
Abstract
Aims:
We previously demonstrated that aryl hydrocarbon receptor (AhR) activation attenuates the cytoprotective effect of hydrogen sulfide (H2S), leading to indoxyl sulfate (IS)-mediated renal tubular damage. However, it is unclear whether this pathway would be present in an in vivo uremic model.
Results:
In a rat chronic kidney disease (CKD) model with 5/6 nephrectomized (Nx), we found that poor renal filtration is associated with accumulation of IS and homocysteine (Hcy), an H2S precursor. Compared with controls, the protein and mRNA levels of H2S-producing enzymes, including cystathionine β-synthase, cystathionine γ-lyase, and 3-mercaptopyruvate sulfurtransferase, were attenuated in Nx kidneys. Since the transcription factor, specificity protein 1 (Sp1), acts as an upstream regulator of these enzyme expressions, we found that the protein level and activity of Sp1 were significantly decreased in Nx kidneys. Interestingly, employing the blocker of the AhR CH-223191 not only reverses the decrease in H2S-producing enzymes and Sp1, but it also reverses H2S reduction in Nx rats. These are associated with the mitigation of plasma Hcy accumulation, renal excretion, perfusion insufficiency, and tubular damage. Moreover, the oxidative stress in Nx kidneys due to increased superoxide formation and decreased glutathione contents was also attenuated by AhR inhibition.
Innovation:
Our findings highlight the deleterious effect of AhR activation on renal H2S formation may be due to IS accumulation and underline AhR blockade as a novel therapy for CKD.
Conclusion:
AhR is detrimental to Sp1 function in vivo, leading to impeding renal H2S generation and exacerbating oxidative stress during CKD progression. Antioxid. Redox Signal. 43, 448–464.

Introduction
Hydrogen sulfide (H2S) is naturally produced by three main enzymes—cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST)—along with cystathionine aminotransferase (Feng et al., 2022). It acts as a gasotransmitter and can freely diffuse across cell membranes because of its lipophilic properties. In the kidneys, H2S plays a pivotal role, serving as a vasodilator that enhances renal blood flow (RBF), inhibits tubular sodium transport, and impacts renin release-functions with repercussions for systemic blood pressure (Feng et al., 2022). The signaling function of H2S not only plays physiological regulatory roles but also protects organs against pathophysiological deteriorations in nephropathy caused by ischemia/reperfusion, urinary tract obstruction, diabetes, and hypertension (Bos et al., 2009; Aminzadeh and Vaziri, 2012).
CBS catalyzes the conversion of homocysteine (Hcy) and serine into cystathionine, which CSE then utilizes to generate cysteine, a common substrate used by all H2S-generating enzymes, including 3-MST, for H2S production (Sen et al., 2010). Epidemiological studies have revealed that elevated plasma levels of Hcy increase cardiovascular risk, especially for patients with chronic kidney disease (CKD) (Mallamaci et al., 2002). The decreased glomerular filtration in CKD likely contributes to elevated plasma Hcy levels, a condition known as hyperhomocysteinemia (HHcy) (Wollesen et al., 1999; Veldman et al., 2005). HHcy can induce vascular injury via atherosclerotic remodeling in kidney diseases (Weber et al., 2016). This can, however, be ameliorated after kidney transplantation, indicating that impaired renal metabolism is responsible for HHcy in patients with CKD (Weber et al., 2016). It is unclear whether HHcy is related to a deficiency in intrarenal H2S-producing enzymes in CKD.
Innovation
Through an unknown mechanism, H2S deficiency in chronic kidney disease (CKD) patients is known to be associated with the accumulation of uremic toxins, leading to oxidative stress and worsening kidney function. Our results reveal a potential mechanism of AhR activation and thereby impairs the regulatory function of the transcription factor specificity protein 1 (Sp1) with respect to H2S-producing enzymes as well as the beneficial effects of AhR blockade on renal H2S system and antioxidant defense. These findings suggest that AhR inhibition may be a promising therapeutic strategy for slowing the progression of CKD.
Besides the precursors for H2S generation, the factors that affect H2S-producing enzyme expression or activity also have profound effects on H2S formation. Diminished CBS expression in patients with CKD leads to H2S deficiency, which triggers oxidative stress under conditions of deteriorating renal function (Song et al., 2014; Yuan et al., 2017b; Lu et al., 2021b; Scammahorn et al., 2021). H2S can reduce oxidative stress, directly scavenging reactive oxygen species (ROS) and boosting reduced glutathione (GSH) levels as a means of self-protection (Whiteman et al., 2004). However, in the context of CKD, a reduction in renal H2S production could weaken the kidneys’ capacity for antioxidant defense (Scammahorn et al., 2021).
As an environmental sensor, the aryl hydrocarbon receptor (AhR) can recognize a variety of exogenous and endogenous molecules, including uremic toxins that usually accumulate in patients with CKD because of reduced kidney function. Increased AhR expression and activation have been observed in patients with CKD and in the kidneys of 5/6 nephrectomized (Nx) rats (Miao et al., 2020). Indoxyl sulfate (IS), one of the uremic toxins, was found to be a potent AhR ligand with a potency for AhR activation similar to that of two well-known exogenous ligands, 2,3,7,8-tetrachlorodibenzo-p-dioxin and benzo[a]pyrene (Schroeder et al., 2010; Stanford et al., 2016). Once activated, AhR is known to modulate gene expression patterns that promote renal fibrosis, endothelial dysfunction, and mitochondrial imbalance via oxidative stress in CKD (Sahebnasagh et al., 2021; Grishanova and Perepechaeva, 2022; Nguyen et al., 2022; Yang et al., 2022). Previous studies have shown that AhR interacts with transcription factor Sp1 and acts on the promoter of the cytochrome P450 1A1 (CYP1A1) gene to initiate transcription (Kobayashi et al., 1996; Ye et al., 2019). Moreover, several studies have revealed that the Sp1 regulates the expression of H2S-producing enzymes (Ge et al., 2001; Ge et al., 2002; Maclean et al., 2004). Our previous results also revealed that decreased Sp1 activity is associated with low CBS and 3-MST expression in IS-treated renal tubular cells (Lu et al., 2022). Restoring Sp1 activity by blocking AhR led to increases in CBS and 3-MST expression, indicating the direct effect of Sp1 on the regulation of the expression of H2S-producing enzymes.
It remains unclear whether the deleterious effects of AhR activation on H2S production can be observed in an Nx rat CKD model. In this study, we aimed to investigate the underlying mechanism of how AhR affects the regulatory effect of Sp1 on the regulation of intrarenal H2S-producing enzyme expression and determine whether AhR blockade may ameliorate functional insufficiency caused by oxidative stress in CKD in vivo.
Results
5/6 Nephrectomy induces renal function impairment and uremic toxin accumulation
After 6 weeks of induction, the plasma levels of creatinine (Cr) in the Nx rats were significantly higher than those in the control rats (Table 1). This phenomenon was associated with poor renal filtration, as estimated based on creatinine clearance (CrCl) and severe proteinuria, indicating abnormalities in kidney function caused by the 5/6 nephrectomy (Nx), meeting the clinical criteria for CKD. This poor renal function was also associated with high plasma and renal levels of IS in the Nx rats. Compared with the controls, renal excretion of IS was significantly lower in the Nx rats. Interestingly, the levels of the precursor for H2S formation, Hcy, were higher in the plasma and the renal tissue of the Nx rats.
Functional Deteriorations and Uremic Toxin Accumulation in 5/6 Nephrectomy (Nx)-Mediated Chronic Kidney Disease
N = 6 in each group.
*p < 0.05, Nx vs. control.
CrCl, creatinine clearance; Hcy, homocysteine; IS, indoxyl sulfate; PCr, plasma creatinine level.
Nx impairs Sp1 and H2S-producing enzyme expression and H2S formation
Compared with that in the control kidneys, the protein expression of the transcription factor Sp1 was markedly decreased in the Nx kidneys (Fig. 1A). This was associated with low Sp1 activity in the renal tissues of the Nx rats compared with that in the controls (Fig. 1B). As Sp1 regulates the gene expression of H2S-producing enzymes, we observed that the protein levels of CBS, CSE, and 3-MST were significantly lower in the Nx kidneys compared with those in the controls (Fig. 1C–E). These findings were also associated with low mRNA levels of H2S-producing enzymes in the Nx kidneys (Fig. 1F–H). Moreover, the H2S levels in the renal vein, renal tissue homogenate, and urine of the Nx rats were all significantly lower than those of the control rats (Table 2).
