
Abstract
Significance:
Cellular senescence is a critical process underlying aging and is associated with age-related diseases such as Alzheimer’s disease. Lipids are implicated in cellular senescence. Fatty acids, particularly eicosanoids, have been associated with various forms of senescence and inflammation, and the associated reactive oxygen species production has been proposed as a therapeutic target for mitigating senescence. When overactivated, calcium-dependent phospholipase A2 (cPLA2) catalyzes the conversion of arachidonic acid into eicosanoids such as leukotrienes and prostaglandins.
Recent Advances:
With a growing understanding of the importance of lipids as mediators and modulators of senescence, cPLA2 has emerged as a compelling drug target. cPLA2 overactivation plays a significant role in several pathways associated with senescence, including neuroinflammation and oxidative stress.
Critical Issues:
Previous cPLA2 inhibitors have shown potential in ameliorating inflammation and oxidative stress, but the dominant hurdles in the central nervous system-targeting drug discovery are specificity and blood–brain barrier penetrance.
Future Directions:
With the need for more effective drugs against neurological diseases, we emphasize the significance of discovering new brain-penetrant, potent, and specific cPLA2 inhibitors. We discuss how the recently developed Virtual Synthon Hierarchical Enumeration Screening, an iterative synthon-based approach for fast structure-based virtual screening of billions of compounds, provides an efficient exploration of large chemical spaces for the discovery of brain-penetrant cPLA2 small-molecule inhibitors. Antioxid. Redox Signal. 41, 1100–1116.
Introduction to Cellular Senescence
Cellular senescence is a key component of the aging process, and is defined as a state of cell cycle arrest accompanied by a variety of morphological and genetic changes (Campisi and d’Adda di Fagagna, 2007). Despite not dividing, these “zombie”-like cells can affect other cells through their impaired function and secretions (Kuilman and Peeper, 2009; Mosteiro et al., 2016). This set of secretions, termed the senescence-associated secretory phenotype (SASP), can reinforce a senescent state and promote the immune clearance of senescent cells (Krizhanovsky et al., 2008; Mosteiro et al., 2016). Other features of cellular senescence include DNA damage, telomere shortening, increased senescence-associated β-galactosidase (SA-β-gal) levels, mitochondrial dysfunction, and metabolic dysregulation (Table 1) (González-Gualda et al., 2021; Gorgoulis et al., 2019). However, senescence is a complex cellular phenomenon, making it challenging to study in vitro and in vivo. The senescent phenotype can vary depending on the stimulus (Özcan et al., 2016) and cell identity—with even different subpopulations of senescent cells being identified (Chen et al., 2020). With this immense heterogeneity, there is no single marker for senescence. Common markers for senescence are also not exclusive to senescence. SASP secretions, composed of cytokines and chemokines, can become elevated as part of wound healing, inflammatory responses, and other biological processes (González-Gualda et al., 2021). Importantly, while senescence is a hallmark of aging, aging itself is a broader progressive biological decline, defined by various other components (Tenchov et al., 2024). Senescent cells are important for embryonic development, regeneration, and wound healing (Demaria et al., 2014; Storer et al., 2013), and normally are cleared by the immune system. However, the number of senescent cells in the body accumulates with age (Hudgins et al., 2018; Tuttle et al., 2020), partly due to impaired immune surveillance (Ovadya et al., 2018), and this prolonged accumulation of senescent cells can promote inflammation, tissue dysfunction, and the pathogenesis of aging-related diseases such as Alzheimer’s disease (AD) (Childs et al., 2017; Olivieri et al., 2018; Tripathi et al., 2021). Considering these challenges, the standard for assessing cellular senescence consists of defining and measuring sets of cell-specific and well-accepted senescence markers (Casella et al., 2019), such as those listed in Table 1. Senescence has been identified in nearly all tissues, and in recent years, senescence-associated phenotypes of the central nervous system (CNS) cells have been better characterized. Although various contributors to senescence have been identified, there remains a large gap in our understanding of how lipids contribute to cellular senescence in the CNS. In this study, we broadly describe CNS cell-specific senescence phenotypes. Focusing on changes in the lipidome and lipid metabolism, this review presents calcium-dependent cytosolic phospholipase A2 (cPLA2) as a valuable therapeutic target for preventing cellular senescence.
ROS, reactive oxygen species; H2AX, H2A histone family member X; HMGB1, high mobility group box 1 protein; IL, interleukin; TNF-α, Tumor necrosis factor alpha; MMP, matrix metalloproteinase; CXCL, C-X-C motif chemokine ligand.
Cell-Specific Senescence Phenotypes in the Brain
Biological aging and accumulating pathologies are thought to enhance senescence in the brain, eventually leading to neurodegeneration and cognitive decline (Lau, Ramer and Tremblay, 2023). The main cells of the CNS—neurons, microglia, and astrocytes—display classical senescence phenotypes, with some key differences reflective of their distinct functional roles (Fig. 1).
