
Abstract
Significance:
Endothelial cells (ECs) are specialized cells lining the interior surface of blood vessels, playing a crucial role in vascular biology. They exhibit remarkable versatility, adapting to various tissue requirements. Their ability to respond to physiological and pathological stimuli ensures proper tissue function and homeostasis.
Recent Advances:
Hypoxia is when the oxygen level in a given organ, tissue, or cell type drops below the physiological level and is insufficient to maintain adequate homeostasis. ECs respond to hypoxia by activating various mechanisms. Hypoxia-induced changes in ECs can promote survival in low-oxygen environments by altering cellular metabolism and inducing neoangiogenesis. However, hypoxia-induced EC responses can also be detrimental, leading to increased production of reactive oxygen species, heightened inflammation, changes in vascular tone, increased permeability of the endothelial barrier, and a higher risk of coagulation.
Critical Issues:
Hypoxia-induced EC responses contribute to the pathogenesis of various diseases, including metabolic diseases (e.g., diabetes, chronic kidney disease), infectious diseases, chronic inflammation, neoplastic diseases, cardiovascular diseases (e.g., atherosclerosis, myocardial infarction, and stroke) lung diseases (e.g., chronic obstructive pulmonary disease and pulmonary hypertension), eye diseases (age-related macular degeneration and retinopathy), and neurodegenerative diseases (e.g., Alzheimer’s disease and Parkinson’s disease).
Future Directions:
Detailed, disease-specific investigations are essential to delineate how endothelial hypoxia responses contribute to various pathologies. Understanding these mechanisms could reveal whether targeting endothelial hypoxia holds therapeutic potential. Antioxid. Redox Signal. 43, 849–868.
Introduction
Physiological oxygenation differs widely throughout the organs of the human body, and even within a given organ, there can be big variations. Therefore, hypoxia is a relative term that defines a condition when the oxygen level in a given organ, tissue, or cell type drops below the physiological level and is insufficient to maintain adequate homeostasis (Lee et al., 2020). Hypoxia can manifest in varying intensity, ranging from mild to severe oxygen depletion (anoxic condition <0.5% O2). Based on the duration, hypoxia can be transient/acute, persistent/chronic, or intermittent with alternating periods of normoxia and hypoxia (Lee et al., 2020).
Endothelial cells (ECs) line the entire vasculature, from the aorta to the smallest capillaries in the skin, and function in different tissues with various oxygen levels. ECs are exposed to hypoxia in diverse physiological and pathological conditions. This review aims to summarize our current knowledge about the effect of hypoxia on multiple biological processes in ECs, including metabolism, redox homeostasis, cell growth and differentiation, cell death, immune response, angiogenesis, production and secretion of vasoactive and procoagulant molecules, and the endothelial barrier function. We also discuss the numerous underlying mechanisms of how hypoxia drives atherosclerotic plaque progression.
Hypoxia and Hypoxia-Inducible Factor Signaling
General considerations
Aerobic species use molecular oxygen in intracellular energy production and for diverse essential biochemical reactions. Hypoxia, a condition with suboptimal oxygen availability in the tissues, is a significant stressor that endangers life. Oxygen partial pressure (pO2) varies largely between organs, and therefore, hypoxia is a relative term representing a pO2 level that is lower than the physiological pO2 (“physioxia”) and indicative of a lack of oxygenation in the tissue (Carreau et al., 2011).
Systemic and cellular adaptive mechanisms have evolved to cope with hypoxia. Systemic adaptive responses include increased alveolar ventilation, elevated heart rate and cardiac output, increased blood pressure, angiogenesis, neoangiogenesis, and enhanced erythropoiesis. At cellular levels, hypoxia triggers a metabolic switch toward pathways with low oxygen demand and suppresses ATP-consuming processes. These complex responses are regulated by hypoxia-inducible factors (HIFs; HIF-1 and HIF-2) (Fig. 1) (reviewed in Lee et al., 2020; Semenza, 2012).

HIFs are heterodimeric transcription factors composed of an oxygen-sensitive alpha (HIF-1α, HIF-2α, or HIF-3α) and a constitutively expressed beta (HIF-1β) subunit (Pugh et al., 1997; Wang et al., 1995). HIF-1 subunits contain basic-helix-loop-helix-PAS domains (Wang et al., 1995) that trigger heterodimerization and DNA binding (Jiang et al., 1996). HIFs recognize and bind to hypoxia-response elements (HREs) located in the promoter or enhancer regions of target genes and initiate their transcription. These HIF target genes promote cellular adaptation and survival in the hypoxic environment (Ratcliffe et al., 1998).
HIF-1β, the obligatory subunit of the HIF complex, is located in the nucleus in excess amounts, and therefore, the transcriptional activity of the HIFs is regulated by the protein expression of the HIF-α subunits (Jiang et al., 1996). Under well-oxygenated conditions, the alpha subunits have a short (∼10 min) half-life. Upon sufficient oxygen availability, the HIF-α subunits undergo enzymatic hydroxylation at highly conserved prolyl residues by prolyl hydroxylase domain (PHD) enzymes (Ivan et al., 2001). Prolyl hydroxylation is followed by polyubiquitylation and rapid degradation of HIF-α subunits by the von Hippel–Lindau tumor suppressor E3 ligase complex (Fig. 1) (Jaakkola et al., 2001). PHD activity depends on the availability of Fe(II) and 2-oxoglutarate as cofactors and molecular oxygen as a cosubstrate, and therefore, PHD acts as a cellular oxygen sensor (Jaakkola et al., 2001).
