
Abstract
Ferroptosis, a distinct form of regulated cell death (RCD), has emerged as a promising approach for cancer treatment owing to its potential to inhibit tumor malignancy. Research indicates that non-coding RNAs (ncRNAs) regulate ferroptosis susceptibility in cancer cells through epigenetic modifications. ncRNAs play essential roles in cancer initiation, metastasis, and drug resistance. Findings indicate that small-molecule compounds (SMCs) target ncRNAs to regulate ferroptosis, providing new opportunities for precision cancer therapy. Therefore, this review aims to elucidate current molecular mechanisms underlying ncRNA-mediated ferroptosis regulation in cancer and investigate the potential of SMCs as therapeutic agents to modulate this process, offering a new strategy for precision in cancer treatment. This review also summarizes the innovative strategy of targeting ncRNAs with SMCs, a therapeutic approach for regulating ferroptosis and transforming the landscape of cancer treatment. Overall, it highlights a novel strategy for cancer therapy by pharmacologically targeting the ncRNA-ferroptosis axis with SMCs. Antioxid. Redox Signal. 43, 345–362.
Background
Cancer is the second leading cause of mortality globally, following cardiovascular diseases, with a poor prognosis for patients with advanced stages (Adhikari et al., 2022; Sung et al., 2021). A high mortality rate persists owing to late diagnoses and ineffective or toxic therapies. Despite significant advancements in targeted treatment and immunotherapy, most conventional therapies remain ineffective in cancers that do not respond to existing treatments (Coan et al., 2024). Additionally, several tumors rapidly develop resistance to standard care, highlighting an urgent and unmet need for new therapeutic strategies that are safe, effective, and long-lasting, particularly as additions to existing treatments.
One of the most important discoveries in molecular oncology is that cancer can result from mutations in both protein-coding and non-protein-coding genes (Monroig et al., 2015). For over two decades, ncRNAs have been recognized as key regulators of cancer hallmarks, acting as both oncogenes and tumor suppressors. Dysregulated ncRNAs are associated with all cancer types, influencing essential cancer-related processes (Calin and Croce, 2006; Gutschner and Diederichs, 2012; Hanahan, 2022; Lenkala et al., 2014; Schmitt and Chang, 2016). These findings indicate that targeting ncRNAs could provide novel therapeutic opportunities in cancer treatment. Since the identification of lncRNA Xist in 1992 (Brockdorff et al., 1992; Brown et al., 1992) and the miRNA gene lin-4 in 1993 (Lee et al., 1993; Wightman et al., 1993), an increasing number of ncRNAs have been discovered and classified. From their initial discovery, ncRNAs were recognized for their therapeutic potential, even before their biological mechanisms were fully elucidated. Over the past decade, significant progress has been made in developing RNA-based therapeutics for clinical application, primarily utilizing antisense oligonucleotides (ASO) and small interfering RNAs (siRNA) (Winkle et al., 2021). Several RNA-based therapies, including anti-miRNAs and miRNA mimics, are in phase II or III clinical trials (Nappi, 2024; Winkle et al., 2021). However, two major challenges in developing ncRNA drugs remain: the RNases-mediated rapid degradation of RNA and the challenges of delivering charged nucleic acid analogs across hydrophobic cell membranes (Lieberman, 2018). Since the observation in 1978 that 13-mer DNA oligonucleotides could inhibit sequence-specific Rous sarcoma virus (RSV) proliferation, it took nearly half a century to develop techniques to overcome these challenges (Stephenson and Zamecnik, 1978; Zamecnik and Stephenson, 1978). ncRNAs are crucial in both cancer pathogenesis and its protective mechanisms. Therefore, SMCs that modulate ncRNA-associated pathways, primarily by upregulating regulated cellular death (RCD) pathways, represent a novel and promising therapeutic strategy for developing novel anticancer therapies.
Ferroptosis, a distinct type of iron-dependent regulated cell death induced by lipid peroxidation (LPO), was introduced in 2012 by Stockwell Lab (Lei et al., 2022). Marked progress has been achieved in elucidating its role in tumor biology, and ferroptosis targeting holds great potential for cancer therapy, including the potential to overcome drug resistance (Dai et al., 2024; Dixon et al., 2012; Elgendy et al., 2020; Friedmann Angeli et al., 2019; Lei et al., 2021; Liu et al., 2022; Ozkan and Bakar-Ates, 2022; Stockwell, 2022; Wang et al., 2021; Wang et al., 2023d; Zhang et al., 2022; Zhang et al., 2022a). ncRNAs are recognized as crucial epigenetic regulators of ferroptosis (Balihodzic et al., 2022; Ensoy et al., 2023; Luo et al., 2021; Valashedi et al., 2022; Wang et al., 2022; Xie and Guo, 2021; Zuo et al., 2022). Given that SMCs can target ncRNAs and ncRNAs can regulate ferroptosis in cancer, modifying the ncRNA-ferroptosis axis with SMCs may provide new opportunities for cancer therapy.
Emerging research indicates that SMCs can target ncRNAs involved in ferroptosis regulation. This review aims to provide a comprehensive overview of the molecular mechanisms of ferroptosis, summarize the current understanding of the role of ncRNAs in tumorigenesis, and explore novel therapeutic strategies targeting ncRNAs, with the potential to expand cancer treatment paradigms. This review underscores the innovative therapy of targeting ncRNAs with SMCs, a promising approach to regulating ferroptosis and transforming the landscape of cancer treatment. This review highlights novel strategies for pharmacologically targeting the ncRNA-ferroptosis axis using SMCs for cancer treatment.