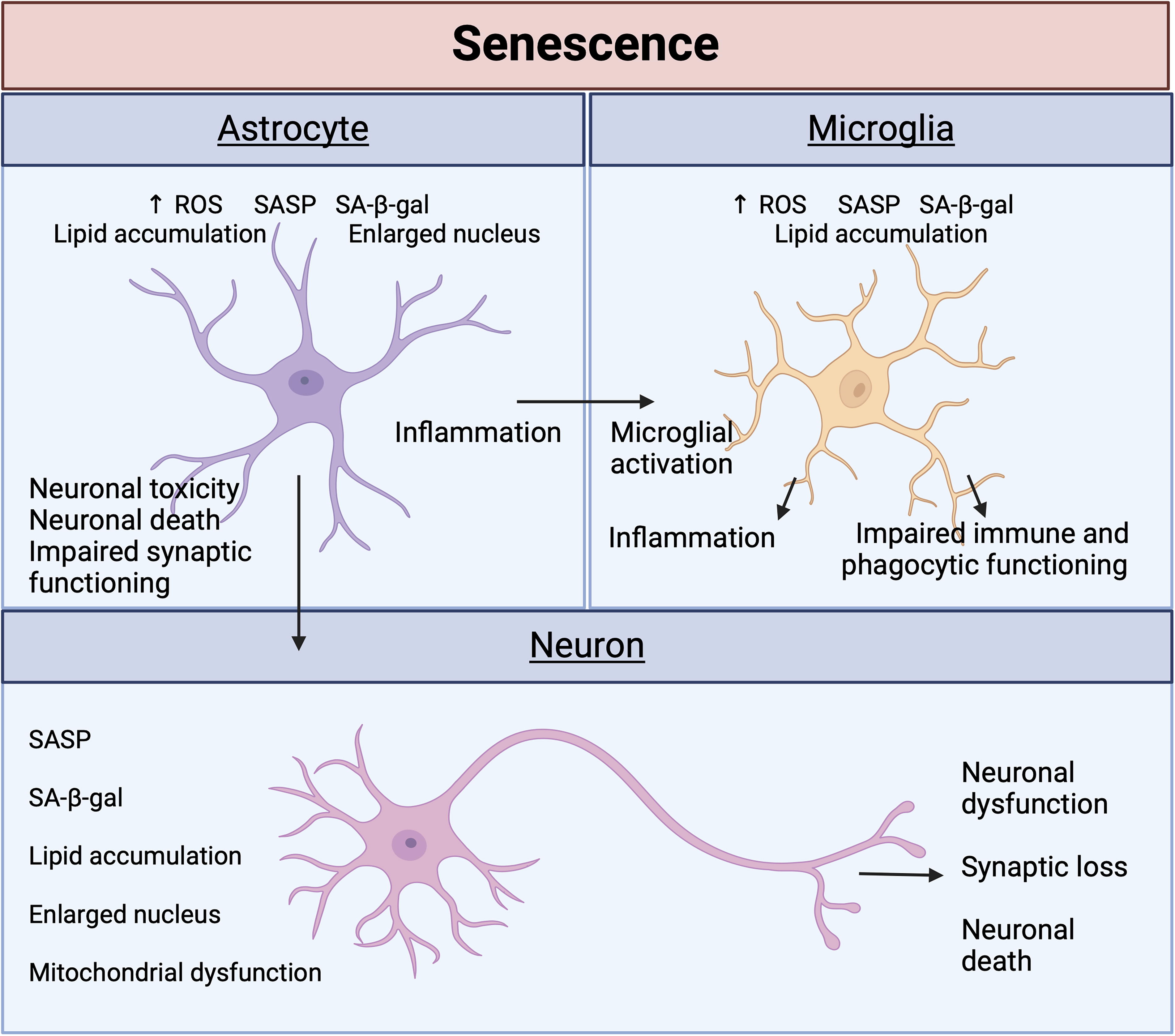
Mature neurons that do not divide conflict with the dominant definition of cellular senescence involving cell cycle growth arrest. However, neurons grown under senescent conditions appear to exhibit other classical markers of senescence, including increased SA-β-gal, lipofuscin accumulation, and gene dysregulation (Mattson and Arumugam, 2018; Moreno-Blas et al., 2019). Regarding lipid metabolism, senescence stimuli appear to upregulate lipid accumulative genes, while downregulating genes that promote lipid homeostasis (Wang et al., 2023). One study suggests that brain regions with more reactive oxygen species (ROS) correspond with a greater number of senescent neurons (Jurk et al., 2012). Accumulation of senescent neurons can lead to neuronal dysfunction, synaptic loss, and cell death, ultimately driving neurodegeneration and cognitive decline (Sikora et al., 2021). Neurons are also greatly affected by senescence-related secretions of other neighboring cell types and can induce changes in surrounding cells. Increased ROS levels in neurons result in lipid droplet (LD) formation in glial cells (Moulton et al., 2021), highlighting how senescence can be accelerated from cell to cell. This also implicates lipid accumulation and mitochondrial dysfunction as the drivers of senescence.
Astrocytes have essential neuroprotective functions (Farina, Aloisi and Meinl, 2007; Pihlaja et al., 2008). During senescence, the neuroprotective properties of astrocytes are diminished, leading to neuronal toxicity and death (Limbad et al., 2020; Marsan et al., 2023). Hippocampal neurons cultured with aged astrocytes exhibit impaired synaptic maturation and transmission (Kawano et al., 2012). Further highlighting the importance of these neuron–astrocyte networks in neuronal functioning, senescent astrocytes secrete less neurotrophic factors (Turnquist et al., 2016). Morphologically, senescent astrocytes have an altered morphology characterized by a flattened, expanded cell body and an enlarged nucleus (Bang et al., 2019; Chinta et al., 2018). Driven by ROS, which are produced by mitochondria and are a critical pathway for senescence, astrocytes undergo dysregulated lipid metabolism and LD formation (Mou et al., 2020; Varela et al., 2021). In addition to SASP secretion, this can promote a proinflammatory state.
Microglia are the primary resident immune cells of the brain and are activated in response to stimuli, such as inflammation or neuronal damage (Kreutzberg, 1996). As such, SASP secretions from other cell types, such as astrocytes, activate microglia and reinforce an inflammatory state (Taylor et al., 2018; Zhang et al., 2022). Recently, lipid droplet-accumulating microglia (LDAM) were identified in aged mice. LDAMs exhibit impaired phagocytic clearing ability and increased ROS and proinflammatory cytokine secretion (Marschallinger et al., 2020). This further implicates lipids and mitochondrial activity in cellular senescence and in driving neurodegeneration.
Different cells of the CNS work synergistically to promote healthy brain functions. Hence, persistent cellular senescence can reinforce and enhance the senescent state with potentially pathological effects.
Lipid Inducers of Senescence
The dysregulation of lipid metabolism is a major hallmark of senescence. Senescent cells exhibit upregulated fatty acid synthase and nonenzymatic β-oxidation of lipids (Quijano et al., 2012) to yield many proinflammatory components that drive senescence in other neighboring cells (Fafián-Labora et al., 2019; Flor et al., 2017). Because of the downregulation of key enzymes such as stearoyl-CoA desaturase, which converts saturated fatty acids into monounsaturated fatty acids, the accumulated fatty acids undergo β-oxidation to drive the hyperphosphorylation of p-tau (Maeda, Scaglia and Igal, 2009). Alongside an increase in fatty acid synthesis, the metabolism of arachidonic acid (AA) by-products—sphingomyelins, ceramides, and fatty acids—is largely increased in senescent cells (Flor et al., 2017). The release of free polyunsaturated fatty acids (PUFAs) from the plasma membrane is associated with multiple forms of senescence (Lizardo et al., 2017; Wiley et al., 2021). In addition to accumulating triglycerides (TAGs) in LDs, free PUFAs undergo enzymatic oxygenation to yield senescence-promoting oxylipins (Wiley et al., 2021) such as leukotrienes (LTs) (Catalano et al., 2005; Wiley et al., 2019) and prostaglandins (Martien et al., 2013; Wiley et al., 2021). Acetyl CoA, a precursor to fatty acids that is increased in response to fatty acid oxidation (Shi and Tu, 2015), is an important carbon source for histone acetylation (McDonnell et al., 2016), suggesting that lipids are important regulators of epigenetic and transcriptomic machinery with probable roles in senescence programs.