However, HIF-1α and HIF-2α show 48% amino acid sequence identity and similar protein structures, they are nonredundant; their expression shows tissue specificity and has distinct target genes and mechanisms of regulation (Tian et al., 1997). In most cells, HIF-1α expression increases fast and peaks early (2–24 h) following the onset of intense hypoxia or anoxia (<0.1% O2), whereas HIF-2α activation is triggered by mild or physiological hypoxia (<5% O2) and continues to be active even after 48–72 h of the onset of hypoxia (Holmquist-Mengelbier et al., 2006; Koh and Powis, 2012). Although HIF-1 and HIF-2 bind to a common consensus DNA‐binding motif HRE, studies showed that they bind different but overlapping sets of sites in chromatin and transactivate the expression of only partially overlapping sets of genes (Hu et al., 2003; Schödel et al., 2011). A recent study found that HIF-1 and HIF-2 show inherent chromatin binding preferences. HIF-1 prefers to bind closer to promoters, whereas HIF‐2 is more likely to bind to distal enhancers. These chromatin binding preferences are independent of the cell type or the degree and duration of hypoxia (Schödel et al., 2011).
HIF-regulated genes are involved in (i) metabolism (e.g., glucose transporter-1, glycolytic enzymes, carbonic anhydrase-9), (ii) vascular biology, and angiogenesis (e.g., vascular endothelial growth factor (VEGF), VEGF receptor (VEGFR), nitric oxide (NO) synthase-2, endothelin (ET)-1, heme oxygenase-1 (HO-1), (iii) erythropoiesis and iron metabolism (e.g., erythropoietin, ceruloplasmin, transferrin, transferrin receptor), and (iv) cell proliferation and survival (Semenza, 2001).
EC-specific features of HIF signaling
ECs are highly specialized cells and show remarkable phenotypic heterogeneity in different tissues and organs. EC morphology (shape and size), monolayer structure (e.g., continuous nonfenestrated, continuous fenestrated, discontinuous/sinusoidal), function (e.g., basal and induced permeability, leukocyte trafficking) transcriptomes, and proteomes vary dramatically across different vascular beds (Aird, 2012, 2007; Cleuren et al., 2019; Kalucka et al., 2020).
EC phenotypes are mainly shaped by their extracellular milieu, which can vary between different organs and nearby ECs of the same organ (reviewed in Aird, 2012, 2007). EC phenotype and function are fine-tuned to serve the underlying tissues’ diverse needs and ensure that ECs survive in the local environments (reviewed in Aird, 2012, 2007).
One such local factor, ECs must adapt to, is the oxygen level in the different tissues (Fig. 2). There is a huge variation in pO2 values between organs, having the highest in the inspired air in the trachea (150 mmHg) and the lowest in the superficial region of the skin (8 mmHg) (Carreau et al., 2011; Wang et al., 2003). Moreover, pO2 can vary in different parts of an organ. For example, while the pO2 in renal cortical tissue is 20–60 mmHg, the pO2 value in the inner medulla is below 15 mmHg (O’Connor, 2006). Another example is the liver, where the pO2 level is 60–65 mmHg in the periportal zone, while below 35 mmHg in the perivenous zone (Kietzmann, 2017). Subsequently, ECs are exposed to profoundly low oxygen levels in the inner medulla of the kidney and the liver sinusoids. Recent results show that ECs not only survive this low oxygen concentration, but hypoxia also plays an essential role in maintaining the phenotype and function of these cells. For example, hypoxia promotes proliferation and plays a role in maintaining the fenestrated structure of liver sinusoidal ECs through the sentrin/small ubiquitin-like modifier-specific protease 1/HIF-1α axis (Qing et al., 2021).

HIF-1 and HIF-2 orchestrate EC adaptation to the hypoxic environment by activating specific or overlapping target genes. A comprehensive study on 10 different primary human ECs from various vascular beds revealed that HIF-1 governs the acute adaptation to hypoxia, whereas HIF-2 is activated later and is responsible for adaptation to chronic hypoxia (Bartoszewski et al., 2019). Angiogenesis is a critical EC-dependent hypoxia response regulated by HIF-1α and HIF-2α (Ben-Shoshan et al., 2009). Studies showed that HIF-1 initiates angiogenesis, but the newly formed vascular network’s maturation is regulated by HIF-2 (Koh and Powis, 2012).
Hypoxia-Induced Alterations of Endothelial Function
As discussed above, ECs live and function in various oxygen levels depending on the tissue environment. Still, ECs can experience hypoxia in several pathological conditions where oxygen delivery to the tissues is compromised, and the oxygen tension drops below the physiological level. These conditions include metabolic diseases (e.g., diabetes, hypoglycemia, nonalcoholic fatty liver disease, chronic kidney disease, obesity), infectious diseases (e.g., infectious pneumonia, viral hepatitis), chronic inflammation, neoplastic diseases (e.g., colon cancer, lung cancer, gastric cancer, breast cancer, pancreatic cancer, prostate cancer), cardiovascular diseases (e.g., atherosclerosis, cardiomyopathy, arrhythmia, congenital heart disease, myocardial infarction, stroke, peripheral artery disease), lung diseases (e.g., chronic obstructive pulmonary disease, pulmonary hypertension), eye diseases (age-related macular degeneration and retinopathy), and neurodegenerative diseases (e.g., Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis) (Luo et al., 2022).