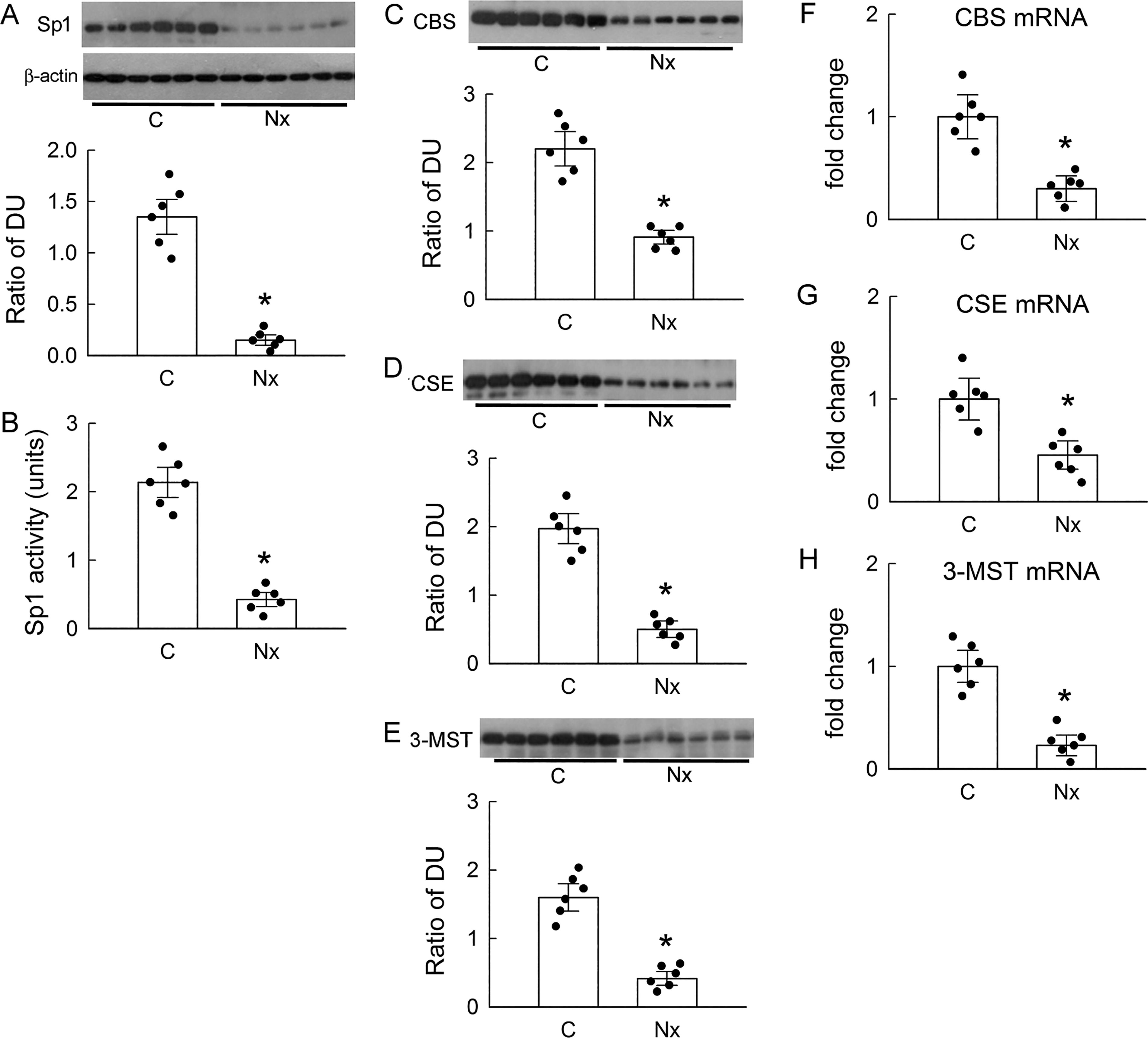
5/6 Nephrectomy (Nx) Decreases Renal Hydrogen Sulfide (H2S) Levels
N = 6 in each group.
p < 0.05, Nx vs. control.
Chronic inhibition of is receptor ameliorates Nx-induced hemodynamic changes and kidney injury
Chronic administration of the specific AhR antagonist CH-223191 (CH) to block its activation in Nx rats partially mitigated the filtration deficiency by lowering plasma creatinine (PCr) levels (Fig. 2A), enhancing the glomerular filtration rate (shown as CrCl, in Fig. 2B), and attenuating kidney injury by ameliorating proteinuria (Fig. 2C) and reducing the abundance of the biomarker kidney injury molecule-1 (KIM-1) in renal tissues and urine (Fig. 2D and E), thus highlighting the detrimental effect of IS, through AhR activation, on renal tubules. CH alone, however, had no effect on these functional indexes and tubular damage markers (Fig. 2A–E).
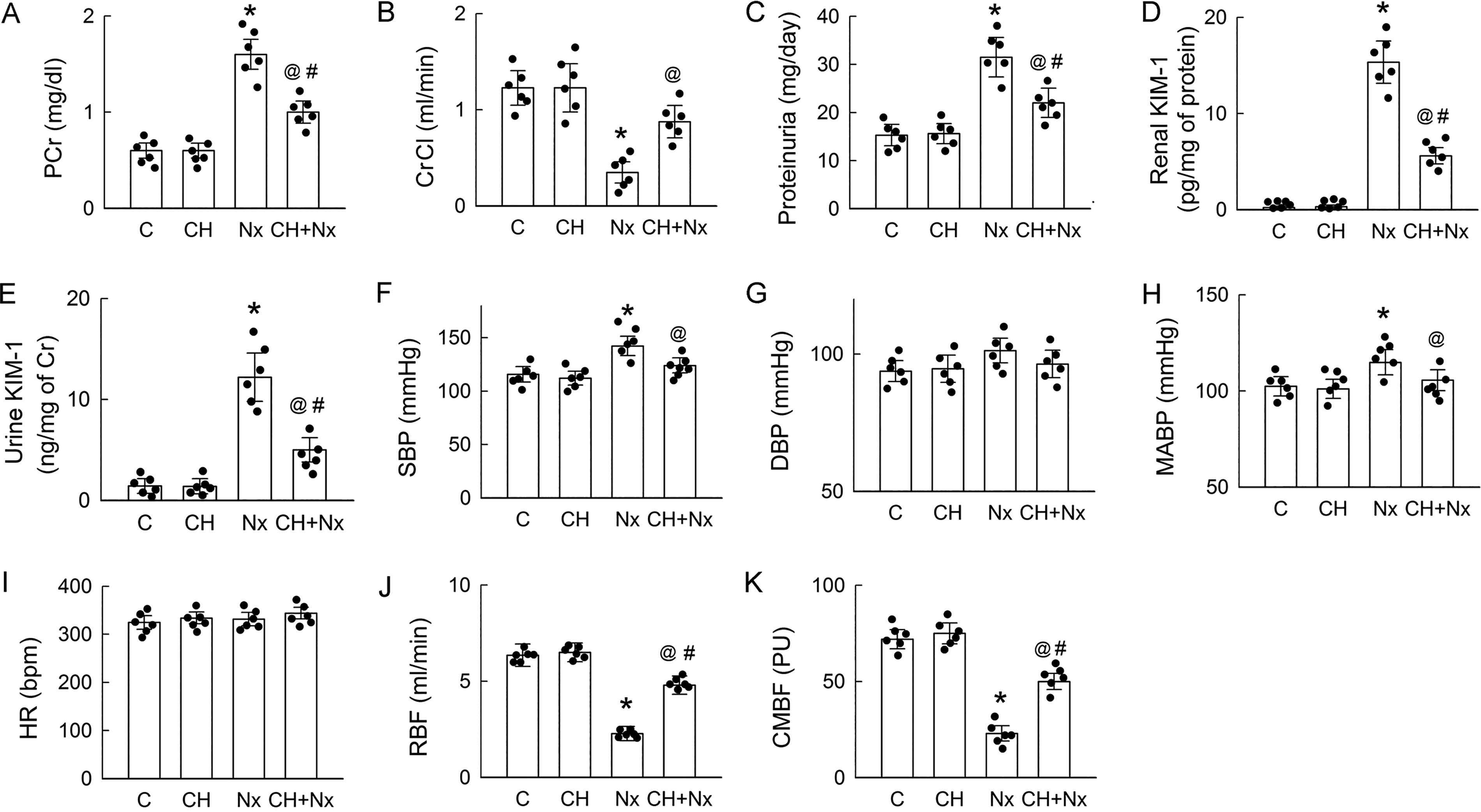
Furthermore, noticeable surges in systolic blood pressure (SBP) and MABP but not in diastolic blood pressure and HR were observed in the Nx rats (Fig. 2F–I). These were associated with decreases in both the RBF and cortical microvascular blood flow (CMBF) in the Nx kidneys compared with those in the control kidneys (Fig. 2J and K). These changes can be attributed to the reduction in the nephron number after Nx. The administration of CH effectively attenuated increases in SBP and MABP caused by Nx while concurrently enhancing RBF and CMBF. CH showed little effect on systemic and renal hemodynamics when treated alone (Fig. 2F–K).
