
Abstract
Aims:
Cathodal transcranial direct current stimulation (C-tDCS), a noninvasive physical therapy, has potential neuroprotective effects in acute ischemic stroke. However, the rational timing of its application and the underlying mechanisms remain inadequately understood. This study aims to investigate its neuroprotective effects and the involved mechanisms.
Results:
Our in vivo results indicated that C-tDCS applied during the reperfusion phase but not during the ischemic phase significantly improved neurological outcomes, reduced infarct volume, and mitigated histopathological damage in middle cerebral artery occlusion/reperfusion rats. C-tDCS during the reperfusion phase suppressed ferroptosis, activated nuclear factor erythroid 2-related factor 2 (Nrf2), and inhibited mitophagy. In vitro, the ferroptosis inducer RSL3 negated the protective effects of cathodal direct current stimulation on HT22 neuronal cells subjected to oxygen-glucose deprivation/reoxygenation injury. Furthermore, the Nrf2 inhibitor ML385 and the mitophagy activator FCCP reversed the inhibitory effects of C-tDCS on ferroptosis, with FCCP also affecting Nrf2 activation by C-tDCS.
Innovation and Conclusions:
These results demonstrate that C-tDCS during reperfusion attenuates cerebral ischemia-reperfusion injury by coordinating mitophagy inhibition and Nrf2 activation to counteract ferroptosis, which provides new evidence for its potential translational clinical applications. Antioxid. Redox Signal. 43, 693–708.
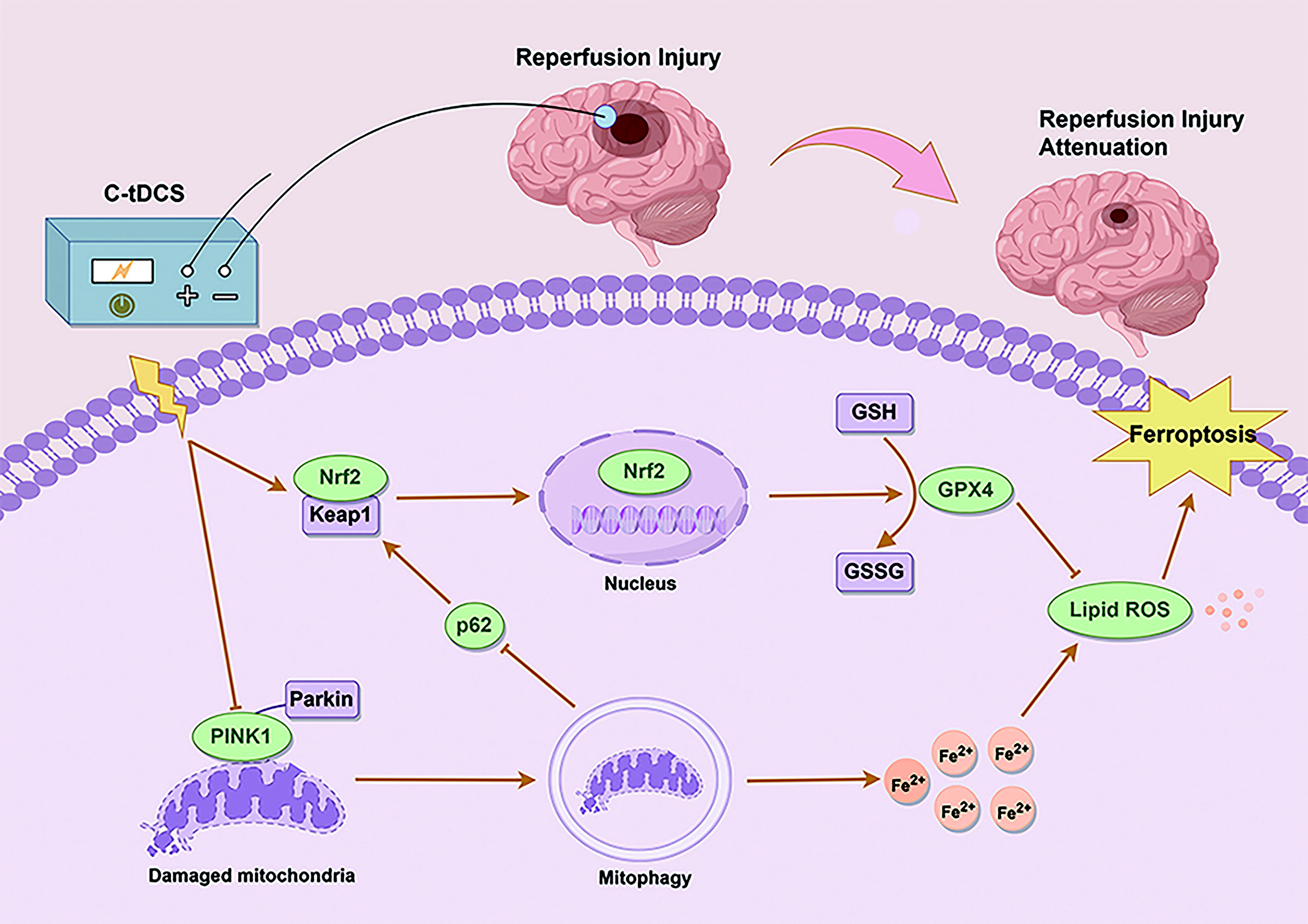
Introduction
Ischemic stroke is a common cause of death and disability worldwide, with its prevalence growing rapidly (Feigin et al., 2023). Revascularization therapies, represented by thrombolysis and thrombectomy, have emerged as the mainstay of ischemic stroke treatment, given their capacity to salvage ischemic penumbra effectively (Powers et al., 2019). However, along with the restoration of blood supply, the damaged brain tissue will suffer reperfusion injury (Wechsler et al., 2023). To date, most neuroprotective interventions have not successfully translated into clinical practice (Chamorro et al., 2021). Therefore, the search for new neuroprotective approaches assumes a pivotal role in advancing the treatment of stroke.
Transcranial direct current stimulation (tDCS), a noninvasive neuromodulation approach, can impact the physiological functionalities of neural and vascular tissue within the brain (Stagg et al., 2018). By delivering a weak, constant direct current, tDCS offers the benefits of ease of use and the absence of chemical drug-related side effects. Recently, several animal studies have indicated that early application of cathodal tDCS (C-tDCS) may have a positive impact on attenuating ischemia-related neurological injury (Cheng et al., 2021; Kaviannejad et al., 2022). Notably, discrepancies in methodologies and parameters across these studies obscure the definitive efficacy and mechanisms of tDCS in the acute stroke phase. Furthermore, a pilot clinical trial has demonstrated that early application of C-tDCS for acute ischemic stroke (AIS) is safe and practicable. However, the main outcomes did not show significant differences between groups (Pruvost-Robieux et al., 2021). Therefore, how to rationalize the use of C-tDCS to attenuate cerebral ischemia-reperfusion injury (CIRI) remains a challenge. Further preclinical investigations are required to elucidate the neuroprotective effects and potential mechanisms of C-tDCS in AIS.
Innovation
Currently, effective neuroprotective strategies to mitigate reperfusion injury following revascularization in AIS remain lacking in clinical practice. This study identifies the reperfusion period, rather than the ischemic phase, as the optimal window for C-tDCS to attenuate CIRI. Notably, we present evidence for the first time that C-tDCS exerts its neuroprotective effects through coordinating mitophagy inhibition and Nrf2 activation against ferroptosis (Fig. 1). These findings suggest that C-tDCS holds promise as a neuroprotective intervention.
Ferroptosis is an iron-dependent mode of cell death, characterized by the generation of reactive oxygen species (ROS) and lipid peroxidation (Stockwell et al., 2017). Previous studies have shown that ferroptosis is critically involved in the pathophysiology of CIRI and that targeted inhibition of ferroptosis can effectively attenuate CIRI (Guo et al., 2021; Tuo et al., 2022). Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor in the endogenous antioxidant response of cells, and its activation can attenuate CIRI by inhibiting ferroptosis (Wang et al., 2023). Nevertheless, it remains unknown whether the Nrf2-ferroptosis pathway contributes to the neuroprotective effects of C-tDCS.