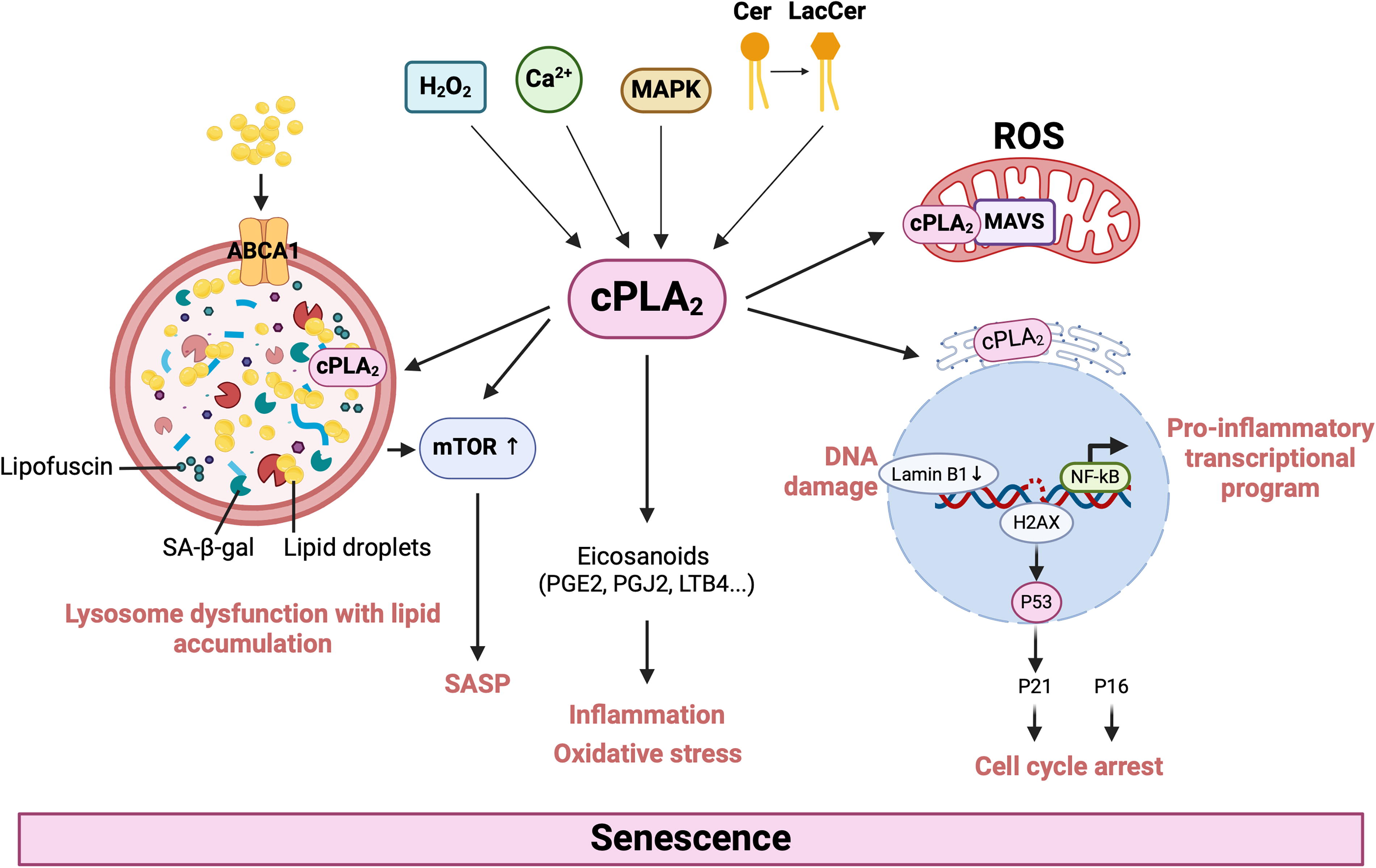
Lysosomes are also important regulators of lipid metabolism, allowing for lipid recycling. In senescence, lysosomes have an enlarged mass and an altered proteome that contributes to SASP (Robbins, Levine and Eagle, 1970; Rovira et al., 2022) and impaired autophagy. Senescent cells exhibit leaky lysosomal membrane permeability, resulting in cytosolic acidification (Johmura et al., 2021). To neutralize increased acidity, senescent cells increase the expression of glutaminase 1, leading to increased ammonia production (Johmura et al., 2021). Cells use autophagy to break down their organelles and macromolecules using lysosomes; however, this process is dysregulated in senescent cells. This dysfunction may also extend to the turnover of metallic cofactors that catalyze reactions within cells. In addition, senescent cells can accumulate higher levels of transition metals (including iron, manganese, zinc, and copper) than nonsenescent cells (Killilea et al., 2003; Masaldan et al., 2018a; Masaldan et al., 2018b). This accumulation may be due to defective lysosomal autophagic degradation of carriers or chaperones such as transferrin (Fig. 2). Accumulation of these metals drives senescence by inducing cell cycle arrest. Enhanced cPLA2 activity can promote senescence through different routes, including lipid oxidation in dysfunctional lysosomes, release of proinflammatory eicosanoids, and ROS production (Fig. 3). Lysosomal cholesterol, rerouted to lysosomes through ATP-binding cassette transporter 1 (ABCA1), can induce mammalian target of rapamycin (mTOR) signaling to drive SASP (Roh et al., 2023).