Besides these various pathologies, hypoxia can occur naturally, for example, at high altitude and during endurance training. In general, hypoxia and activation of the HIF signaling pathway influence multiple biological processes, including metabolism, redox homeostasis, cell growth and differentiation, cell death, and the immune response (Fig. 3) (Luo et al., 2022). Besides these processes, hypoxia provokes EC-specific responses, such as the induction of angiogenesis, the modulation of the production and secretion of vasoactive and procoagulant molecules, and the alteration of endothelial barrier function (Fig. 3).
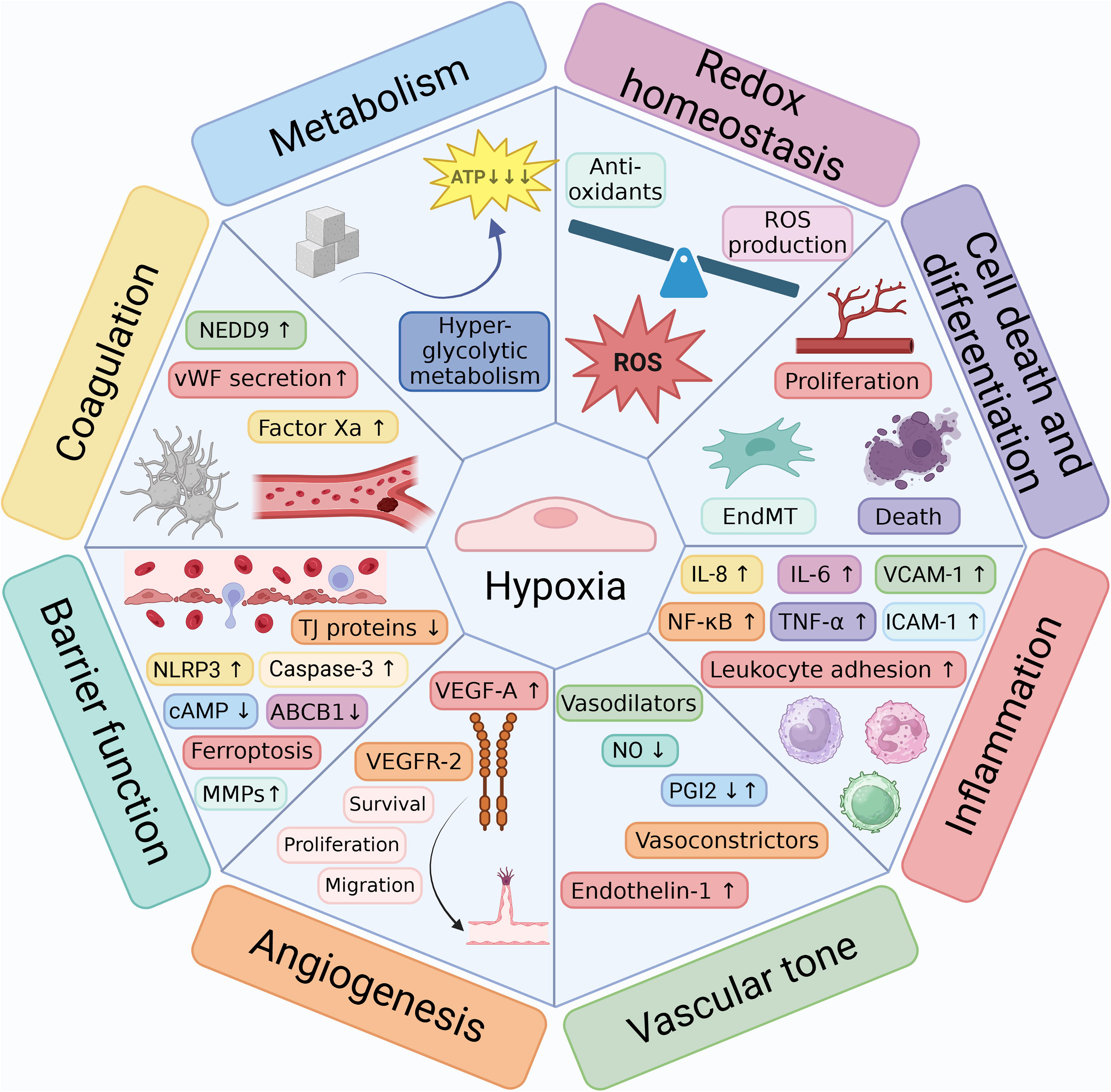
Hypoxia-induced changes in EC metabolism
In general, ECs have relatively low mitochondrial content, and even though most ECs live in a well-oxygenated environment, 85% of ATP is generated through anaerobic glycolysis (De Bock et al., 2013; Eelen et al., 2018, 2015; Li et al., 2019b). One of the important benefits of glycolysis-dominant energy metabolism over oxidative phosphorylation (OXPHOS) is that ECs are resistant to oxygen deprivation; therefore, they can vascularize hypoxic tissues with low oxygen availability (Li et al., 2019b). Moreover, ATP is produced faster from glucose in the glycolytic pathway, allowing the quick formation of new vessels in the hypoxic environment (Leung and Shi, 2022; Li et al., 2019a). The low oxygen demand of ECs is also beneficial, allowing more oxygen transfer to the perivascular tissues (De Bock et al., 2013; Li et al., 2019a). Importantly, the low OXPHOS rate in ECs is associated with a lower incidence of the formation of mitochondria-derived reactive oxygen species (ROS) (Alhayaza et al., 2020). Studies showed that hypoxia, depending on the severity and length of oxygen deprivation, provokes changes in ECs’ metabolism, which is discussed in this chapter (Cohen et al., 2020).