Core Mechanisms of Ferroptosis
In 2012, ferroptosis was identified as a novel type of RCD triggered by iron-dependent LPO (Dixon et al., 2012; Gu et al., 2023; Huo et al., 2023; Stockwell, 2022; Wang et al., 2023a; Yin et al., 2022). Initially, it was described as a non-apoptotic RCD characterized by depletion of glutathione (GSH), iron-dependent LPO, and impaired cystine uptake into cells (Dixon et al., 2012). The discovery of specific small-molecule inhibitors contributes to elucidating its precise molecular mechanisms (Dixon and Olzmann, 2024). Initiation and induction of ferroptosis are mediated by three essential elements: reactive oxygen species (ROS), oxidizable lipids, and LPO (Dai et al., 2024) (Fig. 1). The imbalance between ferroptosis-promoting factors and cellular defense systems leads to the accumulation of lethal lipid hydroperoxides (also known as lipid peroxides) on cellular membranes, causing membrane rupture and subsequent cell death (Chen et al., 2021a; Hadian and Stockwell, 2020; Lei et al., 2022; Pope and Dixon, 2023).

Ferroptosis prerequisites
Iron metabolism, mitochondrial metabolism, and the synthesis or peroxidation of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs) are the key prerequisites that induce ferroptosis (Gan, 2021; Gao et al., 2019; Jiang et al., 2021; Lei et al., 2022).
Iron-dependent LPO
Phospholipid peroxidation, a process dependent on the transition metal iron, PUFA-PLs, and ROS, induces ferroptosis (Dixon et al., 2012; Stockwell et al., 2017; Zhang, 2024). The primary substrates for LPO during ferroptosis are PL-PUFAs owing to their high susceptibility to oxidative damage (Hadian and Stockwell, 2020). These PL-PUFAs are generated by enzymes, including ACSL4 (Acyl-coenzyme A synthetase long-chain family member 4) and LPCAT3 (lysophosphatidylcholine acyltransferase 3), which activate and incorporate free PUFAs into phospholipids. PUFAs can be obtained from dietary sources and the environment or synthesized from the basic building block acetyl-CoA through the action of acetyl-CoA carboxylase (ACC). AMPK and energy stress inhibit ferroptosis by suppressing ACC (Lee et al., 2020). During ferroptosis, PUFA-PLs undergo LPO through enzymatic and non-enzymatic pathways (Hadian and Stockwell, 2020). PUFA-PL peroxidation is primarily induced by non-enzymatic autoxidation, which occurs through a Fenton reaction with iron acting as a catalyst (Conrad and Pratt, 2019; Gaschler and Stockwell, 2017; Shah et al., 2018). The ALOX15 (12/15-lipoxygenase), POR (oxidoreductases cytochrome P450 reductase), NOX (NADPH oxidase), and CYB5R1 (NADH-cytochrome b5 reductase) enzymes promote LPO through enzymatic reactions (Anthonymuthu et al., 2021; Chu et al., 2019; Poursaitidis et al., 2017; Shah et al., 2018; Yan et al., 2021; Yang et al., 2019, 2016; Zou et al., 2020). The interaction between iron and lipids facilitates LPO, producing peroxidized PUFA-PLs (PUFA-PL-OOH) and derivatives such as 4-HNE (4-hydroxynonenal) and MDA (malondialdehyde) (Pope and Dixon, 2023). The lipid hydroperoxides (oxidized lipids) incorporated into the cell membrane trigger ferroptosis (Doll et al., 2017; Kagan et al., 2017).
Iron in ferroptosis
Iron promotes LPO and induces ferroptosis through two primary mechanisms: mediating the non-enzymatic Fenton reaction and serving as an essential cofactor for iron-dependent peroxidases such as ALOXs and POR, which enhances LPO. Additionally, mitochondrial ROS production enhances LPO (Chen et al., 2021b; Hassannia et al., 2019; Lei et al., 2022; Liang et al., 2022). Iron exists in two oxidation states: ferric iron (Fe3+) and ferrous iron (Fe2+), with Fe2+ reacting with peroxidized PUFA-PLs to generate hydroxyl radicals (HO•) (Jacquemyn et al., 2024). These radicals subsequently interact with PUFAs to propagate LPO (Jacquemyn et al., 2024).
Fe2+ directly initiates PUFA-PL peroxidation and triggers ferroptosis through the non-enzymatic Fenton reaction in the non-enzymatic LPO pathway (Conrad and Pratt, 2019). This reaction converts H2O2 (hydrogen peroxide) into HO•. PUFA-PLs react with Fenton reaction-mediated production of ROS (including LO• or HO•) to produce peroxidized PUFA-PLs, thereby inducing LPO (Ayala et al., 2014; Dos Santos et al., 2023; Ryter et al., 2007). In the presence of labile iron, peroxidized PUFA-PLs that are not neutralized rapidly propagate peroxidation to neighboring PUFA-PLs. Cellular processes such as autophagic degradation of ferritin (Gao et al., 2016; Hou et al., 2016), the inhibition of iron exporter ferroportin (Bao et al., 2021; Chen et al., 2020a; Geng et al., 2018), and increased transferrin uptake (Gao et al., 2015) elevate labile iron pool (LIP) in cells, facilitating ferroptosis (Liang et al., 2022).
In the enzymatic LPO pathway, Fe2+ initiates the dioxygenation of PUFA-PLs in the cell membrane by serving as an essential cofactor for iron-dependent peroxidases to facilitate their activity (Chen et al., 2020b; David et al., 2022). During this process, ACSL4 catalyzes the ligation of free PUFAs with CoA, producing PUFA-CoAs, which are subsequently re-esterified and incorporated into PLs by LPCAT3 (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017). With the involvement of labile iron and O2, these incorporated PUFA-PLs undergo peroxidation via PORs and ALOXs, leading to the formation of PUFA-PLs-OOH (Hadian and Stockwell, 2020; Shah et al., 2018; Zou et al., 2020). For further details on lipid sources in ferroptosis, readers may refer to a recent comprehensive review (Conrad and Pratt, 2019; Dai et al., 2024; Stockwell and Jiang, 2020).