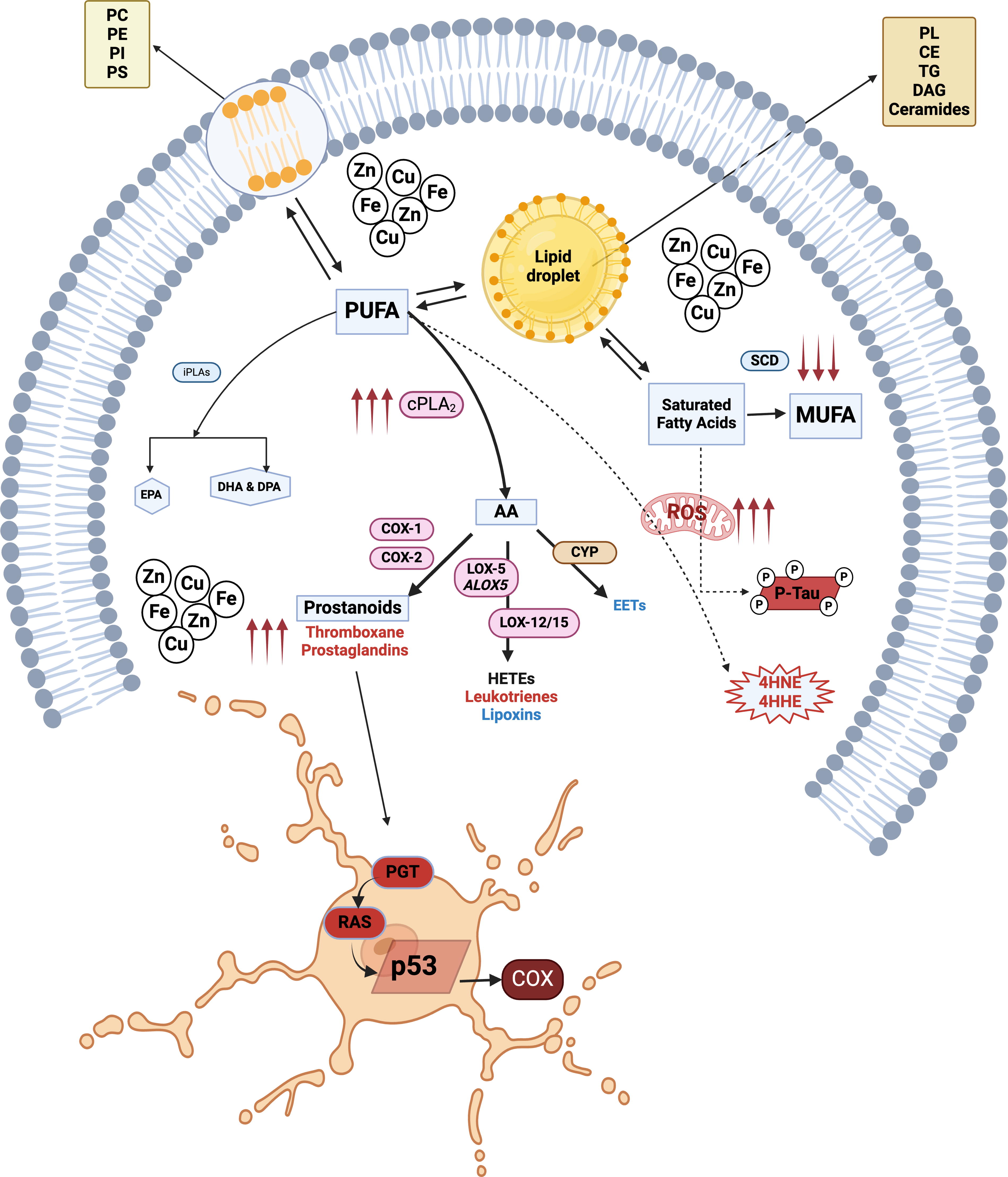
Senescent cells undergo lipid metabolic reprogramming and accumulate LDs (Flor et al., 2017; Hamsanathan and Gurkar, 2022). LDs consist of cholesterol, TAGs, and phospholipids (Fujimoto and Parton, 2011). Treating irradiated fibroblasts with cholesterol enhanced senescence compared with irradiation alone, suggesting that modulating lipid levels accelerates senescence (Roh et al., 2023). More studies are needed to further expand our understanding of which lipids drive senescence. Several lipid species are implicated in senescence (Table 2).
Lipid Inducers of Senescence
Phospholipids
Lysophospholipids are derived from hydrolyzed phospholipids, including lysophosphatidylcholine (LPC). Inducing senescence in fibroblasts resulted in an increase in LPCs. LPC also enhances the secretion of chemokines, a component of SASP. LPC-treated epithelial cells also showed greater senescence, as measured by SA-β-gal, SASP, ROS, and DNA damage (Ohigashi et al., 2019).
Under oxidative stress, phospholipids are oxidized through lipid peroxidation, and are termed oxidized phospholipids (OxPLs). Through oxidative stress, cells become senescent, undergo mitochondrial dysfunction, and increase ROS production. This leads to various lipid changes, including lipid peroxidation (Ademowo et al., 2017). OxPLs are proinflammatory and are associated with various diseases (Nie et al., 2020; Palmieri et al., 2021). Aldehydes, a product of lipid peroxidation, have been found to drive therapy-induced senescence in melanoma cells. The peroxidation-derived aldehyde, 4-hydroxynonenal (4-HNE), was associated with DNA damage and enhanced the senescent phenotype in these cells. Rescuing this with an aldehyde oxidation inhibitor prevented senescence (Flor, Doshi and Kron, 2016).
Sphingophospholipids
Sphingophospholipids, including ceramides and sphingomyelins, are important components of the plasma membrane (van Meer, Voelker and Feigenson, 2008). Numerous studies have shown that sphingolipids increase under senescent conditions, and that exogenous treatment with these lipids can increase senescence (Flor et al., 2017; Giusto, Roque and Ilincheta de Boschero, 1992). For instance, endothelial cells treated with ceramides show elevated markers of senescence (Venable and Yin, 2009). Inhibition of ceramide synthesis reduced the number of therapy-induced senescent cells, and reversing this phenotype accelerated senescence again, further implicating ceramide synthesis as having a major role in cellular senescence (Millner et al., 2022).
Glycerolipids
TAGs are among the most abundant lipids in the body. In senescent fibroblasts, numerous lipid compositions and metabolic changes have been observed, including genes responsible for glycerolipid metabolism, suggesting that TAG synthesis may play a significant role in driving senescence (Tighanimine et al., 2024). In support of this, mesenchymal stem cells treated with TAGs exhibited elevated levels of senescence, namely reduced proliferation, SASP, and ROS. These senescent cells could be rescued by impairing ROS production, suggesting that TAGs may drive senescence through oxidative stress (Xiang et al., 2020).
Sterols
Cholesterols are important cellular components, but excess cholesterol can have negative effects. Cholesterol accumulation can increase cholesterol-rich LDs (Makino et al., 2016). Several studies have observed cholesterol accumulation and increased cholesterol efflux under senescent conditions (Maeda, Scaglia and Igal, 2009; Sene et al., 2013). Treating irradiated fibroblasts with cholesterol enhanced senescence compared with irradiation, suggesting that cholesterol may function as a modulator of senescence (Roh et al., 2023). Oxysterols are oxidized forms of cholesterol generated from enzymatic reactions or from free radicals and ROS (Zerbinati and Iuliano, 2017) and implicated in neurodegenerative disease (Gamba et al., 2015; Gamba et al., 2021). 7-Ketocholesterol, a component of oxidized low-density lipoprotein, is largely composed of fatty acids and promotes apoptosis in macrophages through cPLA2 activation (Freeman et al., 2005). A similar finding is observed with oxysterol 25-hydroxycholesterol, implying that oxysterols activate cPLA2 to upregulate apoptotic pathways.
Oxidized fatty acids
Eicosanoids are derived from AA, a product of cPLA2. The oxidation of AA by lipoxygenases (LOX), cyclooxygenases (COX-1 and COX-2), and cytochrome P-450 produces eicosanoids, which are a category of fatty acids involved in inflammation and senescence (Harizi, Corcuff and Gualde, 2008; Pils et al., 2021; Yamaguchi, Botta and Holinstat, 2022). These by-products include LTs, prostaglandins, and epoxyeicosatrienoic acids, which are potent inflammatory mediators (Leslie, 2015; Six and Dennis, 2000). Proinflammatory lipid mediators are major secondary messengers used by senescent cells to induce senescence in neighboring cells. Senescence-associated LT release drives the profibrotic features of chronic inflammation (Schafer et al., 2017; Wiley et al., 2019). The increased production of prostaglandins from inflammatory PUFAs such as AA reinforces senescence-associated cell cycle arrest. Senescent cells accumulate prostaglandins by upregulating the expression of prostaglandin transporters, but do not alter the metabolism of prostaglandins. Prostaglandin influx activates Ras, leading to activation of p53, a major modulator of the senescence program (Mijit et al., 2020), which then promotes cell cycle arrest and the sequential upregulation of COX-2 to trigger the cascade of more oxylipin biosynthesis (Fig. 2).