Mitochondria are the energy centers of the cells and major targets of reperfusion injury. In response to excessive ROS, cells activate mitophagy, a mitochondrial quality control mechanism primarily mediated by the PTEN-induced putative kinase 1 (PINK1)/Parkin pathway, to remove dysfunctional mitochondria and maintain mitochondrial homeostasis (Zhong et al., 2022). While moderate mitophagy offers neuroprotection, prolonged ischemia can induce excessive mitophagy, which in turn accelerates neuronal death following CIRI (Cai et al., 2019). It has been reported that inhibition of mitophagy can prevent ferroptosis induced by O-GlcNAc transferase small molecule inhibitor 1 (OSMI-1) and Ras-selective lethal 3 (RSL3) (Yu et al., 2022). However, the specific role of mitophagy in CIRI-induced ferroptosis remains unclear. Moreover, autophagy inhibition is linked to Nrf2 activation through the autophagy receptor p62 (Hu et al., 2022). As a form of selective autophagy, mitophagy may also modulate Nrf2 expression through p62, which potentially influences ferroptosis (Li et al., 2022c). Notably, whether C-tDCS can attenuate reperfusion injury by regulating mitophagy has not been reported. The role of mitophagy in the regulation of ferroptosis and Nrf2 within the context of C-tDCS attenuating CIRI needs to be elucidated.
In this study, we initially utilized the middle cerebral artery occlusion/reperfusion (MCAO/R) model to assess the protective effect of C-tDCS on CIRI when applied during either reperfusion or ischemia. Our findings indicate that C-tDCS, when applied during reperfusion but not ischemia, effectively mitigates CIRI. Since ischemia-reperfusion injury involves mitochondrial dysfunction, oxidative stress, and ferroptosis, we hypothesized that C-tDCS exerts neuroprotective effects through the regulation of mitophagy, Nrf2, and ferroptosis. To test this hypothesis, we utilized both in vivo MCAO/R and in vitro oxygen-glucose deprivation/reoxygenation (OGD/R) models to investigate the involvement of these pathways in C-tDCS-mediated neuroprotection.
Results
Timing-dependent neuroprotection of C-tDCS in MCAO/R rats
We first investigated the effects of tDCS with different polarities following reperfusion on neurological outcomes in MCAO/R male rats. Neuro-scores, infarct volume, and cerebral histopathology were measured 24 h after MCAO (Fig. 2A). Sham-operated rats showed no neurological deficits. Both Zea Longa and Petullo scores were significantly lower in the MCAO/R + C-tDCS group compared with the MCAO/R group, whereas no significant improvement was observed in the MCAO/R + anodal tDCS (A-tDCS) group (Fig. 2B and C). Subsequent 2,3,5-triphenyltetrazolium chloride (TTC) staining showed no infarct presence in the sham group, whereas the relative cerebral infarct volume was 46.33% ± 2.67% in the MCAO/R group. In contrast, C-tDCS effectively reduced the infarct volume to 22.52% ± 2.00%, whereas A-tDCS intervention had no significant effect, with an infarct volume of 44.82% ± 3.61% (Fig. 2D and E). A similar neuroprotective effect was observed in female MCAO/R rats, with postreperfusion C-tDCS leading to improved neuro-scores and decreased infarct volumes (Supplementary Fig. S1).
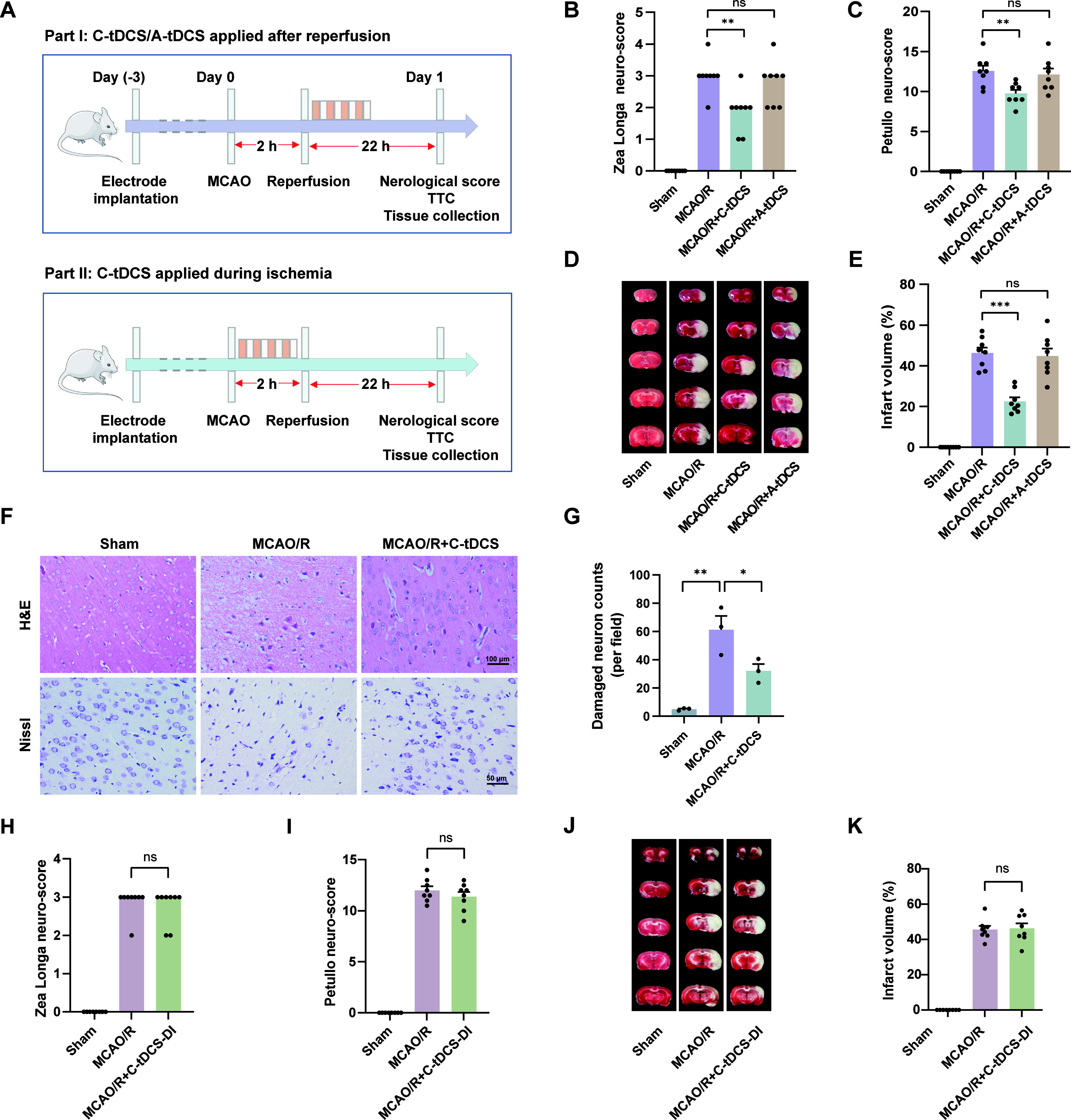
Furthermore, we conducted hematoxylin and eosin (H&E) staining and Nissl staining to evaluate the neuroprotective effects of C-tDCS from a histopathological perspective. H&E staining revealed that neuronal cells in the cortex of the sham group were well-organized and morphologically normal. In contrast, the MCAO/R group exhibited disordered neuronal arrangement, extensive neuronal loss, and vacuolar degeneration. Compared with the MCAO/R group, the MCAO/R + C-tDCS group showed a significant improvement in both the number and morphology of neuronal cells (Fig. 2F). Nissl staining supported these findings, showing that neurons in the MCAO/R group were misshapen with fewer Nissl bodies. These histopathological changes were significantly improved by C-tDCS (Fig. 2F and G). The above results suggest that administering C-tDCS after reperfusion effectively mitigates brain injury in MCAO/R rats.