AhR inhibition enhances H2S formation and Hcy catabolism
Chronic administration of CH had no observable effects on IS levels in the plasma, renal tissues, and urine in the control rats (Fig. 3A–C). CH did not affect the increased levels of IS in the plasma and renal tissues, and the decrease in the IS levels in the urine was not caused by Nx either, indicating that signals downstream of AhR induce renal injury. We performed linear regression analysis to explore the correlation between plasma Cr (Fig. 2A) and IS levels in the rats’ plasma and kidneys (Fig. 3A and B). As shown in Supplementary Data, there were positive relationships between plasma Cr and IS levels (Supplementary Fig. S1A) and plasma Cr and renal Cr levels (Supplementary Fig. S1B) in the vehicle- (control and Nx) and CH-treated (CH and CH + Nx) groups, and all correlations were significant (p < 0.05). A lower R-squared value was found for the CH-treated group, indicating a weaker relationship between the variables. However, when the correlation coefficients of the regression lines were compared between the vehicle- and CH-treated groups within the same variances, no significant differences were found.
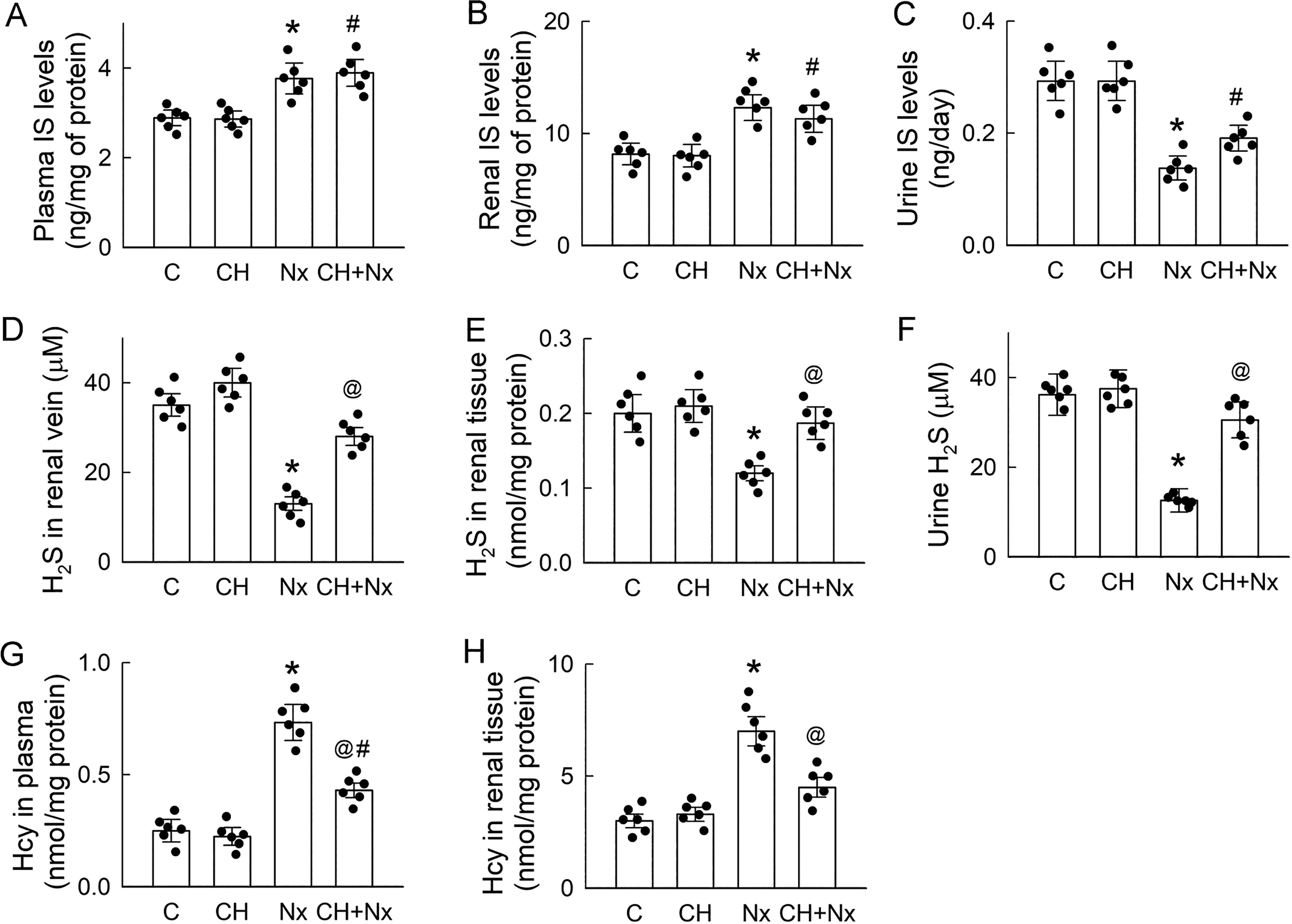
Considering the crucial role of H2S in renoprotection, we assessed the blockade effect of AhR in renal H2S production. The substantial reductions in H2S levels in the renal vein, renal tissue, and urine of the Nx rats were completely reversed by cotreatment with CH (Fig. 3D–F). These results clearly indicate there is a renal H2S deficiency in Nx rats. Moreover, the accumulation of Hcy in both plasma and renal tissues following Nx was effectively reversed using CH (Fig. 3G and H). These findings demonstrate that the elevated IS levels in the Nx rats were associated with poor H2S production through AhR activation; this condition probably leads to the accumulation of the H2S precursor Hcy.
Based on the presence of H2S deficiency in the Nx kidneys, one might ask whether H2S donors can mitigate Nx-induced renal injury. Our results show that chronic supplementation of H2S by GYY-4137 (GYY) ameliorated renal insufficiency (compared with that in the control rats) in the Nx rats. GYY improved glomerular filtration and mitigated tubular damage and oxidative stress in the Nx rats. However, GYY had no effect on Nx-induced proteinuria and HHcy (Supplementary Fig. S2).
Blockade of AhR reverses low Sp1 activity and H2S-producing enzyme expression caused by Nx
Since chronic inhibition of AhR enhanced renal H2S formation in the Nx rats, we wondered whether this improvement was dependent on changes in H2S-producing-enzyme levels. Considering that Sp1 serves as an upstream transcription factor in the regulation of H2S-producing enzyme expression, our results demonstrate that the Nx-mediated low Sp1 protein expression and DNA-binding activity were reversed by CH to levels similar to those in the control kidneys (Fig. 4A and B). This finding confirmed that the binding of Sp1 to the promoter region and its transcriptional activity can be influenced by the IS receptor, AhR. Interestingly, the recovery of Sp1 function caused by AhR blockade was associated with elevated expressions of CBS, CSE, and 3-MST in the Nx kidneys at both the protein (Fig. 4C–E) and mRNA (Fig. 4F–H) levels. These findings imply that the reduced expression of H2S-producing enzymes in the Nx kidneys following the impairment of the transcriptional function of Sp1 may be attributed to AhR.

