ATP levels decrease alongside mtDNA levels and oxidative phosphorylation (OXPHOS) complex activity declines with age. Renal tubular cell mitochondrial homeostasis imbalance results in an abnormal increase in ROS production, leading to kidney fibrosis. More studies confirm that dysfunction in mitochondrial function is closely related to renal aging (Rosa et al., 2023). Research has found that functional decline occurs early on before any morphological tissue change occurs during aging (Docherty et al., 2019).
In the early stages of renal aging, mutations in key genes or signaling mechanisms may be involved in the imbalance of MQC. TUG1, identified from m6A lncRNA microarray analysis, has been shown to be downregulated in human aged kidney samples, as well as in an accelerated aging model. Importantly, both in vivo and in vitro assays supported that the overexpression of TUG1 inhibits the expression of aging-related proteins, reduces SA-β-gal deposition, and alleviates kidney fibrosis. Based on these findings, we elucidate that TUG1 plays a critical role in MQC during renal aging process. PGC-1α is a tissue-specific transcription factor primarily expressed in tissues with high energy demands or abundant mitochondria such as heart, skeletal muscle, kidney, and liver (Huang et al., 2017; Sin et al., 2016; Qin et al., 2020; Wu et al., 2020a). Considering that the expression level of PGC-1α is directly correlated with mitochondrial biosynthetic activity, we silenced TUG1 in HK-2 cells and found that the level of PGC1-α is significantly decreased. The results indicated that PGC1-α may act as the target of lncRNA TUG1. As expected, bioinformatics analysis and ML predicted that TUG1 formed potentially strong interaction with PGC1-α. We further performed the RIP and ChIP-PCR assays and confirmed that TUG1 can directly bind to the TBE regulatory region of PGC1-α. Furthermore, PGC1-α plays a crucial role in regulating signaling pathways for MQC. It mediates transcription and replication of mtDNA through promoting Nrf1/2, as well as TFAM, thereby preventing oxidative damage caused by injury and inflammation(Ye et al., 2022). Our study demonstrates that the overexpression of TUG1 effectively restores decreased expression of PGC1-α in an accelerated aging model, resulting in increased mtDNA quantity and ATP content, along with elevated expressions of COX I and COX IV. These findings suggest that PGC1-α serves as an important regulatory factor for mitochondrial biogenesis driven by TUG1 during renal aging. However, the human kidney samples we collected were obtained from the distal normal tissue of cancer patients. Although they were histologically confirmed to be composed of normal cells, the availability of human aging tissue samples remained a limitation in this study.
M6A is the most common type of RNA modification in eukaryotic cells and plays a crucial role in various diseases (Sendinc and Shi, 2023). METTL14 is a key component of the m6A methyltransferase complex, catalyzes methylation reactions, and has been reported to be involved in kidney diseases (Liu et al., 2014). For instance, METTL14 affects HDAC5-mediated epithelial–mesenchymal transition in diabetic nephropathy tubular cells through the phosphoatase and tensin homolog (PTEN)-regulated PI3K/Akt signaling pathway (Xu et al., 2021). In addition, by downregulating Sirt1 through m6A-dependent mechanisms, METTL14 exacerbates podocyte injury and progression of glomerulopathy (Lu et al., 2021). Our study demonstrates that knockdown METTL14 decreasing the methylation and RNA levels of TUG1, accelerating cellular senescence and renal fibrosis through regulating MQC. Therefore, we conclude that METTL14-induced m6A modification on TUG1 is an important molecular mechanism that regulates TUG1 RNA during kidney aging.
As a newly discovered “reader” protein, IGF2BP2 can target and recognize m6A-modified RNA to enhance its stability (Cao et al., 2023). We found that IGF2BP2 is weakly expressed in the elderly kidney and interacts specifically with TUG1 RNA. Research has shown that IGF2BP2 can selectively bind to various tumor-driving factors such as MYC and differentiation antagonizing non-protein-coding RNA (DANCR), enhancing their RNA stability and influencing post-transcriptional translation processes, thereby promoting tumor cell progression and tumor stem cell characteristics (Weng et al., 2022). Similar to previous studies, we found that siIGF2BP2 inhibited the stability of TUG1 RNA, leading to a decrease in its expression level, indicating that IGF2BP2 is probably involved in enhancing the RNA stability of m6A-modified TUG1.