Enzymatic oxidation of fatty acids yields oxylipins, a lipid component of SASP, which is increased in senescent cells, particularly 1a,1b-dihomo-15-deoxy-delta-12,14-prostaglandin J2 (dihomo-15d-pgj2). The significant release of dihomo-15d-pgj2 from senescent cells into blood and urine may be explored to evaluate the efficacy of senolytic drugs in vivo (Wiley et al., 2021). Other oxylipins such as prostaglandin E2, thromboxanes, and LTs are involved in the inflammation response to cellular senescence (Gorgoulis et al., 2019; Wiley et al., 2019). Increases in COX-2 activity has been shown to be associated with aging (Badawi et al., 2005; Baek et al., 2001; Chung et al., 1999). It has been shown in inducible COX-2 transgenic mice that high expression of COX-2 led to several premature aging phenotypes (Kim et al., 2016) and to lung fibroblasts with higher levels of SA-β-Gal, p16, p53, and phospho-H2Ax (Kim et al., 2016).
cPLA2 Activation and ROS as Drivers of Cellular Senescence and Neurodegenerative Diseases
Several lines of evidence have implicated cPLA2-mediated eicosanoid synthesis in cellular senescence and oxidative stress. In T cell senescence, increased cPLA2 expression was associated with dysregulated lipid metabolism, and inhibiting cPLA2 rescued increases in P21 and P53 expression and prevented these two markers of senescence (Liu et al., 2021). Irradiated senescent fibroblasts showed an increase in AA and AA by-products, namely prostaglandin J2 (PGJ2) and LT B4. Reducing prostaglandin synthesis in these fibroblasts through the inhibition of AA oxidation appeared to reduce senescence, while PGJ2 treatment accelerated several markers of senescence (Wiley et al., 2021). 5-Lipoxygenase (5-LOX), a catalyst in LT production, along with cPLA2, was increased in senescent-like fibroblasts. 5-LOX overactivation was found to initiate growth arrest via p53/p21 by increasing ROS levels (Catalano et al., 2005). Other studies have supported the idea that eicosanoids accelerate senescence, and suggest that targeting eicosanoid synthesis may ameliorate this senescence burden.
Activated cPLA2 has a major role in oxidative stress, where it increases AA metabolism in response to oxidants and changes in intracellular calcium concentration (Chen et al., 1996). Oxidative stressors can activate cPLA2 through mitogen-activated protein kinases (MAPKs), and AA metabolism can further elevate ROS secretions and enable oxidative stress to persist (Yu et al., 2013; Zhou et al., 2019). cPLA2 plays an important role in oxidative and nitrosative stress responses in activated microglial cells (Chuang et al., 2015; Ribeiro et al., 2013). In microglia, cPLA2 activation and AA metabolism pathways contribute to ROS and nitric oxide (NO) production, creating an oxidative environment during cell activation and senescence (Chuang et al., 2015). The presence of ROS facilitates increased intracellular Ca2+ and alternative nonenzymatic oxidation and peroxidation of lipids to form 4-HNE, malondialdehyde, or 4-hydroxyhexenal, as well as other sequential lipid adducts that can reinforce oxidative stress and further aggravate an inflammatory cascade. Reducing downstream products of cPLA2 activation, for instance, lipid hydroperoxides, that is, the intermediates of COX and LOX pathways, and inhibiting cPLA2 alleviate oxidative stress (Chang et al., 2019; Pharaoh et al., 2020).
Ceramide is a lipid secondary messenger that is generated through the hydrolysis of sphingomyelin to form phosphocholine and ceramide or by de novo synthesis from sphinganine by ceramide synthase. Ceramide interacts with the calcium-binding domain of cPLA2 to activate the enzyme that drives inflammatory processes (Huwiler et al., 2001). For instance, in astrocytes, cPLA2 interacts with mitochondrial antiviral-signaling protein (MAVS) to increase the production of eicosanoids and ROS, enhancing neuroinflammation and oxidative stress (Chao et al., 2019; Chuang et al., 2015; Gijón and Leslie, 1999). Following activation by lactosylceramide, cPLA2 is recruited to the mitochondria and promotes nuclear factor kappa B activation (Zhu et al., 2006) through MAVS to drive proinflammatory transcriptional programs (Seth et al., 2005). Infection and ROS have also recently been reported to activate MAVS (Buskiewicz et al., 2016; Galluzzi, Kepp and Kroemer, 2012). However, AA, the byproduct of cPLA2 activity, does not lead to MAVS aggregation, thus supporting the key role of cPLA2 in MAVS activation. Activated cPLA2 eventually leads to lysosomal membrane injury, which may lead to leakage of SA-β-gal, the most widely used biomarker for senescence, into the cell (Lee et al., 2006).
Genetic manipulation and induction or inhibition of cPLA2 activation in in vivo models have implicated a causal role for cPLA2 in mediating numerous disease processes (Table 3). Largely, these studies have assessed cPLA2 activation by its phosphorylation and by-products. The levels of cPLA2 protein and its phosphorylated form are increased in astrocytes surrounding amyloid beta (Aβ) plaques compared with healthy controls (Colangelo et al., 2002; Sanchez-Mejia et al., 2008; Stephenson et al., 1996). Similarly, in the hippocampus of human amyloid precursor protein transgenic mice, cPLA2 activation increased (Sanchez-Mejia et al., 2008). Aβ oligomers can activate cPLA2 to promote neurodegeneration (Palavicini et al., 2017; Sun et al., 2012). In contrast, loss of cPLA2 lessens the memory impairment and hyperactivated glial cells characteristic of AD mouse models (Sanchez-Mejia et al., 2008). Increased cPLA2 activation has also been described following ischemic stroke (Bonventre et al., 1997), spinal cord injury (SCI) (Khan et al., 2015), and status epilepticus (Hartz et al., 2019). Importantly, reducing cPLA2 activity in vivo limits the effects of vascular inflammation on blood–brain barrier (BBB) leakage (Hartz et al., 2019), and may have broader and additional applications for neurodegenerative diseases such as Parkinson’s (Paul et al., 2022), traumatic brain injury (Sarkar et al., 2020), SCI (Liu et al., 2014), epilepsy (Hartz et al., 2019), and stroke (Bonventre et al., 1997). We have shown that apolipoprotein E4 (APOE4) activates cPLA2 through MAPK p38 signaling in cellular and animal models (Wang et al., 2022) and in postmortem human brain tissues (Ebright et al., 2022).