Ferroptosis defense mechanisms
To prevent excessive LPO and inhibit unintended ferroptosis, cells rely on specific protection or surveillance mechanisms (Hassannia et al., 2019). Ferroptosis defense systems function by directly neutralizing lipid hydroperoxides. These defense systems consist of surveillance pathways that operate with or without GPX4 and have distinct subcellular localizations (Gu et al., 2023).
SLC7A11-GSH-GPX4 pathway
The first well-characterized ferroptosis defense system is the SLC7A11-GSH-GPX4 axis (Lei et al., 2022; Sun et al., 2022). As a lipid repair enzyme (Brigelius-Flohé and Flohé, 2020; Brigelius-Flohé and Maiorino, 2013), GPX4 converts and reduces reactive peroxidized PUFA-PLs to non-reactive and non-toxic PUFA-PL alcohols (PUFA-PL-OH) by oxidizing two molecules of reduced GSHs into an oxidized glutathione (GSSG) (Seibt et al., 2019; Ursini et al., 1982). GPX4 serves as a key inhibitor of ferroptosis by preventing the accumulation of peroxidized PUFA-PLs in most cells (Dixon et al., 2012; Forcina and Dixon, 2019; Friedmann Angeli et al., 2014; Ingold et al., 2018; Yang et al., 2014), and it is localized across multiple subcellular compartments, including mitochondria, nuclei, and cytosol. Among these, mitochondrion and cytosol GPX4 play crucial roles in preventing ferroptosis (Dixon and Olzmann, 2024). GPX4 functions in conjunction with System Xc−, a cystine/glutamate antiporter composed of solute carrier family 7 member 11(SLC7A11, also known as xCT) and solute carrier family 3 member 2 (SLC3A2) (Zhang, 2024). xCT facilitates the exchange of intracellular glutamate for extracellular cystine to biosynthesize reduced GSH (Koppula et al., 2018; Sato et al., 1999).
FSP1-CoQH2 system
The second identified endogenous ferroptosis defense mechanism is the ferroptosis suppressor protein 1 (FSP1)-ubiquinone (coenzyme Q10 or CoQ10) system. Independently of GPX4, FSP1 localizes to the plasma membrane and functions as an NADPH-dependent CoQ reductase, converting CoQ10 into ubiquinol (CoQH2), a lipid-soluble antioxidant that inhibits LPO and prevents ferroptosis in cellular membranes (Bersuker et al., 2019; Doll et al., 2019; Nakamura et al., 2023). Additionally, FSP1 suppresses ferroptosis by activating the endosomal sorting complex required for transport III (ESCRT-III), which facilitates the repair of plasma membrane damage (Dai et al., 2020; Pedrera et al., 2021).
GCH1-BH4 system
The third ferroptosis defense system identified as independent of GPX4, which inhibits LPO, is the GTP cyclohydrolase 1(GCH1)-tetrahydrobiopterin (BH4) axis (Kraft et al., 2020; Soula et al., 2020). GCH1 prevents ferroptosis through two mechanisms. First, it remodels the lipid membrane environment. by either depleting PUFA-PLs, which promote ferroptosis, or increasing the abundance of reduced CoQ10 (Stockwell, 2022). Second, it facilitates the production of BH4, an endogenous metabolite with radical-trapping antioxidant activity (Stockwell, 2022). The BH4 produced via GCH1 functions as a cofactor for aromatic amino acid hydroxylases and, analogously to CoQ10, helps suppress LPO (Kraft et al., 2020; Soula et al., 2020).
DHODH-CoQH2 system
The fourth identified ferroptosis defense system, also independent of GPX4, is the dihydroorotate dehydrogenase (DHODH)-dihydroubiquione (CoQH2) axis, which operates within the mitochondria (Mao et al., 2021). DHODH, an enzyme located in the inner mitochondrial membrane, catalyzes pyrimidine biosynthesis and converts CoQ10 to CoQH2. This process maintains mitochondrial CoQ H2 levels, functioning analogously to FSP1 in extramitochondrial membranes (Mao et al., 2021). When GPX4 is inactivated, DHODH activity increases, enhancing CoQH2 production, which neutralizes LPO and suppresses ferroptosis originating in the mitochondrion (Mao et al., 2021).
MBOAT1/2-MUFA system
In 2023, Jiang and colleagues identified a novel ferroptosis defense system that is independent of GPX4 and FSP1. This system consists of a membrane-bound O-acyltransferase domain-containing 1/2 (MBOAT1/2) and phosphatidylethanolamine (PE)-monounsaturated fatty acids (PE-MUFAs) (Liang et al., 2023). PE-PUFAs, which serve as the preferred substrates for LPO, determine the sensitivity of cells to ferroptosis (Doll et al., 2017; Kagan et al., 2017). MBOAT2 functions as a lyso-PL acyltransferase (LPLAT) that selectively transfers MUFAs onto lyso-PE, increasing cellular PE-MUFA levels while decreasing PE-PUFA levels. This process suppresses the initiation and induction of ferroptosis. Additionally, the estrogen receptor (ER) and androgen receptor (AR) directly and transcriptionally modulate MBOAT1 and MBOAT2 expression, respectively (Liang et al., 2023).
SC5D-7-DHC axis
The most recently identified ferroptosis inhibitor is the lathosterol oxidase −7-dehydrocholesterol (SC5D-7-DHC) axis, discovered independently by two research groups in 2024, revealing that 7-DHC functions as a natural ferroptosis suppressor (Freitas et al., 2024; Li et al., 2024b). 7-DHC is synthesized in the endoplasmic reticulum and is present in the cell membrane and mitochondria within the cholesterol biosynthesis pathway. By diverting the LPO pathway away from phospholipids and trapping radicals, 7-DHC suppresses LPO and inhibits ferroptosis in the mitochondria and plasma membrane.