Inhibition of AhR prevents Nx-mediated downregulation of H2S-producing enzymes in renal tissue
In the left part of Figure 5A, the majority of CBS in the control kidneys was found in the tubular epithelium and the lining of the capillary (upper). Less frequent distributions of CBS were found in glomeruli. In the Nx kidney, the tubular expression of CBS was markedly decreased and distributed in clusters in the cytoplasm and the apical membrane of the epithelium and in the capillary (upper middle). Compared with the control kidneys, chronic treatment of CH alone showed little effect on CBS expression (lower middle). The signal for tubular CBS expression in the CH + Nx kidneys was weaker than that for the CH kidneys (lower).
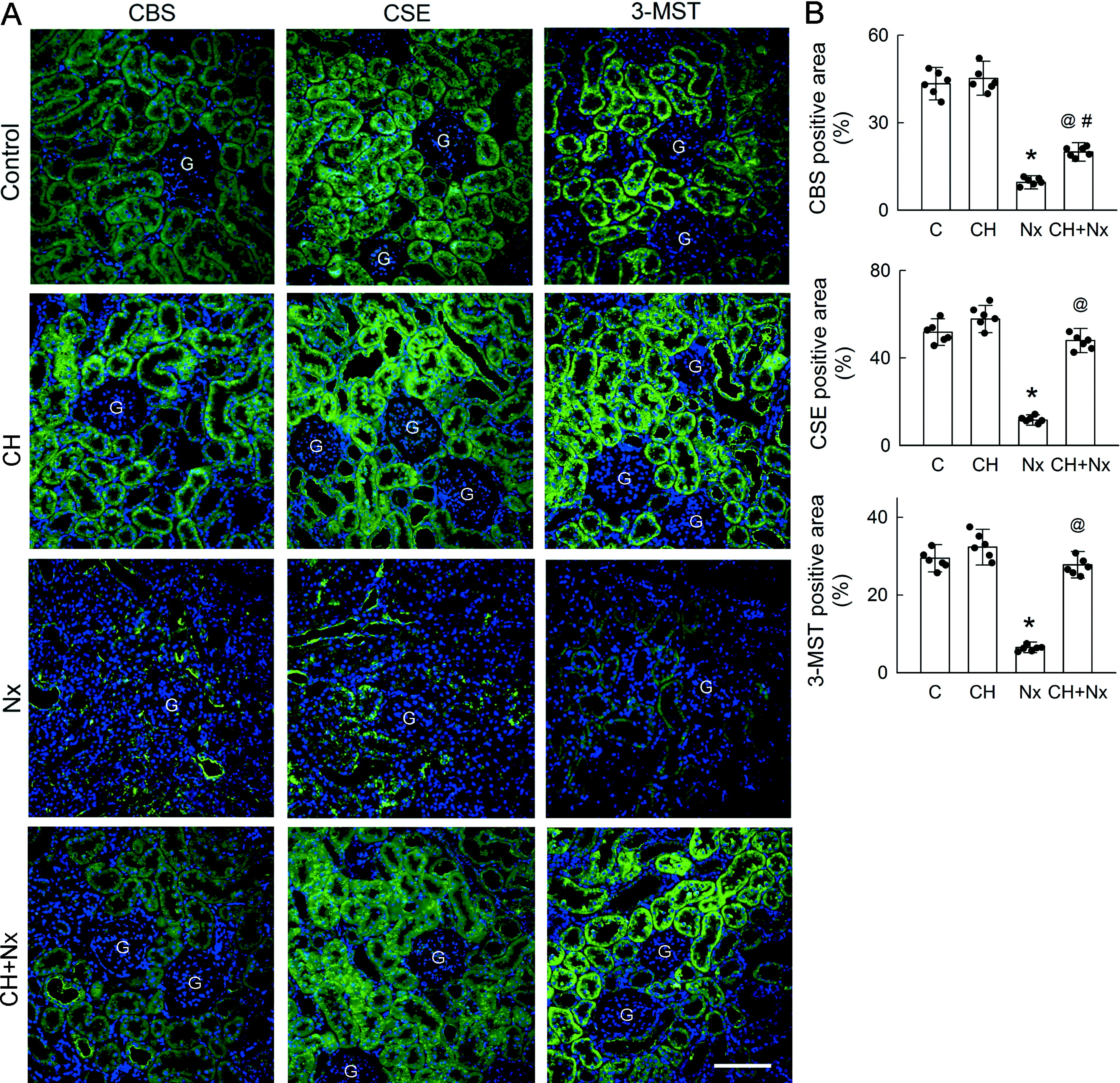
In the middle part of Figure 5A, most of the CSE in the control kidneys was also present in the renal tubules, with a less frequent distribution in the capillary and glomeruli (upper). Similar to CBS, the tubular expression of CSE was greatly reduced in the Nx kidneys, and clustered CSE was found in the epithelial cytoplasm and capillary (upper middle). CH treatment alone had no effect on CSE expression (lower middle). In the CH-treated Nx kidneys, CSE expression was similar to that in the control kidneys, and only some clustered expression was found in the epithelial cytoplasm (lower).
As shown in the right part of Figure 5A, the 3-MST was mainly expressed in the renal tubules of the control kidneys, with low expression in the capillary and glomeruli (upper). The tubular expression of 3-MST was markedly attenuated in the Nx kidneys (upper middle). CH alone showed no effect on tubular 3-MST expression (lower middle). CH somehow increased 3-MST expression in the capillary. CH also reversed the Nx-induced reduction in tubular 3-MST expression (lower).
Providing data after quantification, Figure 5B shows that Nx led to significant decreases in the expression of all H2S-producing enzymes. Blockade of AhR by CH partially reversed the effect of Nx on CBS reduction and completely abolished Nx-induced downregulation of CSE and 3-MST to levels similar to those of the controls.
Inhibition of AhR attenuates oxidative stress in Nx kidneys
Studies have revealed that oxidative stress contributes to the progression of CKD and subsequent renal function loss (Gyurászová et al., 2020; Ling and Kuo, 2018). We, therefore, asked whether the beneficial effect of chronic AhR blockade affects oxidative stress in Nx kidneys. Lucigenin can be excited using superoxide and emits enhanced CL (Tseng et al., 2021); accordingly, our results show that an intrarenal arterial infusion of lucigenin into the Nx kidneys induced an abrupt rise in CL counts on the kidney surface (as shown by the red line of the representative tracing in Fig. 6A), and this increase was more abrupt than that in the control kidneys. Chronic CH treatment decreased the CL counts in the Nx kidneys (the blue line in Fig. 6A). The controls and rats treated with CH only exhibited no effects in the CL recording, as the levels always fluctuated around the baseline (see the light and dark green lines in Fig. 6A). The area under the curve (AUC) results indicate an increase in lucigenin-dependent CL in the Nx kidneys in comparison to that in the control kidneys (Fig. 6B). CH alone showed no effect on CL counts but significantly reduced the CL counts in the Nx kidneys. These results clearly indicate that the excess ROS formation was caused by Nx via the effect of AhR.

Since H2S exhibits antioxidant properties by inducing GSH production (Kimura et al., 2010), we further examined whether the beneficial effect of AhR blockade in terms of relieving oxidative stress in Nx kidneys depends on GSH. The renal content of GSH in the Nx rats was notably lower than that in the controls. This reduction was, however, significantly ameliorated upon co-administration of CH (Fig. 6C). The controls and rats treated with CH alone exhibited no effect in terms of their renal GSH content. Conversely, the renal content of GSSG in the Nx rats was higher than that in the controls, but this elevation was diminished upon cotreatment with CH (Fig. 6D). Compared with the controls, the ratio of GSH to GSSG, a marker of oxidative stress, was significantly lower in the Nx rats. This decrement was, however, effectively reversed in the Nx rats following treatment with CH (Fig. 6E).