Delaying the pathological progression of infarction during the ischemic phase could extend the therapeutic window for thrombolytic or thrombectomy treatments, potentially improving outcomes for stroke patients. To explore this possibility, we conducted an experiment where C-tDCS intervention was applied immediately after MCAO followed by reperfusion (Fig. 2A). However, our findings indicate that the MCAO/R + C-tDCS-DI group did not show a significant reduction in the Zea Longa Score and Petullo Score, as compared with the MCAO/R group (Fig. 2H and I). In addition, there was no noticeable difference in relative infarct volume between the two groups (Fig. 2J and K). These results suggest that C-tDCS intervention during the ischemic phase fails to exert neuroprotective effects.
C-tDCS downregulated ferroptosis and attenuated brain injury by activating Nrf2 in MCAO/R rats
Given the phase-specific neuroprotection of C-tDCS (effective during reperfusion but not ischemia), we first examined ROS, a key pathological factor known to surge during reperfusion but remain relatively low during ischemia. The results showed that C-tDCS after reperfusion effectively reduced ROS levels in the peri-infarct tissues of MCAO/R rats (Fig. 3A and B). We next investigated the ferroptosis phenotype, characterized primarily by lipid peroxidation. Compared with the sham group, the MCAO/R group showed significantly decreased protein levels of glutathione peroxidase 4 (GPX4) and mRNA expression of Solute Carrier Family 7 Member 11 (SLC7A11), which are crucial in counteracting ferroptosis, and elevated mRNA expression of prostaglandin-endoperoxide synthase 2 (Fig. 3C–F; Supplementary Fig.S2). Furthermore, the MCAO/R group demonstrated higher malondialdehyde (MDA) and Fe2+ levels and lower glutathione (GSH) levels in the peri-infarct tissues (Fig. 3G–I). Notably, C-tDCS treatment dramatically reversed these changes induced by MCAO/R, suggesting that it may exert neuroprotective effects by counteracting ferroptosis (Fig. 3A–I).

Given that Nrf2 emerges as a central transcription factor in cellular antioxidant defense, we infer that it may potentially mediate the modulatory effects of C-tDCS on the ferroptosis pathway. The protein levels of nuclear Nrf2, total Nrf2, and Keap1 in rat peri-infarct tissues were measured by immunoblotting analysis. Both nuclear Nrf2 and total Nrf2 levels were increased in the MCAO/R group compared with those observed in the sham group, which indicated that CIRI triggers endogenous antioxidant defenses. Compared with the MCAO/R group, the C-tDCS+MCAO/R group reduced Keap1 levels and further increased the protein levels of nuclear Nrf2 and total Nrf2 (Fig. 3J–K). Immunofluorescence staining supported immunoblot results, showing that C-tDCS intervention increased nuclear localization of Nrf2 relative to the MCAO/R group (Fig. 3L). In addition, to further elucidate the critical role of Nrf2 in mediating C-tDCS’s neuroprotection, we inhibited Nrf2 signaling in rat brain tissues by using ML385, a specific inhibitor that prevents Nrf2 from binding to the antioxidant response element, thereby suppressing its transcriptional activity (Jeong et al., 2024). As shown in Figure 3M–P, there was a significant increase in neuro-scores and infarct volume in the MCAO/R + C-tDCS+ML385 group compared with the MCAO/R + C-tDCS+vehicle group. Collectively, these results indicate that Nrf2 inhibition compromises the neuroprotective benefits of C-tDCS in MCAO/R.
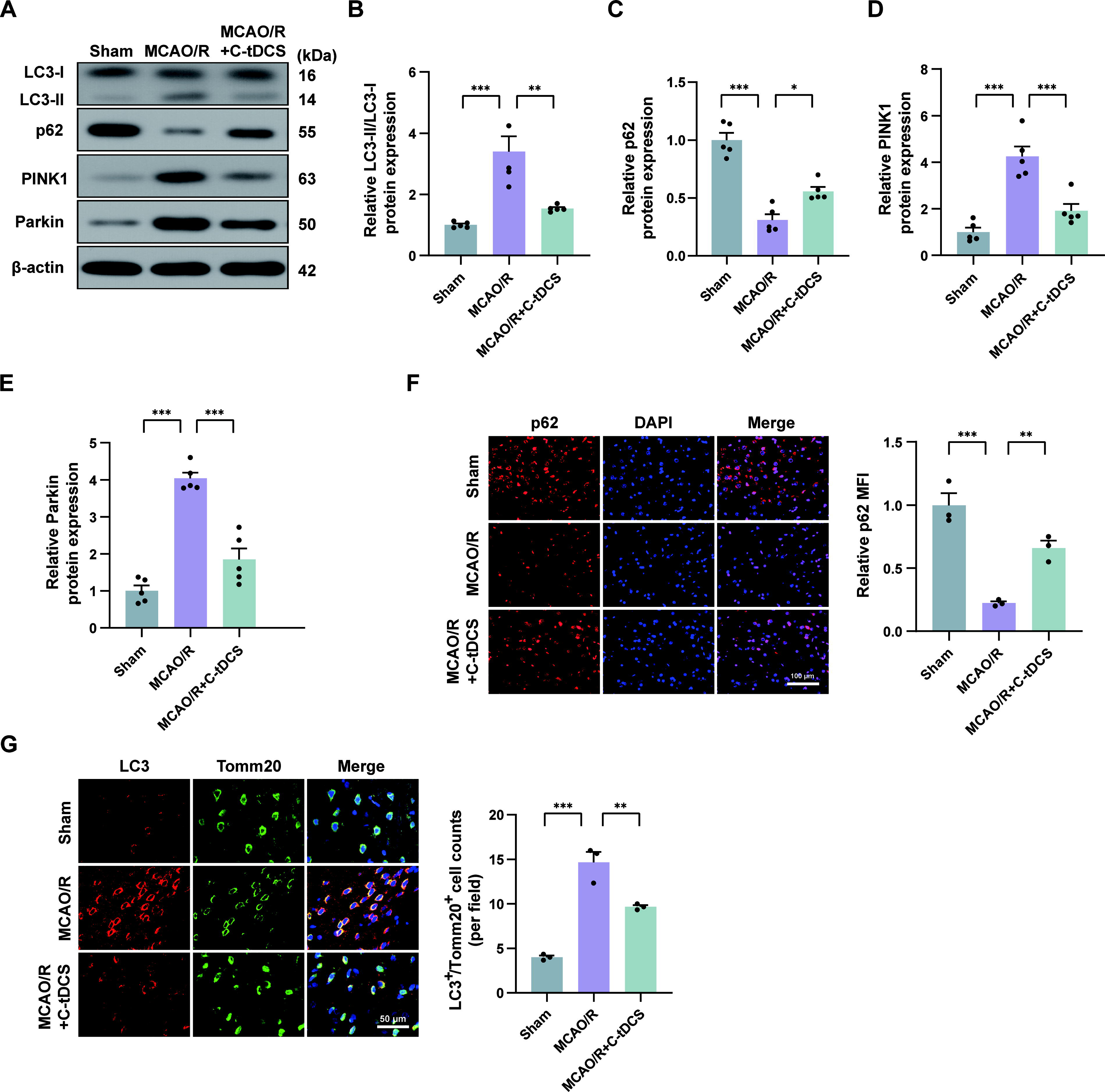
C-tDCS curbed excessive mitophagy in MCAO/R rats
Given that mitochondrial dysfunction is a key feature of CIRI, we next investigated the effect of C-tDCS on mitophagy in the MCAO/R model. We quantitatively analyzed the protein levels of autophagy markers (microtubule-associated protein light chain 3 [LC3]-II/LC3-I and p62) and key components in the classical mitophagy pathway (PINK1 and Parkin) through immunoblotting. The analysis showed a significant upregulation of LC3-II/LC3-I, PINK1, and Parkin in the MCAO/R group compared with the sham group, coupled with a reduction in p62 levels, indicating that MCAO/R may cause excessive mitophagy. Conversely, administration of C-tDCS attenuated the MCAO/R-induced changes in the protein levels of these markers (Fig. 4A–E). The p62 fluorescence intensity also paralleled the p62 immunoblot grayscale across all groups (Fig. 4F). Moreover, we further observed the colocalization of autophagosome marker LC3 with mitochondrial marker Tomm20, which represents mitophagosomes, using double immunofluorescence staining. The results showed that only sparse mitophagosomes were present in the sham group. However, the MCAO/R group exhibited a marked increase in mitophagosomes, which was significantly reduced after C-tDCS intervention (Fig. 4G).