Epigenetic Modification of Ferroptosis by ncRNAs in Cancer Malignancy
Epigenetics is a reversible and dynamic process that modulates gene expression without altering the DNA sequence (Cao and Yan, 2020; Cavalli and Heard, 2019; Wang et al., 2023c). The four major mechanisms of epigenetic regulation include DNA methylation, chromatin structure remodeling, histone modification, and ncRNA regulation (Cao and Yan, 2020; Cavalli and Heard, 2019; Wang et al., 2023c). ncRNAs are functional transcripts with minimal or no protein-coding ability (Chen and Kim, 2024; Lin et al., 2021). Their regulation is a widely studied epigenetic regulatory mechanism (Wang et al., 2023b). The major types of regulatory ncRNAs include microRNA (miRNA), circular RNA (circRNA), and long non-coding RNAs (lncRNAs) (Ashrafizadeh et al., 2022; Entezari et al., 2022; Mirzaei et al., 2022; Saw et al., 2021). ncRNAs regulate signal transduction, cellular behaviors, and tumorigenesis (Chen et al., 2023; Li et al., 2024a; Liu et al., 2021b; Vianello et al., 2024; Xue et al., 2022) and are widely identified as key regulators of multiple cancer hallmarks, including cell death (such as ferroptosis and apoptosis), proliferation, invasion, metastasis, drug resistance, and genomic instability (Li et al., 2024a; Sun et al., 2024a; Wang et al., 2024b). Research indicates that dysregulated ncRNAs are key epigenetic regulators of ferroptosis, regulating iron metabolism, mitochondrial-related proteins, glutathione metabolism, and LPO, thereby modulating cancer onset and progression (Balihodzic et al., 2022; Ensoy et al., 2023; Luo et al., 2021; Valashedi et al., 2022; Wang et al., 2022; Wu et al., 2020; Xie and Guo, 2021; Zuo et al., 2022). Targeting epigenetic modifications that regulate ferroptosis is considered a promising therapeutic approach for cancer (Wang et al., 2023; Yang et al., 2023).
ncRNAs: new Players in Cancer Therapeutic Targets
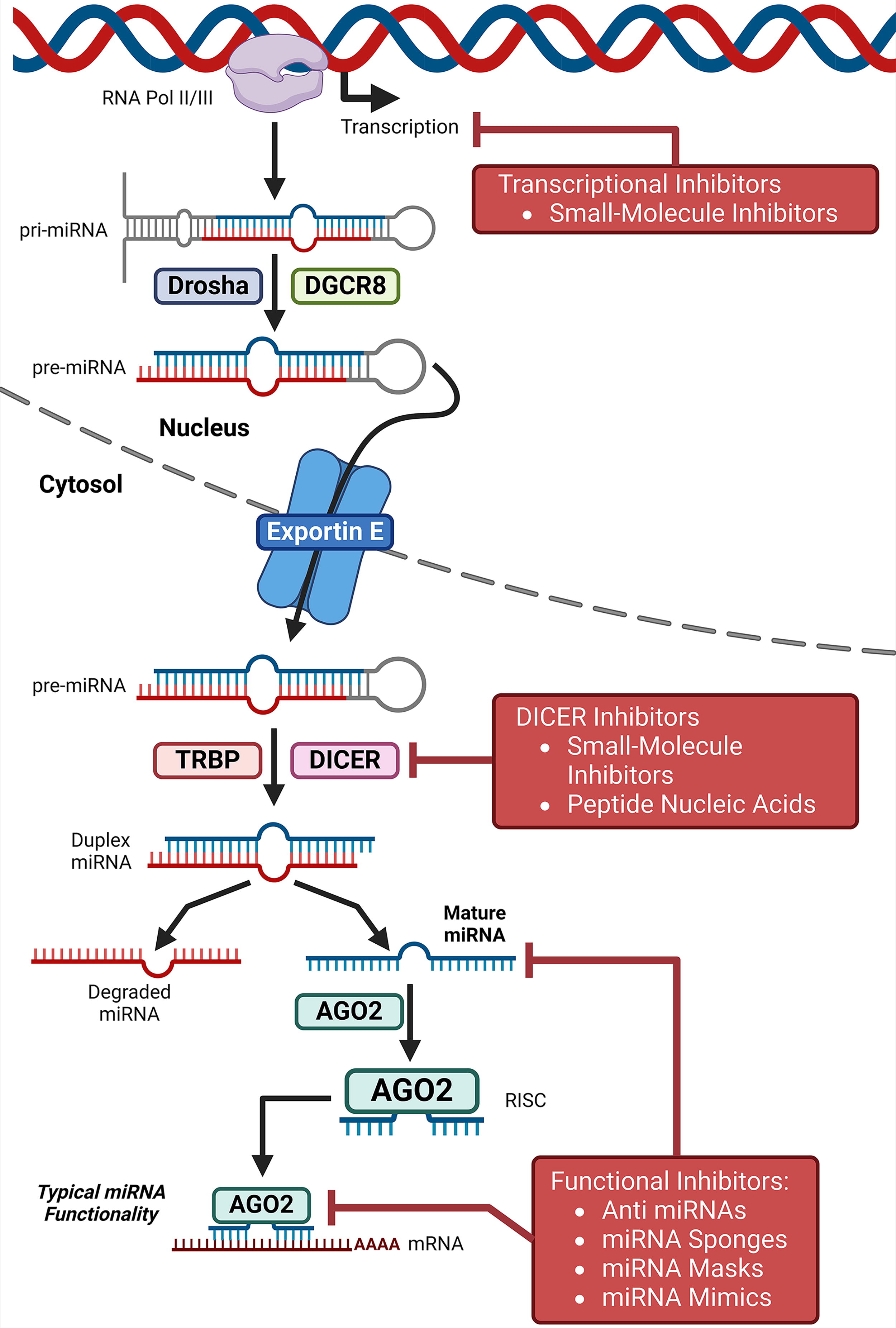
Since the discovery of ncRNAs, scientists have explored their therapeutic potential. Over the past decade, significant efforts have been made to develop clinically applicable RNA-based therapeutics, primarily utilizing siRNAs and ASO (Winkle et al., 2021). miRNAs can be targeted through various approaches, including inhibiting biogenesis, suppression of mature miRNA, miRNA replacement therapy, and preventing miRNA-target interactions (Winkle et al., 2021) (Fig. 2). Owing to their diverse functions, lncRNAs can be targeted by genome-editing techniques, steric blockade of promoter, post-transcriptional RNA degradation pathways, or steric inhibition of RNA-protein interactions and secondary structure formation (Arun et al., 2018) (Fig. 3). In cancer therapy, circRNAs can be targeted by either reducing their biological availability (loss-of-function) or introducing a functional circRNAs (gain-of-function) (Conn et al., 2024; Kristensen et al., 2022) (Fig. 4). While numerous anti-miRNA and miRNA-based therapeutics are in phase II or III clinical trials, no circRNA-targeted or lncRNA-based treatments have reached clinic application (He et al., 2021b; Nappi, 2024; Winkle et al., 2021). We believe that if SMCs can modulate ncRNA activity and promote ferroptosis, they may represent a promising strategy for cancer treatment.


Targeting ncRNAs with Small-Molecule Compounds for Cancer Therapy