Herein, the METTL14/IGF2BP2-TUG1 axis plays a crucial role in renal aging by controlling mitochondrial quality. The downregulation of METTL14 leads to abnormal methylation of TUG1, impacting the stability and expression of TUG1 RNA in an IGF2BP2-dependent manner. Furthermore, TUG1 plays a regulatory role in maintaining mitochondrial homeostasis and cellular integrity, thereby delaying cellular senescence and renal fibrosis through PGC-1α.
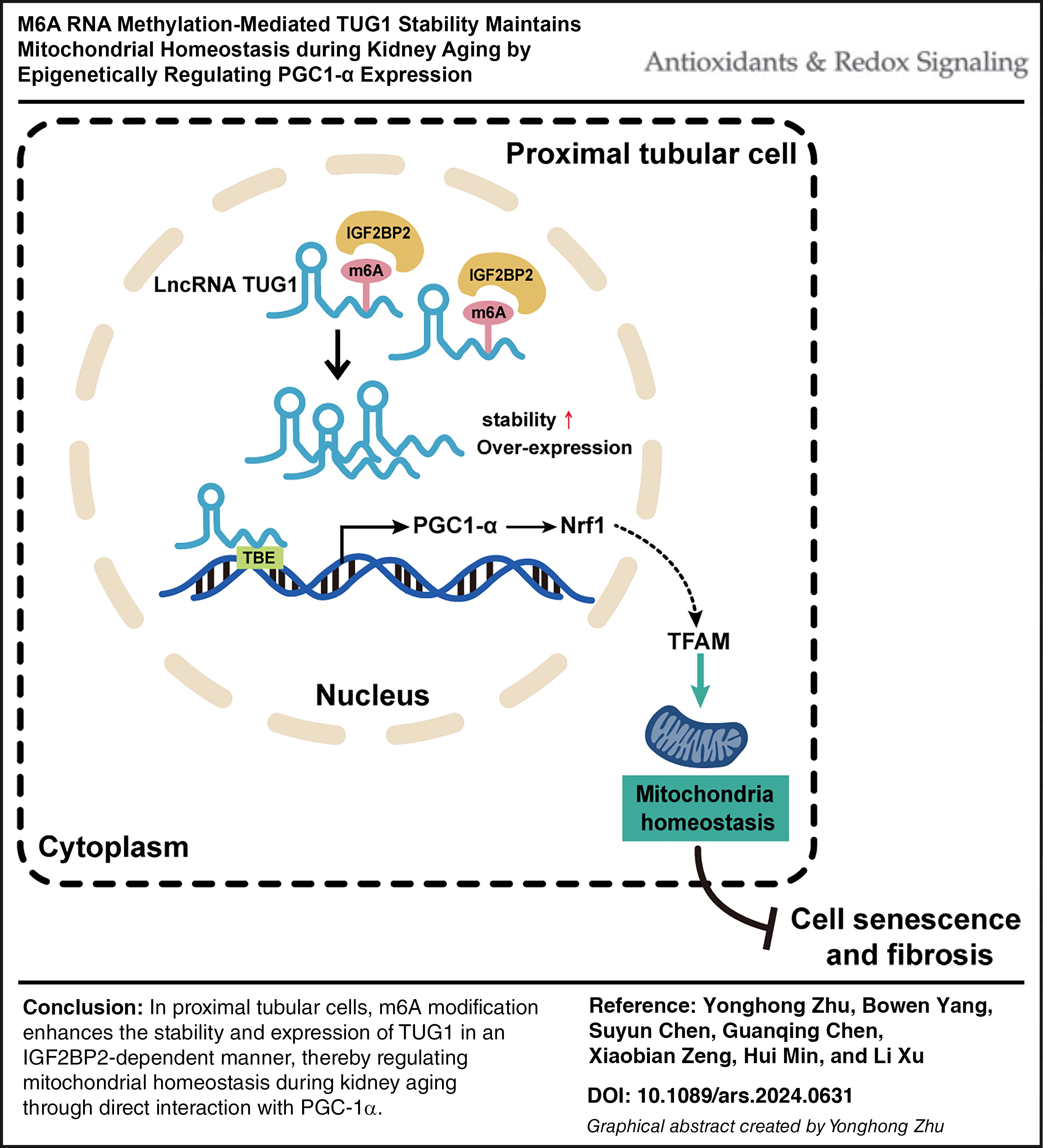
Graphic Summary
We propose a critical role of the METTL14/IGF2BP2-TUG1 axis in renal aging and elucidate the TUG1-driven mechanism for mitochondrial quality control. The downregulation of METTL14 results in abnormal methylation of TUG1, leading to decreased stability and expression of TUG1 RNA in an IGF2BP2-dependent manner. The TUG1-driven mitochondrial quality control targets PGC-1α to regulate mitochondrial homeostasis and cellular integrity, thereby delaying cell aging and kidney fibrosis (Graphic abstract).