Disease Models Featuring Calcium-Dependent Phospholipase A2 Overactivation
APOE, apolipoprotein E; KO, knockout; NF-κB, nuclear factor kappa B.
Challenges for Developing cPLA2 Inhibitors into Clinical Drugs
Treatment versus prevention
The development of cPLA2 inhibitors with potential senescence prevention properties would shift the senolytic field, where drugs have been developed based off senescence biomarkers or repurposed and focused on “removing” senescent cells to prevention. Senolytic prodrugs such as synthesized senescence-specific killing compound 1 and 5-fluorouridine-5′-O-β-D-galactopyranoside are created by modifying senolytics to sense SA-β-gal and activate apoptotic programs only at the site of detected SA-β-gal activity (Chang et al., 2024; Morsli, Doherty and Muñoz-Espín, 2022). While this approach may reduce off-target effects by having the drug become pharmacologically active at the site of the senescent cell, senescence is largely heterogenous with no universal biomarker (Casella et al., 2019; González-Gualda et al., 2021). Although SA-β-gal is one of the most common senescence markers, its usage in the brain is questionable, where quiescent postmitotic neurons have been shown to have high SA-β-gal levels (Musi et al., 2018; Zhang et al., 2019). Other therapeutic interventions include the usage of chimeric antigen receptor T cell therapy cells to recognize cell membrane proteins overexpressed in senescent cells (Amor et al., 2020), but this again raises the questions of biomarker universality and specificity with regard to senescence. Many drug interventions, including polyphenols, such as quercetin, have been shown to exert beneficial effects against inflammatory neurological conditions (Sun et al., 2011) due to their anti-inflammatory and antioxidative properties (Galli et al., 2002; Sun et al., 2008). Earlier developmental efforts have shown that quercetin can mitigate microglial-induced production of NO and ROS upon stimulation by lipopolysaccharide or proinflammatory cytokines (Chuang et al., 2013; Jiang et al., 2014). Quercetin also showed effectiveness in inhibiting oxidative and nitrosative products as well as the suppression of cPLA2-mediated pathways that generate prostanoids, eicosanoids, and leukotrienoids that promote senescence. However, early clinical studies evaluating the brain penetration of dasatinib + quercetin found that only dasatinib, but not quercetin, was detectable in the cerebrospinal fluid 60–90 min after the last dose (Gonzales et al., 2022). Although the regimen appeared safe and tolerable, there were no significant changes in the biomarkers of Aβ, tau, senescence, or cognition after 12 weeks of treatment (Gonzales et al., 2023). The usage of cPLA2 inhibitors presents an option to prevent the accumulation of senescent cell characteristics of many neurodegenerative disorders by reducing cPLA2 activity and hindering cells from inducing a senescence program. This contrasts with many existing senolytic therapies that are designed to clear an already abundant number of senescent cells.
Selective cPLA2 inhibition
It has been challenging to develop selective inhibitors for specific PLA2 isoforms that have advanced into Phase I clinical trials. Many inhibitors have been shown to inhibit the expression of other PLA2 isoforms. For example, AA-trifluoromethylketone has a strong preference for cPLA2 over other PLA2 isoforms but also inhibits group 6A phospholipase A2, thromboxane synthase, and fatty acid amide hydrolase. As a result, in vivo studies on cPLA2 largely make use of gene knockouts rather than pharmacologic inhibition (Sanchez-Mejia et al., 2008). Over the years, various cPLA2 inhibitors have emerged and been subject to further studies to verify their specifities (Magrioti and Kokotos, 2010). Previously, Pfizer had designed indole-based inhibitors that are highly specific for cPLA2. For instance, WAT0196025, blocks cPLA2 but not its β- or γ-isoforms. Merck had also developed heteroaryl-substituted acetone derivatives that are not very potent and inhibit cPLA2 at micromolar concentrations (Magrioti and Kokotos, 2010). In contrast, other similar compounds can inhibit cPLA2 at nanomolar concentrations (Magrioti and Kokotos, 2010). While 2-Oxoamide cPLA2 inhibitors, based on γ-amino acids, have also emerged, these compounds and their high reactivity leads to fast degradation in human plasma, limiting their pharmaceutical potential (Psarra et al., 2018). Among the compounds available, ASB14780 had the most promising profile (Table 4) but appeared to be toxic at higher doses and was discontinued in human trials. In the decade since the development of these compounds, only one has undergone human trials (AK106-001616, Asahi Kasei). A Phase 2 clinical trial explored 100 and 600 mg AK106-001616 in patients with rheumatoid arthritis (RA) in a 28-day trial. In general, adverse effects were similar to those of high-dose naproxen. Although the clinical safety of AK106-001616 has been established in patients with RA, its ability to penetrate the BBB is likely to be limited. We recognize that cPLA2 in the CNS as a drug target for treating patients with AD neuroinflammation will be challenging because a successful clinical candidate must be potent, target-selective, possess acceptable absorption, distribution, metabolism, excretion properties, and be differentially distributed to the brain with sufficient free fraction to inhibit cPLA2 in that organ system. This means that the optimal clinical candidate will have a brain-to-plasma ratio that approaches unity, to avoid dose-limiting peripheral drug-related adverse reactions.
Profiles of Existing cPLA2 Inhibitors That May Serve as Lead Molecules
ATK/AACOCF3, arachidonyl trifluoromethyl ketone; CDIBA, efipladib; BBB, blood–brain barrier penetration; N/A, not available; Pe, permeability efflux ratio; PO, oral; IP, intraperitoneal; ARDS, acute respiratory distress syndrome.
Selecting the right population with brain cPLA2 overactivation and monitoring inhibitor efficacy
In moving the development of cPLA2 inhibitors to in vivo studies, quantitative models for assessing fatty acid kinetics in the brain have been described (Rapoport, 2001; Robinson et al., 1992), and linking directly the incorporation of AA in brain phospholipids to cPLA2 activity using cPLA2 knockout mice (Rosenberger et al., 2003). We have also developed a high-throughput and feasible strategy using positron emission tomography (PET) imaging to measure 18-F AA kinetics in the brain (Van Valkenburgh et al., 2022) as a surrogate of brain cPLA2 activity. Briefly, we have generated [18F]fluoro-AA and we are actively testing mouse models to assess the effects of APOE genotype and cPLA2 inhibition on AA brain uptake and cPLA2 activity. The advantage of PET brain fatty acid imaging is that it can show regional uptake of AA and enable the estimation of uptake kinetics, expanding upon simple measurement and nonimaging methods by capturing temporal and regional information. The information obtained can help identify a population with increased brain cPLA2 activity and monitor the response to cPLA2 inhibition.