Hypoxic modulation of glycolysis and glucose metabolism
Accumulating evidence suggests that hypoxia promotes a metabolic shift in ECs to a hyperglycolytic state. An investigation by Loike et al. revealed that a 3-day hypoxic exposure upregulated glucose transport activity, glucose consumption, and lactate production of bovine aortic and human umbilical vein ECs (HUVECs) (Loike et al., 1992). To see the effect of prolonged hypoxia on ECs, Nauta et al. performed genome-wide RNA-sequencing in primary human microvascular ECs cultured at 20% oxygen and 1% oxygen for 2 weeks (Nauta et al., 2017). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that the upregulated genes in response to prolonged hypoxia were enriched in several metabolic pathways, including glycolysis, gluconeogenesis, and carbon-, fructose-, and mannose metabolism (Nauta et al., 2017). Besides hypoxic culture, the tumor microenvironment is widely used for studying the effect of hypoxia on EC metabolism as neovessels grow into the hypoxic regions of the tumors (Brown and Wilson, 2004). Cantelmo et al. showed that tumor ECs exhibit a hyperglycolytic metabolism characterized by the upregulation of transcripts of most glycolytic genes, including the glucose transporter GLUT1, 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase 3 (PFKFB3), an activator of phosphofructokinase 1, a rate‐limiting enzyme of glycolysis. Besides these, rate-limiting enzymes of glycolytic side pathways, such as glucose-6-phosphate dehydrogenase and hexose-6-phosphate dehydrogenase in the pentose phosphate pathway, as well as key enzymes involved in nucleotide biosynthesis, were also upregulated in tumor-associated ECs (Cantelmo et al., 2016). Glycolytic flux was nearly threefold higher in tumor ECs, as revealed by metabolic pathway analysis, and tumor ECs used more glucose-derived carbon for biomass production than normal ECs (Cantelmo et al., 2016). The reduction of glycolysis by inhibiting PFKFB3 improved vessel maturation and perfusion and increased the vasculature’s barrier integrity (Cantelmo et al., 2016). PFKFB3 inhibition impaired tumor invasion and metastasis and enhanced the sensitivity of the tumor to chemotherapy (Cantelmo et al., 2016).
Hypoxic modulation of the tricarboxylic acid cycle and mitochondrial function
Although mitochondrial content is low in ECs, the tricarboxylic acid (TCA) cycle provides essential substrates for the biosynthesis of nucleotides and amino acids and also the reducing agent NADH (Falkenberg et al., 2019). Beyond this, ECs’ mitochondria have emerged as signaling centers that play a crucial role in regulating a wide range of endothelial functions, including angiogenesis, suggesting the need to reassess the previously neglected role of mitochondria in ECs (Kadlec et al., 2016).
In connection with tissue hypoxia, Chen et al. demonstrated that chronic intermittent hypoxia increased TCA cycle metabolite levels and mitochondrial ROS production in rat aortic ECs (Chen et al., 2024a). Mao et al. induced severe ischemia [hypoxia (1% O2) and nutrient deprivation] in HUVECs, and results of high-throughput proteomics suggested that HUVECs upregulate the TCA cycle and mitochondrial respiratory chain and downregulate glycolysis under extreme ischemic conditions (Mao et al., 2022). Luo et al. found an elevation in malic acid 1 expression in lung tissue in patients and mice with pulmonary hypertension in vivo and in ECs exposed to hypoxia in vitro (Luo et al., 2024). Increased malic enzyme 1 expression was associated with endothelial dysfunction (Luo et al., 2024). A study by He et al. showed that under hypoxic conditions, the reverse TCA cycle was intensified, driving the conversion of α-ketoglutarate to isocitrate in bone marrow-derived ECs (He et al., 2024).
Iron–sulfur (Fe‐S) clusters are essential for mitochondrial metabolism. Chan et al. reported that hypoxia (0.2%−12% O2, 24 h) decreased the expression of Fe-S cluster assembly proteins 1 and 2 (ISCU1/2) in human PAECs through an HIF‐α‐dependent upregulation of microRNA‐210 (miR‐210) that targets ISCU1/2 (Chan et al., 2009). Hypoxia-induced increase in miR-210 and downregulation of ISCU1/2 lead to impaired Fe‐S cluster integrity, repression of mitochondrial metabolism, and increased ROS production. The HIF/miR-210/ISCU1/2 axis is critical in triggering pulmonary hypertension (White et al., 2015).