Materials and Methods
Electronic laboratory notebook was not used
Human specimens
A total of 20 kidney tissues were collected from participants who were diagnosed with renal tumor at the First Affiliated Hospital of China Medical University. The exclusion criteria were as follows: no radiotherapy or chemotherapy was performed before nephrectomy. Participants with other systemic dysfunctions were excluded. Informed consent was provided by all participants before the research started. The protocol for this research was approved by the Research Ethics Committee of China Medical University (Reference number: [2020] 120). People older than 65 years suffered from a high risk of end-stage renal disease and drug-related nephrotoxicity (Hommos et al., 2017). Thus, all participants were divided into two groups according to age (Young: <65 years old and Aged ≥65 years old). Clinical characteristics of the participants are analyzed in Supplementary Table S1
Cell culture and treatments
Human kidney proximal tubular epithelial cells (HK-2 cells) were purchased from Procell Life Science &Technology (Wuhan). The cells were cultured in an incubator with 5% CO2 at 37°C in Minimum Essential Medium (MEM, Procell, PM150410) containing 10% fetal bovine serum and 1% penicillin/streptomycin solution (P/S). HK-2 cells were treated with D-gal (100 mM) for 72 h, referred to as D-gal-treated HK-2 group.
The human overexpressed TUG1 gene was amplified and subcloned into the eukaryotic expression vector pcDNA3.1. The specific targeting sequence of TUG1 siRNA, siMETTL14, and siIGF2BP2 (Syngentech, China) was transfected with Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. The scrambling sequence was used as a control. The siRNA sequences are listed in Supplementary Table S2.
Animal experimental design
The animal experimentation protocol was approved by the Research Ethics Committee of China Medical University (Approval number: KT2020024). All animal experiments were conducted in the specific pathogen free animal laboratory at China Medical University, following the guidelines provided by the NIH Guide for the Care and Use of Laboratory Animals. C57BL/6J mice were sourced from Charles River Laboratories and maintained under standard conditions with ad libitum access to water and chow. For the natural aging model, mice were euthanized at ages of 6 months (representing the young group, n = 8) and 24 months (representing the old group, n = 8) (Miao et al., 2019). To mimic the natural aging process in rodents, we employed an accelerated aging model by injecting D-gal into C57BL/6 mice. D-gal is known to activate senescence-related signals by reacting with amino acids’ free amines, leading to advanced glycation end product formation along with oxidative stress and inflammation. The D-galactose-induced aging model has been widely used in the study of kidney aging (Miao et al., 2019, Gao et al., 2022). In brief, mice received subcutaneous injections of D‐gal at a dosage of 500 mg kg-1 day-1 from weeks 8 to 16. At week 9, adeno-associated viruses (Syngentech, Beijing, China) carrying TUG1 expression vector (AAV2/9-TUG1) were injected into the renal pelvis of D-gal-treated mice. One hundred microliters of pAAV2/9-TUG1-FLAG (NR_152867.2) and empty vector (1 × 1012 virus genome/mL) were injected into the renal pelvis of mice. Three groups were established as follows: 1. control group; 2. D-gal-treated group with AAV-TUG1; and 3. D-gal-treated group with AAV-vector. The number of mice in each group was eight.
IHC staining
The human or mouse kidney tissues were cut into 3-μm-thick sections. For histological observation, well-prepared samples were performed by PAS and Masson staining.