Development of cPLA2 Inhibitors Targeting the Brain
The goal of drug discovery efforts is to identify novel compounds with high potency, specificity, and a favorable brain-to-plasma partitioning coefficient that merit further development. Given the importance of cPLA2 activity on neurotransmission, the goal is not to completely abolish cPLA2 activity but to decrease its overactivation. A successful drug candidate is expected to target patients who exhibit hyperinflammatory states due to their genetic disposition (e.g., APOE4 carriers) and/or other risks assessed with brain PET imaging of AA metabolism. Inhibition of the hyperactivated cPLA2 enzyme thereby ameliorates the heightened hyperinflammation resulting from cPLA2 hyperactivation. The expected systemic adverse event profile of the drug candidate may mirror those of high-dose naproxen as demonstrated in clinical trials on other candidates in the family of cPLA2 inhibitors. Common side effects will result from shared inhibition of thromboxanes, prostaglandins, and prostacyclins. Expected systemic side effects reported in 1%–10% of subjects include gastrointestinal upset or pain, constipation or diarrhea, heartburn, nausea or vomiting, headache, rash, and other flu-like symptoms. Other expected but rare CNS side effects include aseptic meningitis, psychosis, and tinnitus.
Virtual structure-based screening based on computational docking of libraries of small molecules to identify potential hit compounds is a well-established technique for early drug discovery. The advent of giga-scale make-on-demand combinatorial chemical spaces, most prominently the readily available for synthesis (REAL Space) (Grygorenko et al., 2020), presents a great opportunity for drug discovery, providing billions of compounds available for screening using optimized parallel chemistry approaches (Sadybekov and Katritch, 2023). We used the recently developed Virtual Synthon Hierarchical Enumeration Screening (V-SYNTHES) (Sadybekov et al., 2022), an iterative synthon-based approach for fast structure-based virtual screening of billions of compounds, which provides an efficient exploration of large chemical spaces such as REAL. This breakthrough technology accelerates virtual screening by >5000-fold, expanding drug discovery capabilities to tens of billions of REAL chemical space compounds by operating in the space of fragment-like molecules that serve as building blocks for the fully enumerated chemical space (Fig. 4). In the first step, the minimal enumeration library (MEL), which represents the collection of all reaction scaffolds enumerated with representative synthons on one side and a standard methyl or phenyl cap on the other side/(s), is generated. Subsequently, docking of MEL into the protein-binding pocket is performed, followed by selection of the best MEL candidates that fit into the binding pocket and are suitable for growth. Then, the focused library of larger molecules is iteratively generated and screened by enumerating the best MEL candidates with the second and third synthons as needed to grow the full molecules. In the last step, the best predicted full molecules are filtered based on docking scores and poses, chemical novelty, and diversity, and a set of best candidates are selected for synthesis and testing. The hits