Hypoxia-induced upregulation in glutamine metabolism
The TCA cycle primarily utilizes pyruvate, but fatty acids (FAs) and most amino acids can also be transformed into TCA intermediates and enter the TCA cycle. Studies showed the importance of glutaminolysis in endothelial energy production under hypoxic conditions (Falkenberg et al., 2019). During glutaminolysis, glutamine is first converted to glutamate by glutaminase, and then to α-ketoglutarate, a TCA intermediate. Notably, glutamine is a crucial source of glutathione (GSH) to maintain antioxidant capacity under stress conditions. In angiogenic ECs, when cells invade a hypoxic area, glycolysis (glucose to lactate) and glutaminolysis provide the majority of metabolites for biosynthetic pathways and energy to fuel EC sprouting and proliferation (Polet and Feron, 2013). Kim et al. showed that glutamine is essential for EC proliferation but not migration (Kim et al., 2017).
Hypoxia induces the expression of glutaminase 1 in colorectal cancer cells (Xiang et al., 2019). Regarding ECs, vascular stiffness and increased lactate/pyruvate ratio, a marker of tissue hypoxia, were associated with elevated glutaminase 1 expression in PAECs in a rat model of pulmonary arterial hypertension (PAH) (Bertero et al., 2016). Glutamine levels in lung and heart tissues were elevated in the Sugen/hypoxia mouse model of PAH (Izquierdo-Garcia et al., 2018).
Impact of hypoxia on FA utilization
Hypoxia also alters FA metabolism in ECs. ECs can take up FAs directly from the circulation or passively through transporters. In addition, ECs can synthesize FAs endogenously from acetyl-CoA (Falkenberg et al., 2019). To cope with the high FA demand of angiogenesis, hypoxic and proliferating ECs exhibit enhanced FA uptake and increased endogenous FA production. In line with this, Hagberg et al. showed that endothelial FA uptake is upregulated by vascular endothelial growth factor B (VEGF-B) by increasing the expression of FA transport proteins (Hagberg et al., 2010). Furthermore, Singh et al. reported that hypoxia (3% O2, 6 days) increases de novo FA synthesis by upregulating FA synthase (FAS) in human PAECs (Singh et al., 2017). Inhibition of FAS decreased angiogenesis in hypoxic PAECs, suggesting that endogenous FA synthesis is crucial in the hypoxia-induced angiogenic response (Singh et al., 2017). During angiogenesis, VEGF-A upregulates the FA transporter, FA binding protein 4, which regulates FA metabolism, EC proliferation, and migration (Elmasri et al., 2009).
Hypoxia-induced changes in EC redox homeostasis
Controlled ROS formation is essential to regulate redox signaling and maintain vascular function and integrity. Several tightly regulated cellular processes contribute to ROS generation in ECs, including NADPH oxidases (NOX), mitochondria, xanthine oxidases, cyclooxygenases, cytochrome P450 monooxygenases, and uncoupled endothelial nitric oxide synthase (eNOS). ROS production is counterbalanced by antioxidant enzymes, including superoxide dismutases (SODs), catalase, glutathione peroxidase (GPx), peroxiredoxins (Prx), and the thioredoxin (TRx) system in ECs (Fig. 4).

Numerous studies have shown that chronic or acute hypoxia increases ROS generation in different EC types, including HUVECs, PAECs, and microvascular ECs (Chi et al., 2010; Pearlstein et al., 2002; Yu et al., 2010). Both increased ROS production and impaired ROS elimination mechanisms have been implicated in the hypoxia-induced elevation of ROS, the mechanisms of which are discussed in this chapter (Fig. 4).
The effect of hypoxia on NOX-derived ROS production
The NOX enzymes are the major source of ROS in ECs (Ushio-Fukai and Nakamura, 2008). NOX represents a 7-member family of oxidases, whose primary function is the generation of ROS (Kluge et al., 2013; Panieri and Santoro, 2015). In response to stimuli, NOXs transfer electrons from NADPH to oxygen to form a superoxide anion (•O2−) or hydrogen peroxide (Panieri and Santoro, 2015). ECs express four NOX isoforms, including NOX1, NOX2, and NOX5, which generate •O2−, and NOX4, which produces hydrogen peroxide (Drummond and Sobey, 2014). In ECs, NOX4 is the most predominant isoform. NOX4-produced hydrogen peroxide is a hyperpolarizing factor critically involved in vasodilation (Shimokawa and Morikawa, 2005). Supporting this notion, EC-targeted overexpression of NOX4 in mice enhanced vasodilation and decreased blood pressure (Ray et al., 2011). In contrast to the protective features of NOX4, the other endothelial NOX isoforms (NOX1, NOX2, and NOX5) are associated with promoting vascular pathologies (Drummond and Sobey, 2014).