For IHC staining, paraffin sections were placed in a citrate buffer solution after hydration. The sections were performed at high power repair for 8 min, medium power for 20 min in microwave, and then cooled at room temperature for 1 h. The tissue edges were circled using a histological pen and then treated with a droplet of 3% H2O2 solution. After incubating for 30 min, the sections were washed with PBS three times. A blocking solution of 3% BSA was applied for an hour. Subsequently, primary antibody specific for P16INK4A (1:200, Thermo Fisher Scientific Cat# MA5-17142), γ-H2AX (1:200, Proteintech 10856–1-AP), fibronectin (1:500, Abcam ab2413), α-SMA (1:500, Abcam ab5694), collagen type I (1:100, Abcam ab21286), METTL14 (1:500, Abcam ab220030), IGF2BP2 (1:500, Abcam ab124930), PGC1-α (1:300, Proteintech 66369–1-Ig), Nrf1(1:500, Proteintech 66832–1-Ig), and TFAM (1:500, Abcam ab307302) was incubated overnight at 4°C. The next day, secondary antibody was added and incubated at room temperature for 30 min. The sections were added with freshly prepared DAB solution (Solarbio) for 1–10 min before stopping by immersing the slides in tap water. Hematoxylin counterstaining was performed for 2–3 min followed by differentiation with a 1% mixture of alcohol and hydrochloric acid lasting 5–10 s, rinsing with tap water until slides turned blue.
m6A-lncRNA epitranscriptomic microarray analysis
Human m6A-lncRNA epitranscriptomic microarray was performed based on the Arraystar’s standard protocols. Briefly, total RNAs were immunoprecipitated with anti-m6A antibody. The “IP” and “Sup” RNAs were labeled with Cy5 and Cy3, respectively. The RNAs were hybridized to Arraystar Human lncRNA Epitranscriptomic Microarray (8 × 60 K, Arraystar). Raw intensities of IP and Sup were normalized with average of log2-scaled Spike-in RNA intensities.
Transmission electron microscope observation
Relevant sections (80 nm) of the samples were obtained using an ultramicrotome, followed by staining with uranyl acetate and lead citrate. Subsequently, the samples were rinsed and vacuum-dried at room temperature to facilitate observation of the ultrastructure, particularly focusing on foot process effacement and glomerular basement membrane (GBM) through transmission electron microscope. For semiquantitative analysis, ImageJ software was used to measure the thickness of GBM as previously described (Xu et al., 2023).
SA-β-gal staining
Frozen section (8 μm) was prepared for SA-β-gal staining by using Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, 9860).
Quantification of m6A modifications
RNA was extracted from mouse kidney samples using Trizol reagent. After sample quality uniformity, EpiQuik m6A RNA Methylation Quantification Kit (Epigentek P-9005–48) was carried out to assess m6A levels according to the manufacturer’s instructions.
Western blot
Total proteins were extracted using RIPA lysis buffer (Beyotime), while nuclear and cytoplasmic proteins were extracted by Cell Nucleus/Cytoplasm Protein Extraction Kit (Beyotime, P0028). Protein concentration was determined by the BCA kit (Beyotime, P0010). The same amounts of protein samples were loaded in gel plates for electrophoresis and subsequently transferred onto PVDF membranes (Millipore, IPVH00010). Next, the membranes were blocked with 5% nonfat milk for 1 hour and incubated with primary antibody, including P16INK4A (1:1000, Thermo Fisher Scientific MA5-17142), γ-H2AX (1:1000, Proteintech 10856–1-AP), fibronectin (1:5000, Abcam ab2413), α-SMA (1:2000, Abcam ab5694), collagen type I (1:1000, Abcam ab260043), METTL14 (1:2000, Abcam ab308576), IGF2BP2 (1:2000, Abcam ab124930), PGC1-α (1:1000, Proteintech 66369–1-Ig), Nrf1(1:1000, Proteintech 66832–1-Ig), and TFAM (1:1000, Abcam ab307302) at 4°C overnight. After being incubated with HRP-conjugated secondary antibody (1:10000, Abbkine) for 1 h, the membranes were visualized using ECL kit (Tanon, 180–506) and Tanon-4500 Gel Imaging System (Tanon). Quantification of immunoblots was performed with ImageJ.
RT-qPCR
The total RNAs of HK-2 cells were extracted with Trizol Reagent (Takara) Then, RNAs were retro-transcribed with HiScript III 1st Strand cDNA Systhesis kit (Vazyme, R312) and RT-qPCR was performed with ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711). The Thermo Real-Time PCR analysis system is used to complete the reaction through steps with predenaturation, cycling, and melting curve. β-actin was used as an endogenous control.