Protective effects of C-DCS on OGD/R injury and its regulation of GPX4, Nrf2, and PINK1 protein levels
To further investigate the role and mechanism of C-tDCS in mitigating CIRI, we conducted in vitro experiments using conductive 6-well plates. We applied current intensities of 100, 300, 500, and 800 µA to assess the effects of cathodal direct current stimulation (C-DCS) on OGD/R-induced injury in HT22 cells. Experimental results from the cell counting kit-8 (CCK-8) assays showed a significant increase in cell viability across all C-DCS treatment groups compared with the OGD/R control group, and with the increase of current intensity, a trend of incremental enhancement in HT22 cell viability was observed, achieving optimal therapeutic effects at 500 µA. Nevertheless, when the current was increased to 800 µA, cell viability decreased relative to the 500 µA condition (Fig. 5A). Additionally, the lactate dehydrogenase (LDH) release assay, an indicator of cellular damage, corroborated the CCK-8 results (Fig. 5B). The above data suggest that C-DCS intervention at a current intensity of 500 µA offers optimal protective effects against OGD/R damage in HT22 cells.
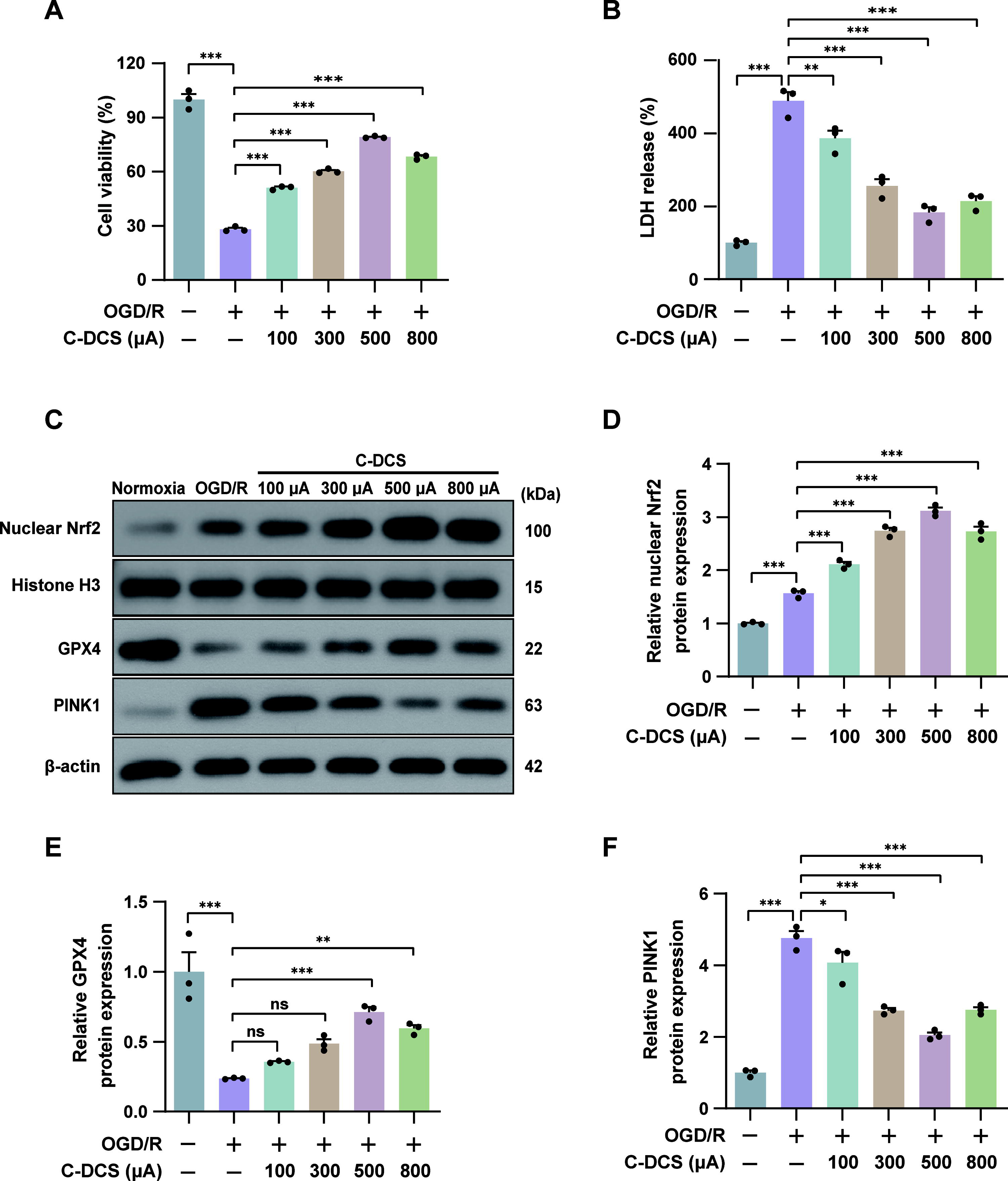
In our in vivo experiments, we identified the possible involvement of Nrf2, ferroptosis, and mitophagy in the attenuation of CIRI by C-tDCS. We subsequently observed the impact of C-DCS on OGD/R-induced changes in protein levels of nuclear Nrf2, GPX4, and PINK1 in the in vitro experiments. Immunoblotting revealed that nuclear Nrf2 and GPX4 levels in cells treated with C-DCS were elevated compared with the OGD/R group, increasing progressively from 100 to 500 µA before declining at 800 µA (Fig. 5C–E). Conversely, PINK1 levels in the C-DCS groups were lower than in the OGD/R group, decreasing from 100 to 500 µA, and then increasing at 800 µA (Fig. 5C and F). This trend paralleled the observed effects of C-DCS on cell viability and injury, further substantiating the potential role of Nrf2, ferroptosis, and mitophagy in the neuroprotective mechanism of C-DCS. Considering that the intervention effect of C-DCS at 500 µA was the most pronounced, we adopted a 500 µA current intensity for subsequent in vitro experiments to better elucidate the underlying mechanism.
C-DCS mitigated OGD/R injury in HT22 cells by inhibiting ferroptosis through Nrf2 activation
To ascertain that ferroptosis mediated the protective effect of C-DCS against OGD/R injury, RSL3, a GPX4 inhibitor that promotes lipid peroxidation, was introduced to induce ferroptosis (Stockwell BR, 2022). The morphological features of mitochondria associated with ferroptosis in each experimental group were observed via transmission electron microscopy. In the Normoxia group, mitochondrial morphology appeared normal. In contrast, the OGD/R group exhibited shrunken mitochondria and increased mitochondria membrane density. After C-DCS treatment, the number of mitochondria with ferroptosis-related features was significantly decreased compared with the OGD/R group. The addition of RSL3 markedly elevated the number of mitochondria associated with ferroptosis, confirming RSL3′s activation of ferroptosis (Fig. 6A). We then assessed cell viability and damage using CCK-8 assay and LDH assay, separately. The results showed that RSL3 prevented the recovery of cell viability, aggravated cell damage in the OGD/R model treated with C-DCS, and undermined the ameliorative effects of C-DCS (Fig. 6B and C).