Exposure of mice to chronic hypoxia (10% O2, 21 days) induced NOX4 upregulation in the medial layer of small pulmonary arteries (Mittal et al., 2007). Regarding endothelial responses, Craige et al. showed that hypoxia induces NOX4 expression in human aortic ECs and that NOX4 overexpression promotes endothelial proliferation, migration, and tube formation (Craige et al., 2011). NOX4 overexpressing transgenic mice showed improved recovery from hind limb ischemia compared with the controls, which further supports the beneficial nature of endothelial NOX4 (Craige et al., 2011). Ghouleh et al. found elevated NOX1 mRNA and protein expressions and increased ROS production in lung tissue obtained from patients with PAH compared with non-PAH subjects (Ghouleh et al., 2017). NOX1 was found to be upregulated in the intima layer of pulmonary resistance arteries in the lungs of patients with PAH (Ghouleh et al., 2017). Hypoxia (1% O2, 24 h) induced the mRNA and protein expressions of NOX1 and induced NOX1-derived ROS production in human PAECs (Ghouleh et al., 2017). NOX1 gene silencing attenuated hypoxia-induced EC proliferation (Ghouleh et al., 2017). A peptide that mimics the activation domain of the NOX1 activator subunit potently inhibited hypoxia-induced NOX1-derived •O2− production and VEGF-stimulated human pulmonary EC migration under hypoxic conditions (1% O2) (Ranayhossaini et al., 2013). Using chromatin immunoprecipitation, Diebold et al. showed that NOX2 is an HIF-1 target gene and that urotensin II-induced angiogenesis is HIF-1 and NOX2 dependent (Diebold et al., 2012). NOX2 also plays a role in the hypoxia-induced mobilization of endothelial progenitor cells in an ROS-dependent manner (Schröder et al., 2009; Urao et al., 2012). The interaction between hypoxia and NOXs is bidirectional (Fig. 5), a notion that is supported by the findings that NOX-derived ROS are involved in HIF-1α and HIF-2α stabilization (Block et al., 2007; Lévigne et al., 2016).
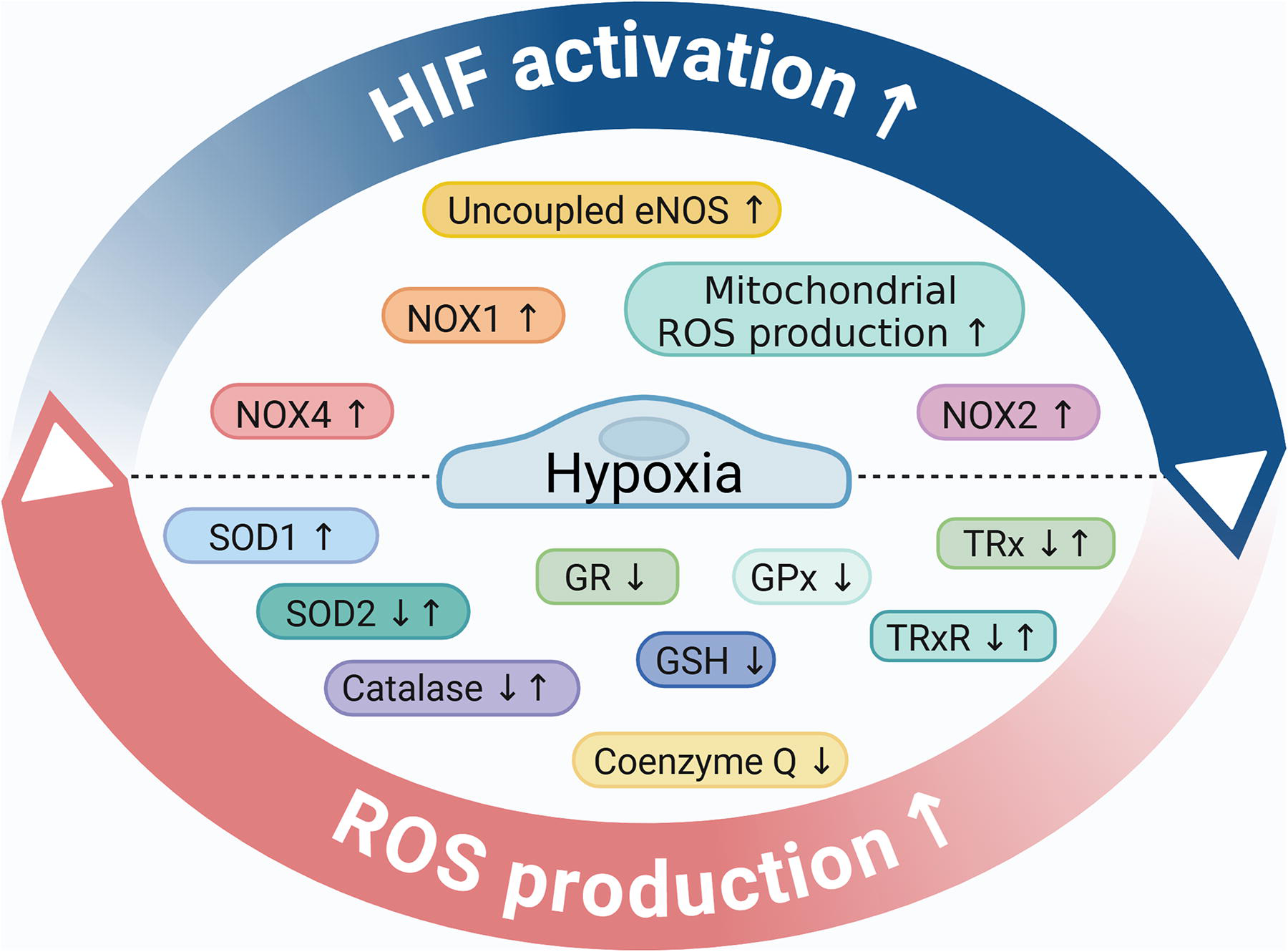
The effect of hypoxia on mitochondrial ROS production
Generally speaking, mitochondria are responsible for most (∼90%) of the cellular ROS production. On the contrary, ECs have a relatively low mitochondrial content, which also shows some heterogeneity. For example, the highly active ECs at the blood–brain barrier have a higher mitochondrial content (8%−11%) than ECs in other capillary beds with a mitochondrial content of only 2%−6%. Mitochondrial ROS generation is a consequence of OXPHOS. Complexes I and III along the cytochrome chain were identified, where electrons derived from NADH or FADH can directly react with oxygen and generate ROS (Balaban et al., 2005). Pearlstein et al. showed that hypoxia (2% O2, 6 h) increases ROS production in HUVECs (Pearlstein et al., 2002). ROS generation is abolished in the presence of diphenyleneiodonium or rotenone, but not in the presence of the NADPH inhibitor apocynin or a xanthine oxidase inhibitor allopurinol, suggesting that hypoxia mainly exacerbates mitochondrial ROS production (Pearlstein et al., 2002). The role of complex I and III has been established in hypoxia-triggered ROS production. Hernansanz-Agustín et al. showed that acute hypoxia (1% O2, 5 min) deactivates the mitochondrial complex I, which can exacerbate ROS production through its Na+/H+ antiporter activity in bovine aortic ECs (Hernansanz-Agustín et al., 2017). ROS generated at the mitochondrial complex III play a key role in HIF-1α stabilization in different cell types (Chandel et al., 