MDA is a polyunsaturated fatty acid peroxidation product and commonly utilized as a biomarker of oxidative stress (Vodošek Hojs et al., 2020). In this study, we observed a significant increase in the urinary excretion of MDA in the Nx rats compared with that of the controls (Fig. 6F). However, chronic inhibition of AhR by administering CH resulted in a marked reduction in urinary MDA levels in the Nx rats. Administering CH alone showed no effect on the urinary MDA levels in the control rats. These results underline the impaired capacity of the antioxidant defense caused by the activation of the IS receptor and its role in increasing the renal antioxidative burden during CKD.
Discussion
Our results demonstrate the potential mechanism, in an in vivo model of CKD, through which AhR activation disrupts H2S production, probably via the accumulation of the uremic toxin IS, leading to oxidative damage in Nx kidneys. Glomerular filtration impairment, poor renal perfusion, and proximal tubular injury probably contribute to IS accumulation by diminishing the renal excretory capacity of the injured kidney. AhR activation further adversely affects the transcriptional regulation of Sp1 and H2S-producing enzyme expression in the remaining nephrons. Along with retarding cytoprotective H2S formation, deficiency in H2S-producing enzyme expression also induces a reduction in the renal catabolism of Hcy, leading to HHcy, which is known to be associated with increased cardiovascular mortality among patients with kidney failure (Mallamaci et al., 2002). Significantly, when a blocker of AhR is administered concurrently, it notably ameliorates the harmful effects of AhR on renal function and systemic hypertension in CKD rats. AhR inhibition beneficially impacts renal Sp1 activity and increases H2S-producing enzyme expression, thus lowering Hcy levels and enhancing H2S formation for the further recruitment of GSH for antioxidant defense. These findings strongly indicate the involvement of the IS/AhR pathway in the renal impairment of the H2S-forming system and in the outcome of nephrectomy-induced kidney damage.
In the rat models of CKD, we found that impaired renal excretion of IS probably causes the accumulation of IS in the plasma and kidneys of Nx rats (Table 1). Uremia changes the diversity of the intestinal flora and epithelial barrier and causes the tryptophan metabolite indole to enter the circulation and thus form IS in the liver, which then accumulates in the plasma of patients with CKD (Lu et al., 2021b). Kidneys, therefore, represent the only way to eliminate IS accumulation in the body through the actions of organic anion transporters 1 and 3 in the proximal tubule (Deguchi et al., 2004). Clinically, patients with progressive renal function decline have been found to have higher baseline serum IS levels than those without progression, particularly in the advanced stages of CKD (Lu et al., 2021b). Moreover, elevated serum IS levels are independently associated with increased all-cause mortality in patients with acute kidney injury (Wang et al., 2019). Thus, serum IS levels can serve as a valuable predictor of renal function deterioration. Scavenging the indole using the charcoal absorbent AST-120 therefore lowers the serum and urine IS concentrations in CKD and delays the initiation of dialysis (Ueda et al., 2007). However, in this study, IS accumulation was not affected by the blockade of its receptor, AhR (Fig. 3A and B), indicating that possible downstream effects are mediated after AhR activation via IS in renal injury.
In the present study, both the plasma and renal tissue levels of Hcy in the Nx rats with H2S deficiency were notably elevated (Table 2); these levels can, however, be partially reversed by means of AhR inhibition (Fig. 3G and H), indicating the possible involvement of IS. In fact, Hcy serves as a substrate for H2S production after catabolism by CBS and CSE (Sen et al., 2010). Consequently, the low expression of these enzymes in Nx kidneys leads to the accumulation of Hcy, a uremic toxin. This observation is consistent with the results of earlier studies on other kidney disease models, where ischemia/reperfusion was shown to decrease CBS activity, resulting in Hcy buildup in the damaged kidney (Xu et al., 2009). Hcy accumulation is known to impair vascular function by reducing nitric oxide production in endothelial cells and promoting proliferation in vascular smooth muscle cells, thereby increasing systemic vascular resistance (Yu et al., 2011). Elevated Hcy levels are also considered a risk factor for cardiovascular mortality among patients with CKD or those undergoing dialysis (Mallamaci et al., 2002). Beyond their impact on the cardiovascular system, high Hcy levels contribute directly to renal injury by inducing oxidative stress, inflammation, endoplasmic reticulum stress, or DNA hypomethylation (Long and Nie, 2016). Moreover, Hcy exacerbates the progression of acute kidney injury by increasing mitochondrial damage, leading to reduced ATP production. Specifically, Hcy decreases ATP synthase β subunit levels and enhances the release of cytochrome c into the cytoplasm (Kaplan et al., 2020). Furthermore, elevated Hcy levels may exert feedback inhibition on CSE enzyme activity, which exacerbates H2S deficiency (Chang et al., 2008). Interestingly, HHcy affects the substrate’s preference for H2S production. At a physiological Hcy concentration, most H2S is produced from cysteine through CBS- and CSE-catalyzed reactions. However, Hcy replaces cysteine as the principal source of H2S in CSE-catalyzed, but not CBS-catalyzed, reactions in HHcy (Yang and He, 2019). Furthermore, in cardiomyocytes, there is negative feedback regulation between CBS and CSE, where HHcy suppresses CBS, leading to the upregulation of CSE via Sp1 induction (Nandi and Mishra, 2017).
Given the reduction in the levels of H2S-producing enzymes, it is important to investigate their upstream signaling, which regulates these enzymes and interacts with AhR. As a transcription factor, Sp1 possesses a DNA-binding domain with three zinc fingers that specifically binds to GC-rich regions within the promoter regions of various target genes that are involved in essential cellular processes, such as cell differentiation, proliferation, and growth (Ge et al., 2001). Upon activation, AhR binds to the xenobiotic response element/GC box, recruiting Sp1 to enhance the transcription of human CYP1A1, which plays a role in metabolizing xenobiotics and regulating oxidative stress, thereby contributing to various diseases (Nguyen et al., 2022). An increase in Sp1 expression has been shown to protect kidneys from ischemia/reperfusion injury (Yuan et al., 2017a). Interestingly, even when Sp1 protein levels are normal, a decrease in Sp1/DNA-binding activity can lead to reduced transcriptional activity, resulting in decreased renal CBS expression. This reduction contributes to kidney injuries induced by ischemia/reperfusion, as previously reported (Wu et al., 2010). In this study, we observed reductions in the levels of Sp1 protein and its DNA-binding activity in Nx kidneys (Fig. 1A and B), and these reductions in transcriptional activity were reversed after AhR inhibition (Fig. 4A and B). Hence, we confirmed that the impairment of H2S production in renal tissues depends on the interaction between AhR and Sp1, which inhibits all H2S-producing enzymes. However, the low Sp1 protein expression observed in this in vivo study contrasts with our previous in vitro findings, which showed that exogenous IS treatment only reduced the activity of Sp1 in renal tubular cells but not its protein levels (Lu et al., 2022). The longer induction time for Nx in rat kidneys and the briefer exposure to exogenous IS treatment in tubular cell cultures may explain the different effects of AhR on Sp1 protein expression. In addition to the presence of its binding sites, the alteration of phosphorylation or synergism with other transcriptional factors also affects the binding of Sp1 to the CBS or CSE promoter and thus gene expression (Wu et al., 2010).