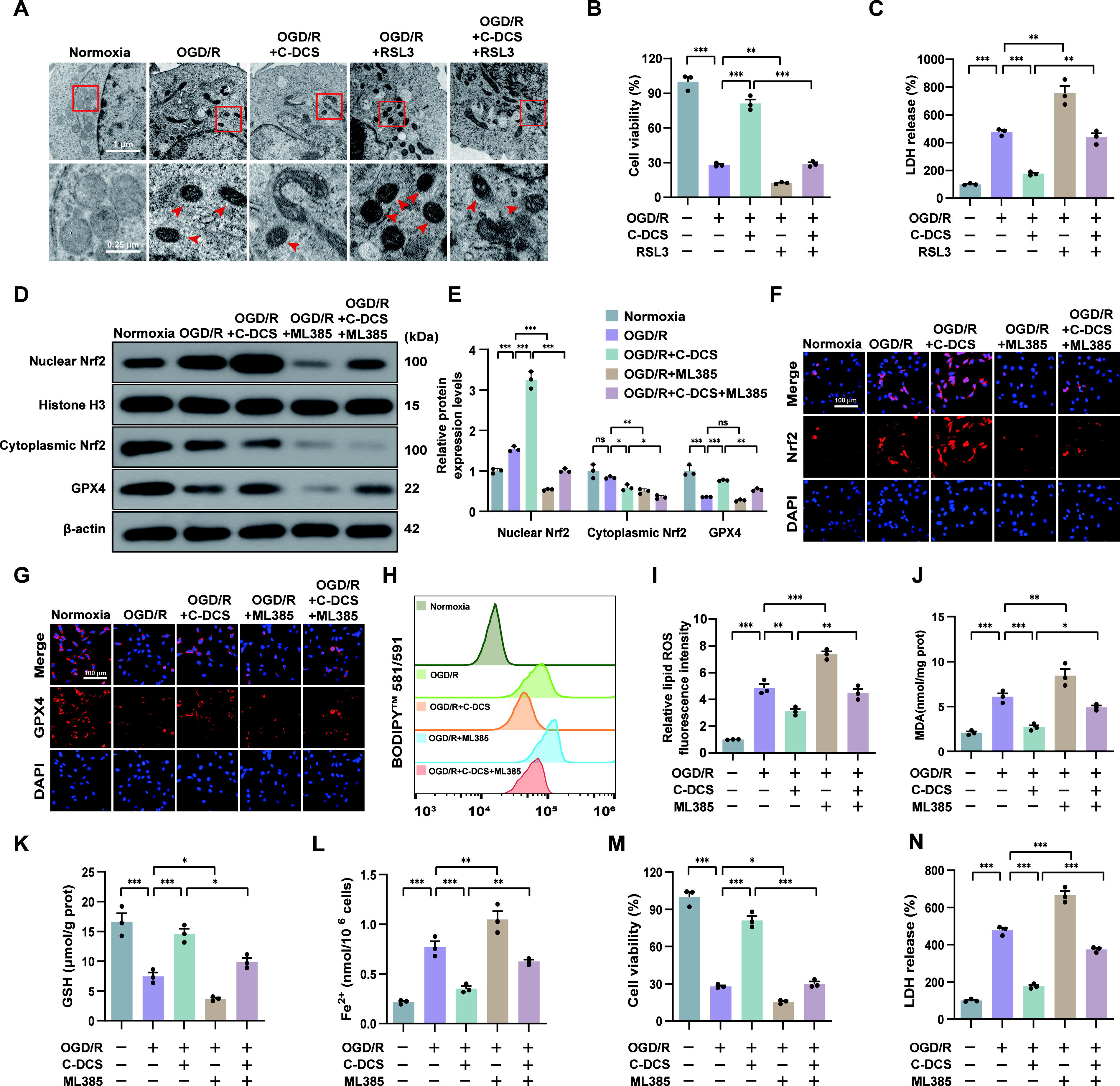
Nrf2 acts as an upstream regulator of ferroptosis. Next, we investigated whether the inhibitory effect of C-DCS on OGD/R-induced ferroptosis was linked to Nrf2 activation through in vitro experiments. Immunoblotting revealed that nuclear Nrf2 protein levels were increased and cytoplasmic Nrf2 levels tended to be attenuated in the OGD/R group compared with the Normoxia group. C-DCS treatment further elevated Nrf2 levels and reduced its cytoplasmic presence, suggesting that C-DCS facilitated nuclear translocation of Nrf2. However, the Nrf2-specific inhibitor ML385 significantly decreased the levels of nuclear Nrf2 and cytoplasmic Nrf2, countering C-DCS’s effects (Fig. 6D and E). Immunofluorescence results also showed that ML385 interfered with Nrf2’s nuclear entry facilitated by C-DCS (Fig. 6F).
In addition, GPX4, a crucial protein in ferroptosis potentially regulated by Nrf2, exhibited expression patterns consistent with Nrf2 trends across experimental groups, as evidenced by immunoblotting and immunofluorescence (Fig. 6D, E, and G). In parallel, other ferroptosis makers were assessed. As shown in Figure 6H–L, the levels of lipid ROS, MDA, and Fe2+ were higher in the OGD/R group than in the Normoxia group, whereas GSH levels were lower. In contrast, C-DCS treatment mitigated these OGD/R-induced changes. Notably, ML385 partially reversed the modulatory effects of C-DCS on these markers. The beneficial effects of C-DCS on cell viability and damage were similarly abolished (Fig. 6M and N). Collectively, these results indicate that C-DCS inhibits ferroptosis through Nrf2 activation, thereby protecting cells against OGD/R injury.
Activation of mitophagy reversed the effects of C-DCS on ferroptosis and Nrf2 in OGD/R cells
Nrf2 inhibition only partly abolished the protective effect of C-DCS against ferroptosis, prompting us to investigate whether mitophagy is also involved in this process. First, we analyzed the levels of mitophagy-related proteins in various cell groups. Immunoblotting revealed an increase in LC3-II/LC3-I, PINK1, and Parkin protein levels, alongside a decrease in p62 protein levels under OGD/R conditions. In contrast, C-DCS treatment reversed these changes, indicating a decrease in mitophagy. These findings align with our in vivo results. In addition, the administration of mitochondrial uncoupler-FCCP (Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone) further activated OGD/R-induced mitophagy and eliminated the inhibitory effect of C-DCS on mitophagy (Fig. 7A and B). These immunoblotting findings were supported by MitoTracker and LysoTracker double labeling revealing alterations in mitophagolysosomes (Fig. 7C). Subsequently, we investigated if mitophagy mediates C-DCS’s effects on ferroptosis. The results showed that FCCP reversed the regulatory effects of C-DCS on cell viability, lipid ROS, MDA, GSH, Fe2+, and GPX4, suggesting that C-DCS downregulated ferroptosis by inhibiting mitophagy (Fig. 7D–M). Interestingly, FCCP also blocked C-DCS-induced Nrf2 activation and nuclear translocation, suggesting that mitophagy may be partially involved in the regulation of Nrf2 by C-DCS (Fig. 7K, L, and N).

Discussion
In this study, we observed that applying C-tDCS during the reperfusion phase attenuated neurological impairment, infarct volume, inflammation, and neuronal apoptosis in MCAO/R rats but was ineffective when applied during the antecedent ischemia. Mechanistically, the Nrf2-ferroptosis pathway and the mitophagy-ferroptosis pathway were found to mediate the protective effects. In addition, mitophagy may play a partial role in the regulation of the Nrf2 pathway by C-tDCS.