IF staining
Cover slips were placed in a 48-well plate and were sterilized under a UV lamp in a biosafety cabinet for 30 min. HK-2 cells were inoculated onto the cover slips in the 48-well plate with different treatment. The cells were fixed by 4% paraformaldehyde for 20 min and were perforated by 0.1% TritonX-100 for 15 min. Then 5% BSA blocking solution (Solarbio) was added into the 48-well plate, incubating for 30 min at room temperature, followed by incubating with the primary antibody P16INK4A (1:100, Thermo Fisher Scientific MA5-17142), α-SMA (1:500, Proteintech, 67735–1-Ig), METTL14 (1:200, Abcam ab308576), and E-cadherin (1:100, Proteintech, 20874–1-AP) at 4°C overnight. On the next day, the cells were incubated with fluorescence secondary antibody working solution (1:100, Abcam) at room temperature for 1 h and stained with DAPI solution (Solarbio) for five minutes. The fluorescence images were observed under a laser confocal fluorescence microscope (Nikon).
Fluorescence in situ hybridization
The HK-2 cells were fixed in 4% paraformaldehyde on the slides and treated with proteinase reagents. The slides underwent incubation at 40°C for 4 h in a buffer solution that had been prehybridized. Overnight hybridization was then carried out using digoxigenin-labeled probes at the same temperature. After washing and blocking, biotinylated anti-digoxigenin antibodies were used to incubate the slides. The slides were then stained with DAPI solution before capturing images through confocal microscopy. The TUG1 probe used was as follows: 5′-DIG-AATCTACCTCCAGTGTTCCTGCCGCATCGTG-DIG-3′.
m6A-LncRNA epitranscriptomic microarray analysis
Human m6A epitranscriptomic microarray and lncRNA microarray analysis were performed based on the Arraystar’s standard protocols. Briefly, total RNAs were immunoprecipitated with anti-m6A antibody. The “IP” and “Sup” RNAs were labeled with Cy5 and Cy3, respectively. The RNAs were hybridized to Arraystar Human LncRNA Epitranscriptomic Microarray (8 × 60 K, Arraystar). Raw intensities of IP and Sup were normalized with average of log2-scaled Spike-in RNA intensities. The raw data have been deposited in the Gene Expression Omnibus database under the accession number GSE232249.
CUT&RUN m6A RNA immunoprecipitation
EpiQuik CUT&RUN m6A RNA Enrichment Kit (Epigentek, MA, USA) was used to perform MeRIP as described in previous studies. Briefly, the extracted RNAs (about 10 μg) were added into the Immune Capture Buffer, rotating at room temperature for 90 min. Then, the solution was added with 10ul of nuclear digestion enhancer and 2 µL of cleavage enzyme mix, incubating for 4 min. Each reaction tube was washed three times with 150 µL of Wash Buffer, digested by 150 µL of protein digestion buffer (PDB) on the magnetic rack. Next, protein digestion solution was prepared by mixing protease K and PDB in a ratio of 1:10 and mixed with the beads, followed by rotation at 55°C for 15 min in a thermal cycler. Twenty microliters of RNA purification solution and 160 µL anhydrous ethanol were added sequentially. The RNA binding beads were resuspended and washed twice with 90% ethanol. Finally, the magnetic beads were captured using a magnetic device until the solution became completely transparent. The mixture was then transferred to a new PCR tube for RT-qPCR.
The selection of aging-related feature genes
This study applied six dimensionality reduction methods: Bagged Trees, Bayesian, RF, Wrapper (Bpruta), Learning Vector Quantization (LQV), and 10-fold cross-validated Least Absolute Shrinkage and Selection Operator (LASSO)-Logistic with 1000 iterations.
The objective function of the LASSO-logistic regression model is as follows:
The RF model conducted 1000 simulations and calculated the importance of the final results, ranking them accordingly. Ultimately, aging-related differentially expressed genes with an importance score greater than 0.2 were selected as the final screened results.
LVQ is a type of artificial neural network introduced by Kohonen and his colleagues. If the synapses are frequently used, they become stronger and function more effectively.
Bagging, or Bootstrap Aggregating, is one of the latest and most successful computational methods for improving unstable classification or classification processes. The final prediction is obtained by averaging (for regression tasks) or voting (for classification tasks) from these base models.
Boruta is a wrapper algorithm for feature selection, which is fully integrated with any classification method that outputs variable importance measures. This method compares the importance of the original attributes with the importance obtained randomly, estimates using their permutation importance, and gradually eliminates irrelevant features to stabilize the test, thus performing top–down selection of relevant features.