According to our immunofluorescent labeling results, the decreased expression of H2S-producing enzymes in Nx kidneys mostly originates from renal tubules (Fig. 5). These findings align with our previous observations regarding tubular cell cultures, wherein the obstruction of H2S generation by administrating the H2S-producing enzyme blocker aminooxyacetic acid worsened IS-induced tubular damage (Lu et al., 2022). In this study, most of H2S-producing enzymes were distributed in the cytoplasm of tubular cells in the control kidneys (Fig. 6). After Nx, tubular H2S-producing enzymes were found to cluster in the cytoplasm. A previous study showed that under stress, CSE can translocate to mitochondria and produce H2S, which can serve as a substrate for ATP production and provide an additional protective mechanism against renal damage (Fu et al., 2012). Further investigation is required to determine whether this mechanism can be found in the tubular cells of residual tissue in Nx kidneys, a tissue that induces the translocation of H2S-producing enzymes to mitochondria. Previous studies have demonstrated that H2S-producing enzymes are not only present in tubular epithelial cells but also expressed in endothelial cells, mesangial cells, and glomerular podocytes (Feng et al., 2022). The tissue distribution of H2S-producing enzymes in this highly blood-perfused organ probably underscores the kidney’s role as the primary source of endogenous H2S production in the body (Feng et al., 2022). Although we did not further clarify which of these enzymes is the major one affecting changes in H2S in Nx rats, a previous study showed that CSE was more dominant than the other two in the kidneys of Sprague Dawley rats, underscoring CSE’s leading role in H2S production (Koning et al., 2015). In our previous in vitro experiment, exogenous IS treatment was used to mimic accumulated IS in CKD (Lu et al., 2022); unlike the persistent decreases in CBS and 3-MST expression, CSE expression only decreased after 24 h of treatment and then recovered, indicating the important role of CSE in the survival of residual tubular cells. In contrast to the in vivo results of this study, there was a complete loss of H2S formation due to decreases in the expression of all three H2S-producing enzymes after 6 weeks, greatly affecting the functional support in the remnant tissues of Nx kidneys. This can be countered by simultaneous treatment with AhR inhibitor (Fig. 4C–H), indicating that IS may suppress H2S production via AhR activation in renal tissues, influencing both H2S release into the renal vein and excretion in urine (Figs. 3D and 4F). Previous studies on patients with CKD or patients with end-stage renal disease also reported reduced levels of H2S, a finding attributed to the downregulation of H2S-producing enzymes (Song et al., 2014; Perna et al., 2009; Yuan et al., 2017b). Similarly, in rats subjected to Nx by means of surgical induction, a reduction in H2S production was observed in both the liver and kidneys. This decrease was accompanied by a significant downregulation of hepatic and renal CBS and CSE expression, suggesting a systemic deficiency that may be linked to the presence of uremic toxins (Aminzadeh and Vaziri, 2012).
In patients undergoing hemodialysis, low sulfhemoglobin levels in erythrocytes have also been shown to indicate H2S deficiency (Perna et al., 2009). Notably, following a standard hemodialysis session, plasma H2S levels significantly increase compared with pre-dialysis levels, suggesting that the removal of uremic toxins may reverse the H2S reduction due to the inhibitory effects of toxins (Perna et al., 2011). These observations underscore the intricacies of the influence of uremia on H2S production. We also explored the beneficial effects of H2S donors and found that chronic delivery of H2S by GYY-4137 ameliorates renal insufficiency in Nx rats by improving glomerular filtration and ameliorating tubular damage and oxidative stress in comparison to control rats (Supplementary Fig. S2). Similar to the previous findings, histological damage to the glomeruli and tubules in Nx kidneys was ameliorated when rats were treated with an H2S donor sodium hydrosulfide (NaHS) for 8 weeks (Askari et al., 2018). NaHS also suppressed renal inflammation and apoptosis by reducing the expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and caspase-3, respectively, and substantially attenuated oxidative stress in the Nx kidneys. Significantly, this supplementation also accelerates the recovery of renal tubular cells in kidneys injured by ischemia (Han et al., 2015).
We previously showed that the supplementation of H2S in tubular cells by means of treatment with the H2S donor NaHS or GYY-4137 significantly increases the cellular GSH content and attenuates superoxide formation (Lu et al., 2022). These findings emphasize the adverse impact of reduced renal H2S production on the exacerbation of oxidative stress in CKD. CKD is widely known for its characteristic feature of increased oxidative stress, which results from either the excessive generation of ROS or the depletion of antioxidants. In patients with CKD, the accumulation of IS in the body induces oxidative stress, which is a key contributor to multiple-organ damage (Lu et al., 2021b). Specifically, IS-induced oxidative stress in the kidneys is associated with podocyte injury, leading to proteinuria, as well as the development of tubulointerstitial fibrosis with vascular damage (Ichii et al., 2014). Furthermore, IS-mediated oxidative stress leads to endothelial dysfunction and senescence, impairing nitric oxide production and worsening renal parenchymal damage through hypoxic induction during the deterioration of renal function (Yu et al., 2011). In this study, we demonstrated a significant increase in oxidative stress in Nx kidneys, primarily attributed to an intensified generation of superoxides, concomitant with a decline in the GSH redox ratio (Fig. 6A–E). Consequently, these heightened oxidative stress levels led to lipid peroxidation in the kidney tissue and an elevation in urinary MDA levels, indicating oxidative damage (Fig. 6F). Kidneys’ high metabolic rate and substantial oxygen utilization make them particularly susceptible to oxidative stress, a key factor in the initiation and progression of CKD. As a crucial intracellular gaseous antioxidant, H2S is known to mitigate oxidative stress in various pathological conditions. In Nx kidneys, an H2S deficiency is likely to impair the antioxidant defenses in residual renal tissues through several mechanisms, as previously reported (Zhang and Hannink, 2003; Kimura et al., 2010; Wang et al., 2019). First, H2S increases GSH production by regulating cystine/cysteine transporters and reallocating GSH to mitochondria. Moreover, H2S enhances the S-sulfhydration of cysteine residues in critical proteins, such as Kelch-like ECH-associated protein 1 (Keap1), which activates nuclear factor erythroid 2-related factor 2 (Nrf2) by freeing it from the Keap1-mediated complex to enhance antioxidant GSH formation in response to oxidative stress. Pretreatment with an H2S donor safeguards against kidney function decline in rodents with ischemia/reperfusion injury by suppressing oxidative stress and enhancing Nrf2 translocation (McFarlane et al., 2023). Second, H2S increases the expression/activity of antioxidative enzymes, including copper-zinc superoxide dismutase and manganese superoxide dismutase, and upregulates antioxidant heme oxygenase-1, NAD(P)H: quinone oxidoreductase 1, and sirtuin 1. Third, H2S reduces ROS production, acting as an oxygen sensor, during mitochondrial oxidative respiration through metabolic modulation. H2S can also inhibit high-glucose-induced NADPH oxidase 4 caused by excess ROS generation by activating AMP-activated protein kinase. Maintaining the redox balance via H2S becomes increasingly important in the face of mounting oxidative stress during CKD progression. Consequently, the disruption of this H2S formation exacerbates oxidative stress, ultimately leading to an irreversible functional decline in Nx kidneys.
Importantly, we have observed that an Nx-induced oxidative stress can be ameliorated by inhibiting AhR (Fig. 6), underscoring a link between heightened oxidative stress and the reduction in H2S levels associated with AhR-mediated toxicity. The protective role of an AhR blockade is increasingly being recognized, extending beyond xenobiotic metabolism to include defense against carcinogenesis and oxidative stress. For example, blocking AhR disrupts the IS-mediated expression of the prooxidative gene CYP1A1, reducing oxidative stress in endothelial dysfunction and preserving the endothelium-dependent vasodilator response to acetylcholine (Nguyen et al., 2022). Additionally, AhR inhibition not only impacts H2S-related antioxidant pathways but also alleviates IS-induced cell senescence and renal fibrosis by reducing the ubiquitination of the proliferator-activated receptor gamma coactivator 1-α during mitochondrial biogenesis in proximal renal tubular cells (Xie et al., 2024). Since AhR is ubiquitously expressed and shows stronger activation potential in both patients with CKD and animal models (Dou et al., 2018), its inhibition may play a crucial role in mitigating adverse effects across various tissues associated with CKD.
The weakening of the antioxidative defense in Nx kidneys may be due to the downregulation of Sp1, as Sp1 has been reported to play a key role in regulating the transcription of various genes in response to oxidative stress. A previous study demonstrated that the expression of the antioxidant protein peroxiredoxin 6 (Prdx6) was reduced in podocytes after they were treated with high amounts of glucose (Zhang et al., 2021). Under these conditions, the overexpression of Sp1 bound directly to the promoter region of Prdx6 enhanced its expression, thereby preventing podocyte injury in diabetic nephropathy. In contrast, silencing Sp1 abolished the beneficial effect of Prdx6 in high-glucose-induced podocyte damage. Moreover, Sp1 is also an oxidative-stress-inducible transcription factor that promotes neuronal cell survival (Ryu et al., 2003). Elevated Sp1 levels, rather than just the Sp1 zinc finger DNA-binding domain, effectively prevent neuronal death in response to oxidative stress. The direct effect of Sp1 on oxidative stress in CKD warrants further detailed study.