Several studies have reported the neuroprotective effects of C-tDCS applied early after reperfusion at various intensities, including 100, 250, and 400 µA (Kaviannejad et al., 2022; Kong et al., 2023; Peruzzotti-Jametti et al., 2013). Considering the potentially significant physiological effects of higher currents and ensuring safety, we selected 400 µA as our intervention intensity. In addition to the parameter settings of C-tDCS, the timing of the intervention is another important factor in its efficacy. Currently, the effective time window for C-tDCS intervention to mitigate reperfusion injury remains undetermined, posing a challenge in setting the intervention time point for future clinical trials. Our results showed that the application of C-tDCS at the onset of reperfusion exerted neuroprotection, corroborating previous findings (Peruzzotti-Jametti et al., 2013). In addition, we concentrated on whether C-tDCS application solely during the ischemic phase could attenuate CIRI as inhibiting the infarct progression during this phase can preserve more salvageable tissue, potentially enhancing recanalization benefits and extending the therapeutic window. However, the absence of a protective effect indicates that the ischemic phase may not be a reasonable time to apply C-tDCS. A previous pilot clinical trial initiated C-tDCS during thrombolysis preparation or execution and during endovascular therapy, which spanned both the ischemic and reperfusion phases, with the result that C-tDCS application did not contribute to the reduction of infarct volume at 24 h after AIS onset (Pruvost-Robieux et al., 2021). In contrast, our study found that C-tDCS intervention during the reperfusion phase significantly reduced infarct volume, whereas intervention during the ischemic phase did not, suggesting that initiating C-tDCS postrevascularization might be more beneficial. Identifying this effective time window for C-tDCS could enhance its therapeutic efficacy and provide new directions for future clinical studies.
Ferroptosis, as a novel mode of cell death, is significantly implicated in the pathogenesis of CIRI. Prior studies suggest the potential role of ferroptosis in CIRI because blocking the cascade of ferroptosis can effectively attenuate CIRI (Liu et al., 2023c). Our results supported the role of ferroptosis in CIRI. Indeed, ferroptosis is primarily triggered by the excessive production of ROS and ferrous iron, both critical factors in the pathogenesis of CIRI (Chen et al., 2021). During the ischemic phase, the deprivation of oxygen damages mitochondria and other organelles, leading to a buildup of metabolic waste. Upon reperfusion, the compromised mitochondria attempt to resume oxidative phosphorylation, inadvertently increasing ROS generation, which exacerbates cellular damage and facilitates free iron release. In turn, the increasing free iron boosts ROS generation through the Fenton reaction, leading to the lipid peroxidation of neuronal membranes and subsequent cell death (Yan et al., 2020). Considering the above pathological characteristics of ischemia-reperfusion injury, it seems reasonable that ferroptosis predominantly occurs during the reperfusion phase but not the ischemia phase. This hypothesis is also supported by literature indicating that ferroptosis inhibitors liproxstatin-1 and ferrostatin-1 attenuated neurological injury in MCAO/R mice but not in permanent MCAO models (Tuo et al., 2022). For CIRI, the effective timing of C-tDCS intervention aligns with the occurrence of ferroptosis, suggesting that modulating ferroptosis may be a key mechanism by which C-tDCS mitigates CIRI. This hypothesis was corroborated by the results of our in vitro and in vivo experiments. Indeed, ferroptosis is an important target for many drugs to exert neuroprotective effects, highlighting the importance of targeting ferroptosis to mitigate CIRI (Li et al., 2022b; Liu et al., 2023a). Our study highlights the potential of noninvasive neuromodulatory approaches in modulating ferroptosis, providing new insights into the neuroprotective mechanisms facilitated by C-tDCS.
Nrf2 is a pivotal transcription factor that resists intracellular oxidative stress injury, with its activation significantly reducing CIRI (Sun et al., 2023). Several ferroptosis-related molecules including GPX4, SLC7A11, and ferritin are regulated by Nrf2, positioning its activation as a crucial strategy against ferroptosis (Dodson et al., 2019). Currently, the relationship between C-tDCS and Nrf2 remains unclear. Huang et al. (2023) conducted experiments to program mammalian gene expression using direct current and discovered that a 10-s exposure of HEK293 cells to a 4.5 V direct current voltage made ectopically expressed Nrf2 hypersensitive to ROS. This implies that electrical stimulation may amplify cellular antioxidant defenses. Corroborating the findings of Huang et al., our reperfusion model demonstrated that C-tDCS intervention significantly activated Nrf2 and promoted nuclear translocation of Nrf2. More significantly, inhibiting Nrf2 diminished the antiferroptotic effects of C-tDCS and exacerbated cell injury, confirming Nrf2’s pivotal role in C-tDCS’s neuroprotective mechanism. Notably, inhibition of Nrf2 did not completely reverse C-tDCS’s impacts on ferroptosis, implying that additional mechanisms also contribute to C-tDCS’s regulation of ferroptosis.
Mitochondria are the primary site for both ROS generation and iron metabolism; therefore, mitophagy may affect ferroptosis. Yu et al. (2022) reported that inhibition of O-GlcNAcylation induced mitophagy, consequently increasing the release of stored iron and amplifying ferroptosis. In Li et al.’s study, β-amyloid disrupted the blood–brain barrier by promoting pericyte mitophagy-dependent ferroptosis (Li et al., 2022a). These studies indicate that mitophagy activation facilitates ferroptosis, primarily by increasing cellular-free iron levels. Contrary to this prevailing view, a recent study showed that activation of mitophagy reduced ROS release and inhibited cisplatin-induced ferroptosis (Lin et al., 2023). Therefore, for the regulation of ferroptosis, the role of mitophagy may be bidirectional. One plausible explanation is that under mild stress conditions, limited mitophagy sequesters iron into mitophagosomes, decreasing free intracellular iron levels. Moreover, moderate mitophagy reduces ROS production by improving mitochondrial function, which may attenuate ferroptosis. Nevertheless, excessive mitophagy may increase the available iron source and exacerbate ferroptosis (Li et al., 2023). Our study focused on the effects of mitophagy on ferroptosis under ischemia-reperfusion conditions and the impacts of C-tDCS on them, which were previously unexplored in the literature. We found that mitophagy exhibited a positive regulatory effect on ferroptosis, whereas C-tDCS suppressed both processes. Our results are reasonable as mitophagy, induced by ischemia exceeding 2 h followed by reperfusion, was detrimental (Zhu et al., 2024). Activating mitophagy partially reversed the inhibition of ferroptosis by C-tDCS, indicating that mitophagy is involved in the regulation of ferroptosis by C-tDCS. This finding is supported by the similar expression patterns of PINK1 and GPX4 in response to changes in current intensity.
In this study, we demonstrated that inhibiting mitophagy by C-tDCS activated Nrf2, providing new evidence for the crosstalk between mitophagy and Nrf2. The interplay between autophagy and Nrf2 may be primarily mediated by p62 (Hu et al., 2022). Inhibition of autophagy can upregulate p62, which then activates Nrf2 by sequestering Keap1 within phagosomes, thereby disrupting Nrf2’s ubiquitination and degradation (Liu et al., 2023b). This activation of Nrf2 in turn promotes further expression of p62, forming a positive feedback loop (Liao et al., 2019). As a specialized form of autophagy, mitophagy also can influence the expression of p62, suggesting that it might also regulate Nrf2 activity. In addition, activating the p62-Keap1-Nrf2 pathway can mitigate ischemia-reperfusion injury by inhibiting ferroptosis (Wang et al., 2023). Drawing on these findings, we hypothesize that under the intervention of C-tDCS, mitophagy inhibition not only mitigates ferroptosis by affecting iron metabolism and ROS release but also counteracts ferroptosis through the activation of Nrf2, thereby protecting neurons against CIRI.
We acknowledge several limitations in our study. First, our study focuses solely on the acute phase of postischemic brain injury, whereas pathological changes following this phase continue to evolve over subsequent days, involving inflammation and secondary neuronal damage. Future studies should extend the observation window to later time points (e.g., 3, 7, and 14 days postreperfusion) to evaluate the long-term effects of C-tDCS, which would provide a more comprehensive understanding of its therapeutic potential in CIRI. Second, due to differences in the three-dimensional arrangement of cortical neurons, the neuroprotective effects of C-tDCS may vary between lissencephalic and gyrencephalic species, potentially limiting the translational applicability of our findings. Third, while the use of HT22 cells in our in vitro study provided valuable insights into the mechanisms of C-DCS-induced neuroprotection, we acknowledge that the absence of glial cells may limit its physiological relevance. Future studies will build on these findings by incorporating more complex cellular models to assess neuronal–glial interactions. Finally, while we demonstrated that C-tDCS inhibits ferroptosis by regulating mitophagy, the specific mechanisms through which C-tDCS affects mitophagy warrant further exploration.