Studies reveal that SMCs can target ncRNAs, making them potential druggable targets for cancer therapy (Zhao et al., 2022). SMCs targeting specific ncRNAs (known as SMCNs) offer significant advantages over oligonucleotides, including better solubility, improved cellular uptake, improved bioavailability, and metabolic stability (Winkle et al., 2021; Zhang et al., 2010). Additionally, SMCNs have the potential for oral administration. Decades of advancements in traditional medicinal chemistry have refined SMC synthesis and optimization, allowing SMCNs to overcome the pharmacological limitations of oligonucleotide-based therapies. SMCNs are not a novel concept; those targeting miRNAs were identified as potential therapeutics over a decade ago (Zhang et al., 2010). SMCNs may influence miRNA processing or regulate miRNAs at the transcriptional and post-transcriptional levels (Vo et al., 2014). Various bioactive compounds can modulate the expression of several tumor-suppressive and oncogenic miRNAs (Jurj et al., 2024; Ruiz-Manriquez et al., 2022). Unlike oligonucleotide-mediated degradation, SMCNs typically carry positive charges and can induce lncRNA loss-of-function through interference with folding or binding partner recognition, similar to the mechanism of steric blocker ASOs (Costales et al., 2020). Furthermore, several bioactive compounds can modulate the expression of several tumor-suppressive and oncogenic lncRNAs (Ruiz-Manriquez et al., 2022). Initially, compound 5 was identified as a molecule that specifically binds to a structured element at the 3′ terminus of the lncRNA MALAT1, promoting its nuclear accumulation and stability(Abulwerdi et al., 2019). The phytochemical quercetin exerts anticancer effects by inhibiting various dysregulated-signaling pathways that induce cancer cells (Rajesh R and Sangeetha, 2024; Vollmannová et al., 2024), and it can bind to and destabilize the lncRNA MALAT1 by interacting with its triple-helical regions (Rakheja et al., 2022). Similarly, compound X1 suppresses tumor growth and metastasis by binding to and destabilizing the lncRNA XIST, disrupting its interaction with the PRC2 epigenetic silencing complex in breast cancer, both in vitro and in vivo (Aguilar et al., 2022).
Small Molecules Targeting ncRNAs to Regulate Ferroptosis in Cancer Therapy
Research indicates that SMCs can regulate ferroptosis in cancer therapy by targeting ncRNAs. This section summarizes the innovative strategy of using SMCs to target ncRNAs (miRNA, lncRNA, and circRNA), enabling novel ferroptosis-centered approaches to anticancer treatment (Table 1 and Fig. 5).

Chemical structures of small molecules targeting ncRNAS which modulate ferroptosis in cancer.
Emerging Small-Molecule Compounds Kill Cancers by Inducing Ferroptosis via Modulating ncRNA
AML, Acute myeloid leukemia; AF, Amentoflavone; CRC, Colorectal cancer; GNA, Gambogenic acid; EGR1, Early growth response 1; ER, endoplasmic reticulum stress; FOXA2, forkhead box protein A2; FTH1, ferritin heavy chain 1; GC, gastric cancer; GPX4, glutathione peroxidase 4; GSH, glutathione; HAND, HIV-associated neurological disorder; HCC, hepatocellular carcinoma; IRG1, immune-responsive gene 1; NSCLC, non-small cell lung cancer; PCa Prostate cancer; ROS, reactive oxygen species; SLC7A11, solute carrier family 7 member 11; TFRC, transferrin receptor.