A Bayesian network is a probabilistic model consisting of two parts: a dependency structure and a local probability model.
Finally, the intersecting feature genes selected by these six ML algorithms were identified as aging-related feature genes. We intersect the genes selected by the six models mentioned above. If a gene is identified as an aging-related feature gene four times or more, we consider it as Hub mRNAs, which are important aging-related feature genes. Visualization of the intersected feature genes can be achieved using the R package UpSet.
RT-qPCR
The cells were collected and resuspended with an equal volume of RIP lysis buffer (Millipore). Protein A/G agarose beads were mixed to homogeneity, and then 50 μL was pipetted into a centrifuge tube for each sample and washed twice with 0.5 mL of RIP washing buffer. The magnetic beads were resuspended in 100 μL RIP washing buffer, added with either 5 µg of specific antibodies against PGC1-α (Thermo Fisher Scientific PA5-72948) or IgG antibody, and incubated at room temperature for 30 min. The mixture was centrifuged at 4°C, at a speed of 4000 rpm for 1 min, and was washed five more times. Then, protein A/G-antibody complex was resuspended in 900 μL RIP washing buffer with RNase inhibitor. The cell lysate was thawed on ice and centrifuged at 14,000 rpm for 10 min. Then 10 μL of the cleared lysate was taken as Input, and stored at −80°C. Next, 100 μL supernatant and 900 μL Protein A/G-antibody complex were incubated overnight at 4°C with gentle rotation. After washing five more times, the complex was resuspended in 150 µL proteinase K buffer and incubated at 55°C for 30 min. RNA was extracted using 1 mL Trizol Reagent (Takara) and precipitated with 6 µg glycogen for further PCR tests.
Chromatin immunoprecipitation assay
The Chromatin Immunoprecipitation Kit (Millipore, 17–371) was utilized for conducting the ChIP experiment. In brief, fixed cells fragmented chromatin was incubated with a PGC1-α antibody (Thermo Fisher Scientific PA5-72948) and protein G magnetic beads. QPCR analysis was performed on the DNA released from the precipitates. The primer sequences used were specific to the upstream TBE element of PGC1-α binding region: forward, 5′-CCTCTTTCCCTTTCAGTCTCC and reverse, 5′-CCACCTTGAACACTATTCCTG-3′. Negative controls included IgG and no antibody condition.
Luciferase reporter and mutation assay
The TUG1-Mut mutants were generated using the Mut Express II Fast Mutagenesis Kit V2 (Vazyme). HK-2 cells were transfected with 250 ng 3’ UTR luciferase reporter plasmid (Promega). The luciferase activity was assessed using the Dual Glo Luciferase Assay kit (Promega), as instructed by the manufacturer.
RNA decay assay
The cells were added with fresh complete medium supplemented with actinomycin D (5 μg/mL) after a different treatment. Then the cells were continued to be cultured for an additional 30 min before being collected at specified time points (0 h, 1 h, 2 h, 4 h, and 6 h). RT-qPCR was performed to detect the remaining levels of lncRNA TUG1 at each time point using 2-ΔΔCt for calculation.
Mitochondrial oxygen consumption rate measurement
The rates of mitochondrial oxygen consumption in HK‐2 cells were measured by using the XF Cell Mito Stress Test Kit (Seahorse Bioscience) with a Seahorse XFe96 Extracellular Flux Analyzer (Seahorse Bioscience). The protocol and analysis methods were executed in accordance with the previously established procedures (Xu et al., 2023).
Statistical analysis
All statistical analyses were performed using the SPSS 15.0 software. Data were presented as mean ± SEM. Statistical analysis was conducted using a two-tailed unpaired Student’s t-test for comparing two groups and one-way ANOVA with Bonferroni test for multiple comparisons. Two-way ANOVA was used to compare multiple variables between multiple groups. p value <0.05 was considered significant.
Footnotes
Authors’ Contributions
L.X. led the project. L.X. and B.Y. designed and conceived the study. Y.Z. drafted the article. Y.Z., S.C., G.C., X.Z., and H.M. performed data analysis. B.Y., L.X., and H.M. revised the article. Y.Z., S.C., and G.C. performed the functional experiments.
Author Disclosure Statement
The authors declare no conflict of interests.
Funding Information
This research was supported partly by the
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.