In conclusion, this is the first study to find that applying C-tDCS during the reperfusion phase, rather than the ischemic phase, produces neuroprotection, which establishes an effective time window for using C-tDCS to attenuate CIRI. Furthermore, the neuroprotective mechanisms of C-tDCS involve counteracting ferroptosis through the coordinated inhibition of mitophagy and activation of Nrf2. These findings highlight the potential for translating C-tDCS into a viable clinical therapy.
Materials and Methods
Experimental animals and grouping
A total of 136 male Sprague–Dawley and 18 female rats (SPF grade, 8–9 weeks) were sourced from Beijing Huafukang Bioscience Co., Inc. (License number: SCXK [Beijing] 2019-0008) for MCAO/R or sham MCAO/R procedures. Seventeen male rats and two female rats were excluded due to subarachnoid hemorrhage, modeling failure, or death, leaving 119 male and 16 female rats in the final study. The rats were maintained at 19–23°C, 50%–70% humidity, and a 12-h light/dark cycle, with free access to food and water. All procedures were approved by the Laboratory Animal Welfare and Ethics Committee of China Medical University (IACUC No. CMU20231460) and complied with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
The in vivo experiments were conducted in three parts. In Part I, we investigated the effects of C-tDCS applied during reperfusion. A total of 56 male rats were randomly allocated to four groups: the sham group (n = 16), receiving sham MCAO/R and sham C-tDCS; the MCAO/R group (n = 16), receiving sham C-tDCS postreperfusion; the MCAO/R + C-tDCS group (n = 16), receiving C-tDCS immediately postreperfusion; and the MCAO/R + A-tDCS group (n = 8), receiving anodal tDCS immediately postreperfusion. Additionally, 16 female rats were randomly allocated to either the MCAO/R-Female group or the MCAO/R + C-tDCS-Female group. In Part II, we examined the effects of C-tDCS during ischemia. A total of 24 rats were randomly divided into three groups (n = 8 per group): the sham group; the MCAO/R group, receiving sham C-tDCS after ischemia; and the MCAO/R + C-tDCS-DI group, receiving C-tDCS immediately after the onset of ischemia. Part III focused on the potential role of Nrf2 in mediating the neuroprotective effects of C-tDCS. A total of 30 rats were randomly assigned to five groups (n = 6 per group): the sham group, the MCAO/R group, the MCAO/R + C-tDCS group, the MCAO/R + C-tDCS+vehicle group, and the MCAO/R + C-tDCS+ML385 group. ML385 was administered at 50 pmol/5 µL and injected into the lateral ventricle 1 h before MCAO. The vehicle group received the same procedure, but the injected fluid lacked ML385. All animal experimental procedures were performed in a blinded manner. Randomization was conducted using assigned random numbers.
MCAO/R models
Animal models were established by experienced animal surgeons. The MCAO/R model was used to induce transient cerebral ischemia as previously described (Tuo et al., 2022). Briefly, rats were anesthetized with 2% sodium pentobarbital (40 mg/kg) administered intraperitoneally and given the analgesic carprofen (5 mg/kg) subcutaneously. A midline incision ∼2 cm in length was made on the neck to expose the right common carotid artery. A silicone-coated nylon monofilament (Beijing Cinontech Co. Ltd., 2438A5, China) was inserted ∼18 mm into the right common carotid artery and gently advanced to the origin of the right middle cerebral artery until slight resistance was felt, thereby blocking distal blood flow. After 2 h of ischemia, the monofilament was slowly withdrawn to restore blood supply. Body temperature was maintained at 37 ± 0.5°C using a heating pad throughout the procedure. Lidocaine gel was applied to the neck incision for local postoperative pain relief.
Lateral ventricle injection
Under 2% sodium pentobarbital anesthesia, a careful incision was made on the rat’s scalp to expose the skull. The rats were immobilized in a stereotaxic apparatus, and a position 1.5 mm to the right and 1 mm posterior to Bregma was designated as the injection site for the lateral ventricle. A small hole was created at the marked location using a cranial drill (RWD Life Science, Shenzhen, China). A Hamilton microliter syringe, controlled by a syringe pump (RWD Life Science, China), was employed to administer 50 pmol/5 µL of ML385 or 5 µL of vehicle to a depth of 3.7 mm at a rate of 0.5 µL/min. After injection, the needle remained in situ for an additional 5 min before being carefully withdrawn.
Neuro-scores
Two neuro-scores were performed 24 h after MCAO onset to assess neurological impairment in rats between groups. The Zea Longa scoring scale was as follows (Longa et al., 1989): score 0, no signs of neurological impairment; score 1, inability to fully extend the left forelimb; score 2, spinning to the left while walking; score 3, paralysis to the left while walking; and score 4, inability to walk and impaired consciousness. Furthermore, a more detailed Petullo score ranging from 0 to 20, with higher scores indicating more severe impairment, was performed as previously described (Zhang et al., 2023).
Cerebral infarction volume analysis
The rats were euthanized at 24 h of MCAO onset. Brain tissue was rapidly transferred and frozen for 20 min. Coronal sections, each 2 mm thick (totaling five slices), were incubated with 2% TTC solution (Solarbio, G3005, Beijing, China) for 10 min in a light-protected water bath at 37°C. Infarcted and noninfarcted right hemisphere volumes were quantified using Image J software (NIH, USA). The relative infarct volume was calculated as (left hemisphere volume—noninfarcted right hemisphere volume)/left hemisphere volume × 100%.
Histological staining
Brain tissues were immersed in 4% paraformaldehyde for fixation, then embedded in paraffin, and sliced into 5 µm thick sections. For H&E staining, the sections were successively immersed in H&E solutions. For Nissl staining, the sections were stained with 0.5% cresyl violet solution for 10 min. Microscopic examination was conducted to observe histological changes in the peri-infarct tissues.
Immunofluorescence staining
Brain sections and cultured cells were fixed with 4% paraformaldehyde. Brain sections were fixed overnight at 4°C, whereas cultured cells were fixed at room temperature for 15 min. Fixed sections or cells were rinsed with PBS and permeabilized with 0.1% Triton X-100 (Beyotime, Shanghai, China) and then were incubated at 4°C overnight with primary antibodies against GPX4 (1:200, Abclonal, A1933, Wuhan, China), Nrf2 (1:200, Wanleibio, WLH3846, Shenyang, China), and p62 (1:200, Affinity, AF5384, Jiangsu, China). After washing, sections and cells were incubated with goat anti-rabbit IgG (1:200, Invitrogen, A27039) secondary antibody at room temperature for 1 h. Then, 4′,6-diamidino-2-phenylindole (DAPI, Aladdin, Shanghai, China) was added to stain the nuclei. In addition, brain sections were double-stained with LC3 (1:200, Wanleibio, WL01506) and Tomm20 (1:200, Proteintech, 66777-1-Ig) to visualize mitochondrial autophagosomes. Cultured cells were double-stained using LysoTracker Green (100 nM, MKbio, L7526, Shanghai, China) and MitoTracker Red (100 nM, MKbio, MX4307, China) to assess mitophagolysosomes. These steps were performed according to the manufacturer’s instructions. Fluorescent signals were observed using a fluorescence microscope.
ROS detection
ROS levels in rat brain tissues were assessed by 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) staining (Beyotime, S0033, China). Briefly, fresh-frozen brain sections were incubated with 10 µM DCFH-DA at 37°C for 30 min in the dark, followed by three washes with PBS. Fluorescent signals were visualized using a fluorescence microscope.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from rat brain peri-infarct tissues using TRIzol reagent (Invitrogen, Massachusetts, USA), and RNA concentration was measured with a spectrophotometer (Thermo, NanoDrop 2000, USA). RNA was reverse transcribed into cDNA using HiScript III RT SuperMix for qPCR (+gDNA wiper; Vazyme Biotech, China). For real-time quantitative polymerase chain reaction (qRT-PCR), we utilized SYBR Green Master Mix (Solarbio, China). The 2−ΔΔCt method was employed to analyze gene expression, with β-actin serving as the internal reference gene for normalization. The promoter sequences are provided in Supplementary Table S1.