Small molecules targeting miRNAs regulating ferroptosis in cancer therapy
Gambogenic acid (GNA), a bioactive compound derived from Garcinia hanburyi resin, exhibits antitumor effects by inducing ferroptosis, apoptosis, necroptosis, and regulating the cell cycle and autophagy (Huang et al., 2024; Liu et al., 2021a; Mi et al., 2024; Wu et al., 2023b; Zhao et al., 2020). Studies show that in colorectal cancer (CRC), GNA induces ferroptosis by upregulating miR-1291 (Ma et al., 2023) (Table 1 and Fig. 6). GNA activates endoplasmic reticulum stress (ERS) by upregulating miR-1291, which targets the forkhead box protein A2 (FOXA2). GNA also induces ferroptosis, evidenced by GSH depletion, decreased GPX4 expression, increased ROS production, and enhanced uptake of transferrin-mediated Fe2+ (Ma et al., 2023). Mechanistic studies show that GNA activates AMPKα and SLC7A11/GPX4 signaling. These findings suggest that GNA induces ferroptosis and activates ER stress via miR-1291 in CRC cells (Ma et al., 2023). Icariin, a major bioactive component of Herba Epimedii, is a natural prenylated flavonol glycoside with diverse pharmacological effects, including antitumor, antioxidant, antiaging, neuroprotective, immunomodulatory, and aphrodisiac effects (Sánchez-Gutiérrez et al., 2024). Icariin induces ferroptosis to eliminate cancer cells (Sheng et al., 2024) and enhances the efficacy of PD-1 inhibitors by inducing ferroptosis (Haoyue et al., 2025). Curcumol, a major terpenoid derived from Curcumae Rhizoma, induces ferroptosis in cancers by inhibiting the Nrf2/HO-1 signaling pathway (Feng et al., 2024; Sheng et al., 2024). Icariin and curcumol inhibit proliferation and increase apoptosis, ROS levels, and miR-7 expression in prostate cancer (PCa) cells (Xu et al., 2023). Their combined treatment enhances antitumor activity. Icariin and curcumol induce autophagy and ferroptosis in PCa cells, which is reversed by silencing miR-7 (Xu et al., 2023). Inhibiting mTOR/SREBP1/SCD1-mediated lipogenesis promotes ferroptosis in cancer cells (Xu et al., 2024; Zheng et al., 2024). Studies show that miR-7 inhibits mTOR and downregulates mTOR/SREBP1 pathway expression in PCa cells. These findings suggest that icariin and curcumol induce ferroptosis in PCa by inhibiting mTOR/SREBP1/SCD1-mediated lipogenesis through miR-7 upregulation (Xu et al., 2023). Studies show that icariin induces ferroptosis in renal cell carcinoma cells by upregulating miR-324-3p, which inhibits the GPX4 axis (Yu et al., 2022). Icariin inhibits RCC cell proliferation, migration, and invasion. Ferroptosis induction is evidenced by the accumulation of Fe2+, MDA, ROS, and reduced GSH/GPX4 levels. GPX4 overexpression reverses icariin-mediated ferroptosis (Yu et al., 2022). Icariin upregulates miR-324-3p, which directly targets GPX4 (Yu et al., 2022). These findings suggest that icariin induces ferroptosis via the miR-324-3p/GPX4 axis in RCC cells. Amentoflavone (AF) is a naturally active phenolic compound derived from Selaginella tamariscina with documented anticancer activity (Tuli et al., 2023). Amentoflavone exerts antitumor activity by inducing apoptosis (Lin et al., 2023; Su et al., 2022; Yang et al., 2022) and enhancing chemotherapy efficacy through AKR1B10 inhibition (Zhao et al., 2022). In GC tissues and cells, miR-496 expression decreases while ATF2 expression increases (Tang et al., 2023). Amentoflavone induces ferroptosis, increases miR-496 expression, and decreases ATF2 expression in GC cells. Silencing miR-496 reverses amentoflavone-mediated GC cell proliferation and ferroptosis and decreases ATF2 expression (Tang et al., 2023). Mechanistically, miR-496 targets ATF2, while miR-496 overexpression inhibits proliferation and induces ferroptosis in GC cells (Tang et al., 2023). These findings suggest that amentoflavone induces ferroptosis in GC cells by increasing miR-496, which inhibits ATF2. Cancer cells develop anoikis resistance, thereby enabling their survival under anchorage‐independent conditions (Wang et al., 2024a). Empagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, is a well-tolerated and highly effective antidiabetic drug (Wu et al., 2023a). Empagliflozin exerts anticancer effects in breast cancer (Abdelhamid et al., 2022; Karzoon et al., 2025; Wu et al., 2023a). Empagliflozin induces ferroptosis in anoikis-resistant cells by synergistically reducing specificity protein1 (SP1) and CD98hc levels while activating miR-128-3p in breast cancer (Nalla and Khairnar, 2024). Metformin triggers ferroptosis in MDA-MB-231 cells by upregulating miR-324-3p expression. Overexpression of miR-324-3p inhibits cell viability, while miR-324-3p inhibitors enhance it (Hou et al., 2021). miR-324-3p binds to and downregulates GPX4 (Hou et al., 2021). Metformin also induces ferroptosis in in vivo xenografts by upregulating miR-324-3p expression (Hou et al., 2021). These findings indicate that metformin induces ferroptosis by inhibiting GPX4 through miR-324-3p upregulation (Hou et al., 2021). Propofol exerts antitumor activity by inducing apoptosis (Sun et al., 2024b; Zhang et al., 2024) and ferroptosis (Jin et al., 2024), modulating m6A modification (Chen et al., 2024), and enhancing chemotherapy efficacy (Sue et al., 2024). Propofol reduces cisplatin resistance and induces ferroptosis in non-small cell lung cancer (NSCLC) cells. Propofol inhibits GPX4 transcription by upregulating miR-744-5p and miR-615-3p (Han et al., 2023). miR-744-5p/miR-615-3p downregulation or GPX4 upregulation attenuates propofol-mediated suppression of cisplatin resistance. Additionally, propofol inhibits tumor growth and reduces cisplatin resistance by inducing ferroptosis by increasing miR-744-5p/miR-615-3p expression to inhibit GPX4 (Han et