2000), including pulmonary ECs (Yegambaram et al., 2024). Mitochondrial ROS are involved in hypoxia-induced upregulation of interleukin-6 (IL-6) and increased endothelial permeability (Pearlstein et al., 2002). Mitochondrial depolarization, fission, and mitochondrial dysfunction were observed in HUVECs exposed to hypoxia (1% O2, 1 h) (Giedt et al., 2012), as well as in microvascular ECs isolated from the Sugen/hypoxia model of PAH (Suresh et al., 2019). Besides the abnormal mitochondrial morphology and function, microvascular ECs derived from the Sugen/hypoxia model are characterized by increased ROS production, elevated basal intracellular [Ca2+], increased migration, and proliferation (Suresh et al., 2018).
The role of hypoxia in eNOS-derived ROS production
Under homeostasis, eNOS produces NO that is responsible for relaxing vascular smooth muscle cells (VSMCs) (Bredt and Snyder, 1990; Ignarro et al., 1987). The normal function of eNOS requires dimerization of the enzyme, the presence of its substrate L-arginine, and the availability of the essential cofactor tetrahydro-L-biopterin (BH4) (Förstermann and Münzel, 2006). NO is a free radical, and its bioavailability is modulated by its fast reaction with a •O2−, which yields peroxynitrite (ONOO−), a reactive peroxide (Radi, 2018). The cofactor BH4 is highly sensitive to ONOO−-induced oxidation. Insufficient levels of BH4 trigger superoxide production by eNOS, referred to as eNOS uncoupling (Förstermann and Münzel, 2006). Chalupsky et al. showed that hypoxia triggers eNOS uncoupling by decreasing the availability of BH4 in human PAECs and isolated murine pulmonary arteries (Chalupsky et al., 2015).
The effect of hypoxia on antioxidant pathways
Several studies addressed the effect of hypoxia on the activity of the components of the antioxidant network in ECs. Kong et al. investigated the regulation of SOD1 in bovine aortic ECs and found that anoxia (0% O2, 72 h) caused an about twofold increase in SOD1 activity (Kong and Fanburg, 1992). Anoxia also increased SOD1 and catalase activity in immortalized rat brain ECs (Rabin et al., 1996). In contrast, Koziel et al. found that chronic hypoxic exposure (6 days, 1% O2) increased intracellular and mitochondrial ROS formation, whereas the levels of SOD1 and SOD2 remained unchanged in HUVECs (Koziel and Jarmuszkiewicz, 2017). In some cases, hypoxia triggered the downregulation of antioxidant enzymes. For example, SOD2 protein expression was decreased, whereas mitochondrial ROS production was elevated in mouse pulmonary ECs isolated from chronically hypoxic diabetic mice (Pan et al., 2017). Exposure of bovine brain capillary ECs to chronic hypoxia (<1% O2, 72 h) resulted in decreased enzyme activities of SOD, catalase, GPx, and glutathione reductase, and reduced levels of GSH (Plateel et al., 1995). The effect of hypoxia (1% O2) was investigated on the components of the TRx system (TRx, TRx reductase, TRx interacting protein) in human endothelial progenitor cells and HUVECs. Hypoxia (1% O2) increased the expression of all three components of the TRx system in endothelial progenitor cells, whereas prolonged hypoxia (72 h) depleted TRx reductase and TRx interacting protein levels in HUVECs (Park et al., 2010). In contrast, hypoxia (3% O2, 24 h) decreased TRx and TRx reductase 1 levels in HUVECs (Bruschi et al., 2023). In addition, Adesina et al. found that hypoxia inhibits the expression and activity of the mitochondrial TRx2, which promotes the pathogenesis of pulmonary hypertension (Adesina et al., 2017).