Cell culture
The HT22 mouse hippocampal neuronal cell line was obtained from Wanlei Biotechnology (Shenyang, China). The cells were cultured in Dulbecco’s modified eagle medium (DMEM, high glucose, Servicebio), supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Cultures were maintained in a humidified incubator at 37°C with 5% CO2 to ensure optimal growth conditions.
OGD/R model and cell grouping
OGD/R experiments were conducted following previously established protocols (Tuo et al., 2022). Briefly, HT22 cells were seeded in pretreated 6-well plates and incubated overnight at 37°C. The following day, the old medium was discarded and replaced with glucose-free DMEM for 2 h incubation in a hypoxic environment (37°C, 94% N2, 5% CO2, 1% O2) to simulate ischemic conditions. Afterward, the medium was replaced with DMEM containing 25 mM glucose, and the cells were reoxygenated in a normoxic incubator (37°C, 95% air, 5% CO2) for 22 h.
The in vitro experiments were divided into two main parts. The first part evaluated the protective effect of C-DCS on OGD/R cells. The groups were as follows: Normoxia, OGD/R, OGD/R + C-DCS 100 µA, OGD/R + C-DCS 300 µA, OGD/R + C-DCS 500 µA, and OGD/R + C-DCS 800 µA. C-DCS was applied in constant-current mode immediately after reoxygenation, with 15 min on followed by 15 min off, repeated for four cycles.
Based on the results from the first part, the second part of the mechanism experiment utilized 500 µA C-DCS. The groups included: Normoxia, OGD/R, OGD/R + C-DCS, OGD/R + RSL3, OGD/R + C-DCS + RSL3, OGD/R + ML385, OGD/R + C-DCS + ML385, OGD/R + FCCP, and OGD/R + C-DCS + FCCP. RSL3 (Selleck, Texas, USA; ferroptosis inducer), ML385 (Selleck, USA; Nrf2 inhibitor), and FCCP (Sigma, Missouri, USA; mitophagy activator) were added at 5 µM 1 h before OGD/R and maintained throughout the experiment.
C-tDCS and C-DCS interventions
C-tDCS was applied in MCAO/R rats (Supplementary Fig. S3). Three days prior to the MCAO/R operation, an electrode fixation device with a bottom diameter of 2.1 mm was implanted on the skull surface of anesthetized rats using nontoxic glass ionomer cement (New Century Dental, China). The center of the device was located 2.5 mm to the right and 0.5 mm posterior to the Bregma. C-tDCS intervention was performed using a programmed constant-current DC power supply (Keithley, 2200-72-1, Ohio, USA). A metal electrode on the head was connected to the cathode, and a 3 cm diameter round silicone electrode placed over the ventral thorax was connected to the anode. Before starting C-tDCS, the electrode fixation device was filled with saline. The current intensity was set to 400 µA, with a stimulation mode of 15 min on and 15 min off, repeated for four cycles.
C-DCS was applied in OGD/R cells (Supplementary Fig. S4). Based on previous literature (Sun et al., 2022), we improved and fabricated conductive 6-well plates. Two circular through holes were drilled between adjacent culture wells of the 6-well plate (Corning, USA), and the holes were filled with 2% PBS agar. Prior to electrical stimulation, the 6-well plates were coated with 10 µg/mL poly-L-lysine. To minimize the toxic effects of metal ions on the cells, the electrodes were made from highly inert platinum discs with a 7 mm diameter. To simulate cathodal stimulation, cells were inoculated in the first well of each 6-well plate and connected to the power cathode, whereas the third well was connected to the anode. A known value of 2 mL of cell culture medium was added to the wells through holes to complete the circuit. OGD/R cells received 15 min of stimulation followed by 15 min of interruption at the start of reoxygenation, for a total of four cycles.
Cell viability and damage
Cell viability was assessed in each group using the CCK-8 (Wanleibio, WLA074, China). Briefly, 10 µL of CCK-8 reagent was dispensed per 100 µL of medium and then incubated for 1 h at 37°C in an incubator with 5% CO2, shielded from light. The absorbance was then measured at 450 nm with a microplate reader (BioTek, USA).
To assess cell damage, LDH activities were assayed in cell supernatants collected from each group. LDH reaction reagent (Wanleibio, WLA072, China) was added to each well in accordance with the manufacturer’s protocol. After incubation for 1 h, the absorbance values were read at 450 nm by a microplate reader.
Lipid ROS detection
Lipid ROS levels in HT22 cells were detected using the BODIPY™ 581/591 C11 probe. The cells were incubated with BODIPY™ 581/591 C11 (MKbio, MX5211, China) at 37°C for 1 h, ensuring thorough mixing. The fluorescence signals were analyzed using a NovoCyte flow cytometer (Agilent, USA).
Fe2+, MDA, and GSH content assays
The contents of Fe2+, MDA, and GSH in rat peri-infarct tissues and HT22 cells were determined using the Iron Assay Kit (Abcam, Ab83366, UK), MDA Assay Kit (Wanleibio, WLA048, China), and GSH Assay Kit (Wanleibio, WLA105, China), respectively, following the manufacturer’s instructions.
Immunoblotting
Protein was extracted from rat brain peri-infarct tissues or HT22 cells using a lysis buffer supplemented with phenylmethylsulfonyl fluoride (PMSF). Protein concentration was quantified with a BCA Protein Assay Kit. The extracted proteins were separated by SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane via electroblotting. The membrane was blocked with 5% nonfat milk in Tris-buffered saline with Tween 20 (TBS-T) buffer for 1 h at room temperature to prevent nonspecific binding. After blocking, the membrane was incubated with the primary antibody overnight at 4°C and then incubated with the secondary antibody (1:5000, Wanleibio, China) for 1 h at room temperature. Visualization of protein bands was performed using an enhanced chemiluminescence assay. The following primary antibodies were used: GPX4 (1:1000, Abclonal, A1933), Keap1 (1:1000, Wanleibio, WL03285), Nrf2 (1:1000, Wanleibio, WL02135), LC3α/β (1:1000, Wanleibio, WL01506), p62 (1:1000, Wanleibio, WL02385), PINK1 (1:1000, Wanleibio, WL04963), Parkin (1:1000, Wanleibio, WL02512), β-actin (1:1000, Wanleibio, WL01372), and Histone H3 (1:1000, Wanleibio, WL0984a).
Transmission electron microscopy
HT22 cells were fixed with electron microscopy fixative (Servicebio, G1102, China) for 2 h at room temperature. After centrifugation, the cells were pre-embedded in 1% agarose and then fixed in 1% osmium tetroxide in the dark for 2 h. Subsequently, the cells were dehydrated using a graded series of ethanol solutions. Following infiltration and embedding, the samples were polymerized at 60°C for 48 h. Ultrathin sections (70 nm) were prepared using an ultramicrotome, stained with 2% uranyl acetate and lead citrate, and observed under a transmission electron microscope (Hitachi, Japan).
Statistics
Statistical analysis was conducted using GraphPad Prism 9.0 (GraphPad Software, USA). The Shapiro–Wilk test was used to assess the distribution of variables. Data with a normal distribution were expressed as mean ± standard error. Student’s t-test was employed for comparisons between two groups, whereas one-way analysis of variance with Bonferroni’s post hoc test was used for multiple group comparisons. For nonnormally distributed data, the Mann–Whitney U test was applied. All statistical tests were two-tailed, with significance set at p < 0.05.
Authors’ Contributions
X.D.L., Y.X.N., Y.F.P., J.Y.N., J.L., Y.N.Z., Q.K.Z., and T.C.X. conducted the experiments. Z.A.Z. and X.W.H. analyzed and interpreted the results. X.D.L. drafted the article and created the graphs. H.S.C. conceived the experiments, revised the article, and provided financial support.
Footnotes
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
Data Availability
Data will be made available on request.