al., 2023). These findings indicate that propofol overcomes cisplatin resistance in NSCLC by inducing ferroptosis through GPX4 inhibition, mediated by upregulating miR-744-5p/miR-615-3p (Han et al., 2023). Sanggenol L, an active agent in Morus Bark, induces ferroptosis by upregulating miR-26a-1-3p, which directly targets the E3 ubiquitin ligase mouse double minute-2 homolog (MDM2) (Fu et al., 2024). miR-26a-1-3p-mediated MDM2 suppression enhances p53 protein levels and decreases its downstream target SLC7A11, ultimately triggering ferroptosis in NSCLC. In summary, these findings suggest that sanggenol L induces ferroptosis by upregulating miR-26a-1-3p, which inhibits MDM2, leading to p53 stabilization and subsequent SLC7A11 downregulation (Fu et al., 2024). α-Hederin suppresses the proliferation and metastasis of cisplatin-resistant NSCLC cells in vitro and in vivo. α-Hederin induces ferroptosis by decreasing SLC7A11 and GPX4 while activating the DNA damage-inducible transcript 3-activating transcription factor 3 (DDIT3-ATF3) pathway (Han et al., 2024). α-Hederin promotes the nuclear expression of early growth response 1 (EGR1), which binds to and inhibits miR-96-5p transcription. miR-96-5p directly inhibits DDIT3 (Han et al., 2024). These findings suggest that α-Hederin induces ferroptosis in NSCLC by activating DDIT3/ATF3 pathway, which inhibits SLC7A11/GPX4. This effect is mediated through EGR1 nuclear expression, leading to miR-96-5p downregulation (Han et al., 2024). Increased NFS1 expression is implicated in acute myeloid leukemia (AML) cells. Silencing NFS1 enhances ferroptosis, evidenced by increased ROS accumulation, an expanded labile iron pool, and decreased GPX4 expression (Liu et al., 2023a). GPX4 overexpression reverses NFS1 silencing-induced ferroptosis. miR-335-5p was identified as an upstream negative regulator of NFS1. Resveratro

Small molecules targeting LncRNAs regulating ferroptosis in cancer therapy
SMCs targeting lncRNAs is a promising strategy in cancer therapy. Cinobufotalin, an active compound derived from the traditional Chinese Medicine ChanSu, exhibits broad-spectrum antitumorigenic activities (Bai et al., 2021; Li et al., 2022; Meng et al., 2021; Xia et al., 2022). Cinobufotalin suppresses lung cancer cell growth by inducing ferroptosis by upregulating TFRC-mediated lncRNA LINC00597 expression. This process decreases miR-367-3p in NSCLC (Huang et al., 2023) (Table 1 and Fig. 6). Curcumenol induces ferroptosis in vitro and in vivo in NSCLC. In lung cancer cells, curcumenol downregulates the lncRNA H19. LncRNA H19 overexpression reverses curcumenol-mediated anticancer effects, whereas lncRNA H19 silencing enhances curcumenol-induced ferroptosis (Zhang et al., 2022b). Mechanistic studies show that lncRNA H19 functions as a competing endogenous RNA (ceRNA), binding to miR-19b-3p, thereby transcriptionally enhancing the activity of ferritin heavy chain 1 (FTH1) (Zhang et al., 2022b). β-elemene enhances erlotinib-mediated cytotoxicity by inducing ferroptosis in EGFR-mutant NSCLC cells with primary resistance to EGFR-TKI (Xu et al., 2023). β-elemene combination with erlotinib upregulates lncRNA H19 expression. Knockdown or overexpression of lncRNA H19 confers resistance or sensitivity to erlotinib, respectively, in in vitro and in vivo studies. Furthermore, lncRNA H19 overexpression facilitates erlotinib-induced cytotoxicity by triggering ferroptosis (Xu et al., 2023). These findings suggest that β-elemene boosts erlotinib-induced antitumor activities in NSCLC by inducing ferroptosis through lncRNA H19 upregulation (Xu et al., 2023). Gambogenic acid induces ferroptosis by reducing SLC7A11 expression through the downregulation of lncRNA NEAT1 (Wang et al., 2022). Ketamine inhibits hepatocellular carcinoma (HCC) cell proliferation by inducing ferroptosis through GPX4 downregulation in both in vitro and in vivo (He et al., 2021a). Ketamine decreases lncPVT1 expression, which interacts with miR-214-3p to inhibit its sponge function on GPX4. Loss of lncPVT1 enhances ferroptosis, an effect reversed by GPX4 overexpression or miR-214-3p silencing (He et al., 2021a). GPX4 overexpression or miR-214-3p silencing also reverses ketamine-induced inhibition of cell growth and ferroptosis. These findings indicate that ketamine induces ferroptosis by downregulating GPX4 via lncPVT1 suppression, thereby impeding miR-214-3p as a sponge for GPX4 (He et al., 2021a).
Small molecules targeting circRNAs regulating ferroptosis in cancer therapy
Dexmedetomidine (DEX), a highly selective α2-adrenoceptor agonist widely used in intensive and anesthetic care for its sedative and anxiolytic effects, exhibits anticancer effects (Carnet Le Provost et al., 2024). A recent study shows that DEX inhibits cell viability and promotes apoptosis in GC cells in vitro while inhibiting tumor growth in vivo. DEX induces ferroptosis in GC, as evidenced by increased iron and ROS decreased GSH, GPX4, and SLC7A11. These effects are abolished by the ferroptosis inhibitor Ferrostatin-1 (Gao and Wang, 2023) (Table 1 and Fig. 6). DEX downregulates circ0008035 levels and E2F transcription factor 7 (E2F7) while upregulating miR-302a in GC cells (Gao and Wang, 2023). E2F7 functions as a ferroptosis inhibitor in cancer (Liu et al., 2023b; Xi et al., 2022). Circ0008035 upregulates E2F7 expression by acting as a sponge for miR-302a. Circ0008035 suppresses DEX-induced ferroptosis in GC cells, an effect that is reversed by miR-302a overexpression E2F7 silencing (Gao and Wang, 2023). Collectively, these findings suggest that DEX triggers ferroptosis in GC by downregulating circ0008035, thereby preventing miR-302a as a sponge for E2F7, eventually decreasing E2F7 in GC (Gao and Wang, 2023).