and leads identified by V-SYNTHES can be naturally expanded by structure–activity relationship (SAR)-by-catalog in the same REAL Space, and then optimized further with medicinal chemistry using simple and well-established chemical reactions, as described (Sadybekov et al., 2022). Beyond the successful validation of cannabinoid receptors and rho-associated protein kinase, V-SYNTHES has shown promising results in a variety of target classes (Sadybekov and Katritch, 2023), and our preliminary results suggest its applicability to the identification of cPLA2 inhibitors.
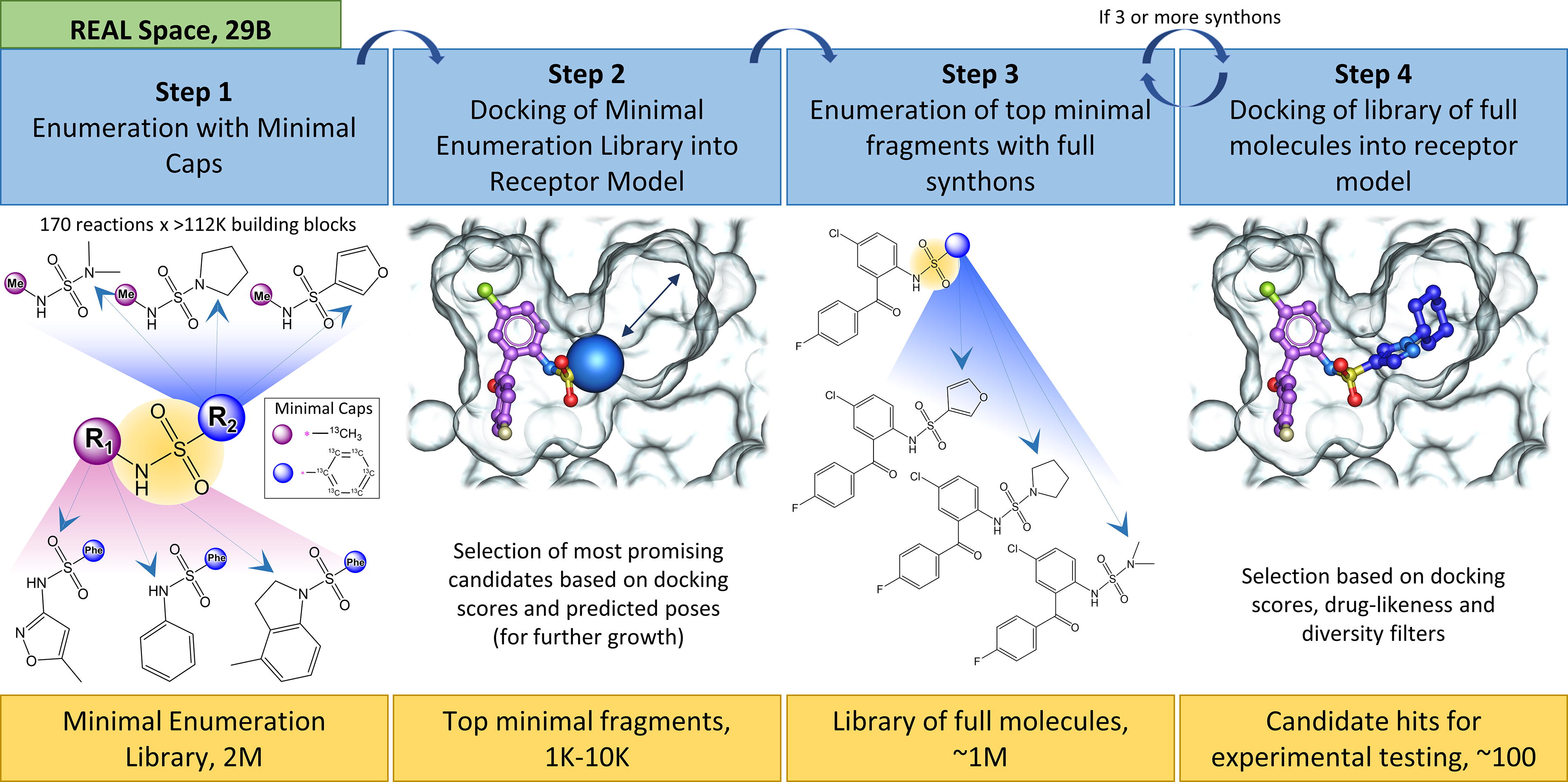
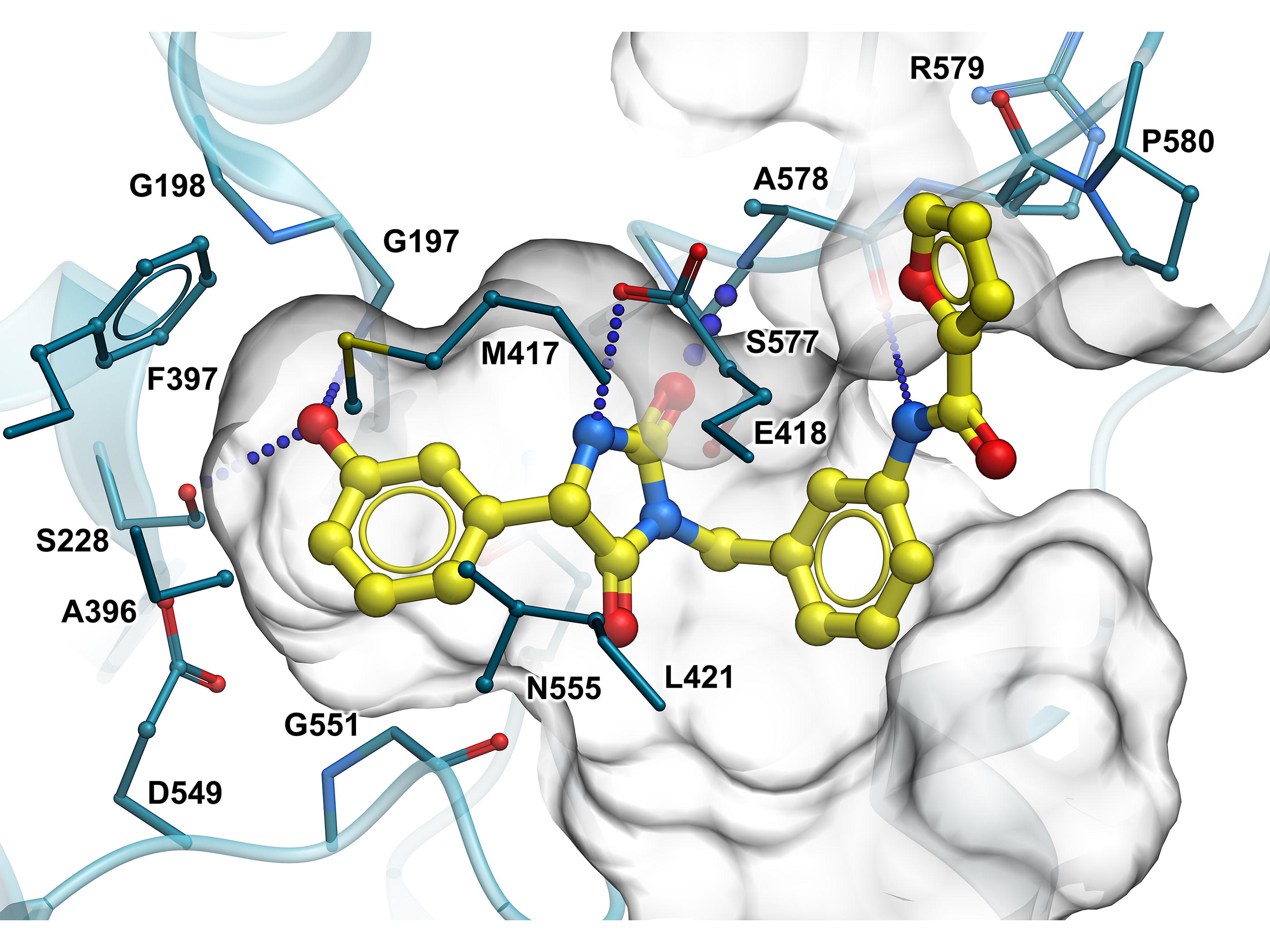
The V-SYNTHES algorithm was applied to screen enamine REAL Space containing more than 20 billion compounds to identify several promising scaffolds for cPLA2 inhibitors. A structural model of the cPLA2 active site binding pocket was prepared from the available apo structure of cPLA2 (PDBID:1CJY) by optimizing the hydrogen atoms in the binding pocket in the presence of previously docked known ligands. The resulting model was used in structure-based V-SYNTHES screening, and 127 molecules were selected for synthesis and testing. The selection was guided by predicted docking scores and poses, chemical diversity, and physicochemical properties, including predicted solubility (LogS), gut absorption (molCACO2), and BBB permeability (BBB Score), based on the accurate regression and deep-learning models of these properties implemented in internal coordinate mechanics (ICM)-Pro drug discovery software package (Molsoft LLC). Molecules with H-bonds to R200, S228, A578, and T680 were prioritized in the selection process (Fig. 5). A total of 117 compounds were synthesized and delivered by Enamine in 6 weeks. The testing revealed several promising scaffolds in the low micromolar range suitable for further SAR-by-catalog optimization in REAL Space. We have an ongoing effort to examine their potency, specificity, BBB penetration, and other pharmacokinetic/pharmacodynamic properties.
In summary, we present evidence for the connection between dysregulated lipid metabolism and cellular senescence, focusing on cPLA2-mediated overactivation as a major driver of cellular senescence, neuroinflammation, and oxidative stress in neurodegenerative conditions. We highlight significant challenges in the development of potent, specific, and brain-penetrant cPLA2 inhibitors. We present a novel pipeline for the discovery of such cPLA2 inhibitors that may have senescence prevention potential and are of particular relevance in AD.
Footnotes
Authors’ Contributions
C.H. wrote the sections on senescence. I.A. wrote the sections on lipids and cPLA2 as a drug target. A.S. and V.K. covered the sections of drug discovery platforms. H.N.Y. designed the review and extensively edited the article.
Author Disclosure Statement
The authors have declared that no conflict of interest exists.
Funding Information
This work was supported by the National Institute on Aging (RF1AG076124, R01AG055770, R01AG067063, R01AG054434, R21AG056518, and P30AG066530 to H.N.Y.; the Alzheimer’s Drug Discovery Foundation (ADDF) (GC-201711–2014197 to H.N.Y.), and donations from the Vranos and Tiny Foundations and Ms. Lynne Nauss to H.N.Y, R01GM147537 to VK and USC CTSI KL2 (UL1 TR000004) to IA.