The current findings were obtained using a rat model of Nx, and such models may not fully reflect the complexity of human CKD. Furthermore, in addition to AhR blockade enhancing H2S production and reducing oxidative stress, potential mechanisms leading to CKD progression, such as inflammation and fibrosis, should also be fully explored. Understanding the overall protective effects of AhR inhibition may provide an effective treatment such as AhR blockade for CKD.
Conclusions
Our results clearly reveal the damaging effects of AhR activation on renal H2S production in Nx rats with IS accumulation when using an AhR antagonist. The decrease in renal H2S production can be attributed to the downregulation of H2S-producing enzymes such as CBS, CSE, and 3-MST, as well as a reduction in their upstream regulator Sp1’s DNA transcriptional activity, all of which can be induced by AhR activation. The Nx-induced decreases in H2S levels make renal tissue more susceptible to oxidative damage, thereby promoting the progression of CKD. Therefore, H2S restoration or replenishment may represent a promising new approach to treating CKD.
Materials and Methods
Induction of CKD model using Nx
Male Wistar rats, aged 6–8 weeks and weighing around 220 g, were obtained from BioLASCO (Taipei, Taiwan). They were housed under controlled conditions with a 12 h light–dark cycle. All the procedures involving these animals adhered to the guidelines set by the Institutional Animal Care and Use Committee at Fu Jen Catholic University (permission no. A10959), and animal care and experimental protocols were performed in accordance with the National Institutes of Health Guidelines for Care and Use of Laboratory Animals (2011). All surgeries were performed under sodium pentobarbital anesthesia (60 mg/kg, i.p.), and all efforts were made to minimize suffering.
The rat CKD model was induced by means of Nx through a two-step procedure as follows: After anesthetization, a right paravertebral incision was made in the dorsal region, followed by the dissection of skin, muscle, and adipose tissue until the right kidney was exposed. The right kidney was removed by completely ligating the renal vessel and hilum with 4-0 silk. One week after the unilateral nephrectomy, a subtotal nephrectomy of the contralateral kidney was performed according to a previous method using double ligation in renal parenchyma (Perez-Ruiz et al., 2006). Briefly, the tissues surrounding the kidney were carefully isolated to avoid damage to the adrenal gland and ureter. Two surgical threads (3-0) were then placed at both poles of the kidney, perpendicular to the major axis and in parallel to each other. Special attention must be taken to leave the ureter free of the ligature. Changes in the degree of paleness in the poles of the kidney confirmed that the ligature was compressed sufficiently. After recovery from anesthesia, the rats were housed individually for 6 weeks to induce CKD. The sham-operated rats (controls) were exposed to the same surgical treatment but without right nephrectomy and tissue ligation of the left kidney (N = 6 in each group).
Delivery of AhR blocker via mini-osmotic pump
To explore the role of IS, another set of control and Nx rats were administered a vehicle solution (dimethyl sulfoxide [DMSO]) or specific AhR blocker CH-223191 (N = 6 in each group) using mini-osmotic pumps (Model 2006; Alzet, Cupertino, CA, USA) for 42 days. This delivery system ensured a stable drug infusion, thus maintaining a consistent drug concentration in the bloodstream. The pumps were incubated in sterile saline at 37°C for 4 h for activation before being subcutaneously implanted into the rats according to the manufacturer’s instructions and our previous method (Lu et al., 2021a). Following this, the rats were treated with Nx or a sham operation in the left kidney. The pumps were carefully filled with CH-223191 prepared in a 50% DMSO solution, at a concentration of 0.21 μg/μL, using a Hamilton syringe to avoid gas residue in the pump. The total amount of chemicals was released over 42 days at a rate of 1 μg per day. For subcutaneous implantation, a small incision was made in the skin between the scapulae. A small pocket was created in the subcutaneous connective tissues using a hemostat. The pump was inserted into the pocket with the flow moderator pointing away from the incision. The mini-osmotic pump was connected to a PE-50 catheter and inserted into the external jugular vein for continuous delivery of the drug or vehicle solution into the circulation. In the control group, the pump was filled with the same volume of DMSO. All the procedures were conducted under sterile conditions. The rats were returned to individual cages after recovery from anesthesia.
Metabolic cage and hemodynamic measurements
After 6 weeks, urine was collected over 24 h, and the total amount was determined using metabolic cages, as previously reported (Lu et al., 2021a). Briefly, urine and fecal pellets fell through the grid floor and were collected in separate tubes. The rats were individually housed during each urine collection session. The cages were thoroughly cleaned with detergent and water between collections.
After urine collection, the rats were anesthetized and underwent surgical preparation for the measurement of systemic and renal hemodynamics as previously described (Lin et al., 2015). The PE-50 catheter was placed into the left carotid artery for blood sampling and continuous measurement of the systemic mean arterial blood pressure (MABP). The heart rate (HR) was measured by means of arterial pulse wave analysis using MP36 AcqKnowledge software (Biopac, Los Angeles, CA). Another catheter was inserted into the left femoral vein to facilitate saline supplementation. The left kidney was exposed via a flank incision, and the RBF was measured using an ultrasound flow probe (Transonic Systems, Ithaca, NY). Regional blood perfusion in the renal cortex of the left kidney, termed the CMBF, was monitored using a flowmeter (Transonic Systems). All hemodynamic parameters were continuously recorded and displayed on a monitor. After 1 h of recording, blood (0.5 mL) from the left renal vein was sampled for creatinine and chemiluminescent assays. Renal tissues were then excised after transcardial perfusion of cold phosphate-buffered saline (PBS, pH 7.4) and postfixed in 10% formalin for immunofluorescent staining. A portion of the kidney was sampled and stored at −80°C to evaluate the protein or mRNA expression as described in the following sections.
Determination of H2S levels in urine and renal tissues
The urine samples were centrifuged at 4°C to obtain the supernatant. Renal tissues were homogenized with cold PBS at a volume of 10 times the tissue weight. A homogenized 200 μL urine supernatant and tissue solution was obtained to determine H2S levels according to the manufacturer’s instructions and previous method (Yang et al., 2008; Lu et al., 2022) for 5 min at 25°C, utilizing an ion-selective electrode (Lazar Research Laboratories, Los Angeles, CA) on a Fisher Accumet Model 10 pH meter (Fisher Scientific, Pittsburgh, PA). Standard solutions were prepared using NaHS with concentrations ranging from 0.1 to 100 μM. The H2S levels in renal tissues were normalized based on the protein amount in each sample and expressed as micromolar per milligram of protein.
Examination of superoxide formation by means of chemiluminescence analysis
The release of superoxide (O2 • −) from the left kidney into the renal vein was determined as previously described (Tseng et al., 2021). A 250 μL blood sample was collected as described above and stored at 4°C in the dark (wrapped in aluminum foil) until measurement. The sample was then mixed with 0.1 mL of PBS (pH 7.4) before chemiluminescence (CL) measurement. During the analysis, the blood was stored in a dark room to detect the emission of photons from CL using a Chemiluminescence Analyzing System (CLD-110, Tohoku Electronic Industrial Co., Sendai, Japan). A lucigenin solution (0.1 mM) was prepared in 1.0 mL of PBS and added to 100 μL of the blood sample. The AUC was used to assess the total CL amount. In each experiment, a duplicate test was performed, and the results are expressed as CL counts per second. All the chemicals in this assay were obtained from Sigma-Aldrich.