Conclusions and Perspectives
Advancement in our understanding of ncRNAs in cancer has driven the development of novel classes of drugs targeting ncRNAs. Findings indicate that dysregulated ncRNAs regulate ferroptosis by modulating glutathione metabolism, iron metabolism, mitochondrial-related proteins, and LPO, thereby modulating cancer initiation and progression. Targeting ncRNA-mediated epigenetic modifications modulating ferroptosis presents a new direction for cancer treatment. Studies show that small-molecule drug-like compounds target ncRNAs to regulate ferroptosis, offering new opportunities for precision cancer therapy. This review highlights the innovative strategy of using SMCs to target oncogenic ncRNAs, an approach that regulates ferroptosis and reshapes the cancer therapy landscape.
Despite significant progress in developing small molecules that target ncRNAs regulating ferroptosis for cancer therapy, challenges remain, especially in developing cancer drug resistance. Further optimization of existing targeted therapies remains possible. First, research is needed to elucidate how ncRNAs mediate epigenetic modifications specific to ferroptosis. Second, further research is required to elucidate the molecular biology of ncRNAs, as their structures, mechanisms, sequences, functions, and modularity are often inferred from a limited subset of experimentally characterized ncRNAs to date. Third, more research is needed to produce SMCN candidates for pharmaceutical development. Fourth, although studies show that certain SMCs modulate microRNAs or lncRNAs, the mechanisms, signaling pathways, and chemical structural properties responsible for these effects remain uncovered. For miRNA, inhibiting miRNA biogenesis disrupts the expression of the mature functional miRNA, which can be achieved with SMCs (Jurj et al., 2024). Studies show that SMCs interact with pre-miRNA species and regulatory protein-RNA complexes. SMCs interact with different miRNA processing stages by binding to miRNA precursors or protein-RNA complexes (Jurj et al., 2024). SMCs can translocate into the nucleus, where they interact with primary miRNA (pri-miRNA) and precursor miRNA (pre-miRNA) molecules, thereby modulating their abundance. Additionally, SMCs can interact with Dicer enzymes and the Argonaute-RNA-induced silencing complex (AGO-RISC), thereby diminishing mature miRNA levels. This reduction in mature miRNA impairs translational repression, mRNA target cleavage, and mRNA deadenylation (Jurj et al., 2024). The potential mechanism by which the SMCs discussed in this review target ncRNAs to regulate ferroptosis in cancer therapy may involve modulating various miRNA processing steps, warranting further investigation. Fifth, further research is needed to identify the toxicity profile of SMCN pharmacophores, enabling the development of anticancer agents with improve patient acceptability. A key challenge to address is the potential off-target effects of SMCNs, which may affect the tissue of interest and other physiological systems (Monroig et al., 2015). Therefore, the discovery of novel SMCNs requires extensive in vitro and in vivo studies to evaluate possible side effects and unintended off-target effects. Cutting-edge drug delivery systems, including nanoparticle carriers and CRISPR-Cas9 system, enhance the specificity and efficiency of SMC delivery to target ncRNAs. Nanoparticles facilitate cancer-specific drug delivery through passive targeting mechanisms and active targeting strategies. Additionally, they improve the pharmacokinetics and bioavailability of encapsulated drugs, leading to enhanced therapeutic efficacy and safety compared to those of conventional treatment modalities. Novel nanoparticle development will add to delivering drug molecules to the specific target site and reduce the adverse side effects of anticancer therapies in humans. As research on SMCNs regulating ferroptosis remains in its early stages, continued investigation and development of chemical matter and innovative technologies are essential to fully realize the therapeutic potential of this emerging strategy in cancer treatment. SMCNs regulating ferroptosis provide a promising therapeutic approach in cancer treatment, aligning with the increasing focus on combination therapies and precision medicine. As our understanding of cancer and the chemistry of SMCNs regulating ferroptosis continually advances, these molecules will likely be vital in the ongoing goal of anticancer therapy. In the near future, small molecules targeting ncRNAs involved in ferroptosis may become a potential component of the pharmacopeia, expanding the landscape of druggable targets to provide a novel therapy for cancer treatments.
Consent for Publication
All of the authors are aware of and agree to the content of the paper and their being listed as a co-author of the paper.
Footnotes
Authors’ Contributions
J.Z., H.W., and Yu.W. designed and conceived the Review. Y.W., F.M., and W.Z. contributed substantially to discussion of the content. J.Z. and Y.W. wrote the article. H.W. generated the figures. J.S.F. and H.W. generated the figures. J.S.F. edited the article. All authors contributed to reviewing and/or editing of article. All authors approved the final article.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Author Disclosure Statement
Funding Information
This work was supported in part by the Hohhot Youth Talent Plan Project (2023003), Hohhot Science and Technology Bureau Applied Research and Development Funds (2023–14), Science and Technology Project of High-level Clinical Specialty Construction in Hohhot Public Hospital of Inner Mongolia Autonomous Region Health Commission (2023SGGZ029), Wu Jieping Medical Foundation (320.6750. 2024–13-59), Science Foundation of Aerospace Center hospital (YN202402 and YN202423), and the Science Foundation of AMHT (2022YK01 and 2024YK04).