Blood and urine biochemical assay
The IS concentrations in the plasma, renal tissue homogenate, and urine were quantified using a commercial enzyme-linked immunosorbent assay (ELISA) kit (MBS3809027, MyBioSource, San Diego, CA, USA) according to the manufacturer’s instructions. The total protein amount in the samples was determined using a commercial kit (BioVision, Milpitas, CA, USA), as previously described (Tseng et al., 2021). The IS levels in the plasma and kidneys are expressed as per milligram of protein. The urinary excretion of IS was then calculated as the daily amount. The Hcy levels in the plasma and renal tissues were measured using a commercial ELISA kit (MBS1600141, MyBioSource) and are expressed per mg of protein. The creatinine levels in the plasma and urine were determined using commercial kits (BioQuant, Reinach, Switzerland) as previously described (Tseng et al., 2020). The CrCl was then calculated using the following formula: [urinary creatinine level]/[PCr level] × [24 h of urine volume amount]/1440. Furthermore, the level of KIM-1, a marker of tubular damage, was assessed using a commercial kit (ab223858, Abcam, Cambridge, MA, USA) following the manufacturer’s instructions to evaluate the individual and combined effects of Nx with CH-223191.
Protein expression of H2S-producing enzymes and Sp1 in renal tissues
The protein expressions of H2S-producing enzymes and Sp1 in renal tissues were determined through Western blotting as previously described (Lu et al., 2022). Kidney tissues were homogenized in ice-cold PBS and centrifuged at 12,000 g at 4°C. The supernatant was immediately used to measure the expressions of CSE, CBS, 3-MST, and Sp1. The supernatant was separated through sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then electrophoretically transferred onto polyvinylidene difluoride membranes, as previously described (Manna and Jain, 2012). The blotted membrane was blocked in fresh PBS containing 5% non-fat milk and subsequently incubated in a primary antibody solution against CSE (clone OTI1E12, lot #H0124), CBS (clone GT5199-1, lot #M0025), 3-MST (clone TMDU-D8, lot #L1522), or Sp1 (clone ARC0128, lot #B1125) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. Next, the membranes were incubated for 1 h at room temperature with a horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA) after rinsing with PBS to remove the unbound antibodies. The secondary antibody was visualized using an enhanced CL procedure (Thermo Scientific, Rockford, IL, USA).
Measurement of Sp1 activity
Nuclear-extracted proteins were prepared from renal tissues using a commercial kit (BioVision, Milpitas, CA, USA), as described above, following the manufacturer’s instructions and previously described (Lu et al., 2022). Nuclear proteins (10 µg) were used to determine Sp1 activity using a high-throughput commercial kit (Abcam, Cambridge, UK). A specific double-stranded DNA sequence containing the Sp1 consensus binding site (5′-GGGGCGGGG-3′) was immobilized onto a 96-well plate. The active Sp1 in the nuclear extract was labeled using an oligonucleotide that specifically binds to Sp1. The epitope of Sp1 is recognized by a primary antibody. Hence, Sp1 can only be detected when it is in its active form or bound to its target DNA. Next, a secondary antibody conjugated to horseradish peroxidase provided the colorimetric readout at optical density 450 nm.
Localization of renal H2S-producing enzymes
Indirect immunofluorescent staining was used to examine the renal localization of CBS, CSE, or 3-MST, as described previously (Lin et al., 2015; Tseng et al., 2020). The postfixed kidneys were stored in 10% sucrose solution containing 4% paraformaldehyde at 4°C, embedded in an O.C.T. compound (Tissue-Tek, Sakura Finetek, Torrance, CA, USA), and frozen at −20°C. The tissues were then cut into 5 μm sections on a cryostat (Microm, Heidelberg, Germany), which were thaw-mounted on coated slides. After rehydration and washing with 0.01 M PBS (pH 7.4), the tissue sections were processed for indirect immunofluorescence using a tyramide signal amplification kit (PerkinElmer, Waltham, MA, USA). After blocking with 5% skimmed milk prepared in PBS for 1 h, the sections were incubated overnight at 4°C with an antibody against CBS, CSE, or 3-MST. The sections were then incubated for 1 h at room temperature with the corresponding fluorescein-conjugated secondary antibody (for tissue slides) and examined on an inverted fluorescent microscope, as described above. Nuclei were counterstained using 0.5 µg/mL of 4’,6-diamidino-2-phenylindole. The specificity of each antibody was tested using preincubation, with the specific blocking peptide provided by Santa Cruz Biotechnology (150 μg/mL), before performing the test. The mean fluorescence intensity per tissue section was quantified using ImageJ v1.54 software (National Institutes of Health, Bethesda, MD, USA) and the positive area was expressed as percentage.
Detection of renal mRNA expression of H2S-producing enzymes by means of real-time quantitative polymerase chain reaction
The RNA from renal tissues was extracted using a commercial RNA extraction kit (RareRNA, Bio-East Technology, Taipei, Taiwan) following a previously established protocol (Tseng et al., 2020; Lu et al., 2020). Starting with 5 µg of total RNA, we synthesized cDNA at 42°C for 45 min using 5 µg of oligo(dT)15 primer (Life Technologies, Carlsbad, CA, USA) and 200 units of reverse transcriptase (Moloney murine leukemia virus; Promega, Madison, WI, USA). A real-time quantitative polymerase chain reaction (RT-qPCR) was performed on an ABI StepOne™ instrument (Applied Biosystems, Foster City, CA, USA) using standard protocols. The cycle threshold (Ct) values of the target genes were adjusted by subtracting the average Ct values of glyceraldehyde-3-phosphate dehydrogenase, providing a reflection of the target gene mRNA levels. Changes in CBS, CSE, and 3-MST gene expression were calculated as 2−ΔCt, and fold changes relative to the control group are presented. The primer sequences used for RT-qPCR are listed in Table 3 as previously described (Wu et al., 2010; Kuksis et al., 2014; Liu et al., 2023; Tseng et al., 2020; Lu et al., 2020).
List of Primer Sequences Used for Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
3-MST, 3-mercaptopyruvate sulfurtransferase; CBS, cystathionine β-synthase; CSE, cystathionine γ-lyase; GADPH, glyceraldehyde 3-phosphate dehydrogenase.
Assessment of oxidative stress by means of GSH/glutathione disulfide and lipid peroxidation
The cellular GSH and oxidized glutathione, i.e., GSSG, contents in the renal tissues were analyzed using a commercial kit (OxisResearch, Portland, OR, USA), and the GSH redox ratio was calculated using the following equation, as indicated by the manufacturer’s instructions: redox ratio (%) = (GSH − 2GSSG)/GSSG × 100. The tissue pellet was lysed by means of treatment with the GSH scavenger supplied in the kit. Tissue lysate equivalent to 100 µg of total protein was added to the assay kit. The GSH and GSSG contents were measured with an ELISA reader at 412 nm based on their individual standard curves. The oxidative stress caused by Nx was further evaluated by measuring the urinary excretion of the lipid peroxidation metabolite malondialdehyde (MDA) using a commercial kit (MDA-586, OxisResearch, Portland, OR, USA), as previously described (Tseng et al., 2021).
Statistics
An electronic laboratory notebook was not used. Continuous variables are expressed as the mean ± SD. Group differences were assessed using unpaired t-tests or one-way analysis of variance. Data were analyzed with Prism 3.0 for Windows (GraphPad Software Inc., San Diego, CA, USA), and statistical significance was set at p < 0.05.
Footnotes
Authors’ Contributions
C.-L.L.: Writing—review and editing, writing—original draft, methodology, investigation, validation, formal analysis, data curation, conceptualization, funding acquisition. Y.-S.T.: Validation, methodology, investigation, data curation, conceptualization, funding acquisition. W.-B.W.: Writing—review and editing, validation, methodology, investigation, conceptualization. C.-H.L.: Methodology, investigation, conceptualization, funding acquisition. M.-C.M.: Writing—review and editing, writing—original draft, methodology, investigation, supervision, resources, validation, formal analysis, data curation, conceptualization, funding acquisition.
Data Availability
The data are listed in Supplementary Data and are available upon reasonable request.
Author Disclosure Statement
All the authors declare no competing interests.
Funding Information
This work was supported by the Ministry of Science and Technology, Taiwan (grant numbers 111–2314-B-030–005-MY3 and 110–2314-B-030–005-MY3); the Far Eastern Memorial Hospital (grant number 104-FEMH-FJU-04); and the Fu Jen Catholic University Hospital (grant numbers PL202008007V and PL-202108008V).
Supplementary Material
Supplementary Data
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.