The enzymatic antioxidant network is supported by antioxidant molecules that can act directly to inactivate ROS and pro-oxidants or can serve as a cofactor for antioxidant and detoxification enzymes. Reduced GSH is one of the most important low-molecular-weight antioxidants synthesized in cells (Forman et al., 2009). A recent metabolomic study on microvascular ECs found that both short-term and chronic hypoxia led to a reduced abundance of GSH. This reduction was attributed to an increased consumption rate rather than a decrease in de novo synthesis of GSH (Cohen et al., 2020). Coenzyme Q is a ubiquitous lipophilic cellular antioxidant distributed in all cell membranes, primarily the inner mitochondrial membrane (Crane, 2007). A recent study by Dominiak et al. showed that chronic hypoxia triggers coenzyme Q deficiency in ECs via decreased hydroxy-methylglutaryl-CoA reductase expression in an HIF-1α-dependent manner (Dominiak et al., 2024).
Although there are a lot of discrepancies in the presented results regarding the effect of hypoxia on the antioxidant capacity of the cells, it is clear that the antioxidant systems fail to compensate for hypoxia-induced increases in ROS production in ECs. Therefore, many attempts were made to understand whether increasing the antioxidant capacity of the cells can attenuate hypoxia-induced detrimental effects. In line with this notion, Wheeler et al. showed that the overexpression of SOD3 inhibited hypoxic tumor vascularization in mice (Wheeler et al., 2003). Overexpression of mitochondria-targeted catalase or SOD3 protects against hypoxia-induced pulmonary hypertension (Adesina et al., 2015; Ahmed et al., 2012). Overexpression of mitochondrial Prx3 protects the heart after ischemia–reperfusion injury and prevents left ventricular remodeling and failure after myocardial infarction and in mice (Kumar et al., 2009; Matsushima et al., 2006).
Hypoxia-induced angiogenic response
One of the most prominent endothelial-specific responses to hypoxia is angiogenesis, which is fundamental for organ growth and repair. Angiogenesis is a complex process that involves several consecutive steps, including degradation of the basement membrane, EC proliferation, migration, and the formation of tube-like structures (Carmeliet, 2005). Accordingly, angiogenesis is orchestrated by the interaction of three endothelial subtypes as follows: tip cells, which are highly migratory to guide and pull the new sprout to the appropriate direction, stalk cells, which elongate the newly formed sprout, and establish the lumen, and phalanx cells, which drive the maturation of the vessel (Eelen et al., 2020).
Hypoxia triggers angiogenesis through HIF-1 activation, which targets numerous genes encoding proangiogenic molecules involved in different steps of neovessel formation. For example, matrix metalloproteinases (MMPs) cleave extracellular matrix proteins. Their activation during angiogenesis facilitates EC migration. Studies showed that hypoxia stimulates the production of MMP-2 and MMP-9 in the vasculature through ROS-dependent activation of the nuclear factor kappa B pathway (Myasoedova et al., 2018). EC survival, proliferation, migration, invasion, sprouting, and tube formation are regulated by different angiogenesis-related growth factors, including VEGF, fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), angiopoietin 1 (Ang1), and cytokines (Davis, 2012; Samanta et al., 2017).
VEGF-A, which is strongly upregulated by hypoxia in ECs, is recognized as a key factor that triggers and sustains angiogenesis through the binding to its receptor, VEGFR2, whereas the binding of VEGF-A to its decoy receptor, VEGFR1, negatively regulates angiogenesis (Carmeliet, 2005; Shweiki et al., 1992; Takahashi and Shibuya, 2005). FGF is upregulated by hypoxia in ECs and is required for sustained induction of HIF-1α and sufficient angiogenesis under hypoxic conditions (Calvani et al., 2006). FGF promotes angiogenesis by activating FGF receptors, and FGF receptor signaling is essential for vascular remodeling after ischemia–reperfusion injury (House et al., 2016). Newly formed blood vessels are stabilized by VSMCs and pericytes. PDGF-B, a subtype of PDGF excreted mainly by ECs, plays a major role in the recruitment of mural cells during angiogenesis and is thereby involved in the maturation of sprouting blood vessels (Andrae et al., 2008). The overproduction of PDGF has been implicated in the pathogenesis of atherosclerosis and PAH, characterized by increased cell proliferation and fibrosis (Heldin and Westermark, 1999). Ang1, a member of the angiopoietin family of growth factors, is the main ligand for the Tie2 tyrosine kinase receptor (Davis et al., 1996). Ang1 is a potent proangiogenic factor that induces endothelial migration, tube formation, and sprouting and triggers the recruitment of mural cells to enhance vessel maturation (Metheny-Barlow and Li, 2003).
Accumulating evidence suggests that a hypoxia-induced increase in the glycolytic flux plays an important regulatory role in angiogenesis. In line with this notion, De Bock et al. showed that inhibition of PFKFB3 decreases vessel sprouting and branching (De Bock et al., 2013). In addition, it has been shown that lactate, a by-product of anaerobic glycolysis, promotes angiogenesis through the inactivation of PHDs, hence triggering an HIF-mediated increase in VEGF production (Kumar et al., 2007). In addition, miRs are emerging components of the complex regulatory circuit of hypoxia-induced angiogenesis, a topic that has been reviewed recently by Greco et al. (Greco et al., 2014).
The effect of hypoxia on EC differentiation, growth, and death
Angiogenesis occurs in hypoxic or even anoxic conditions; therefore, investigations of how ECs behave under such circumstances are of particular interest. ECs can respond to hypoxia/anoxia in two opposing directions. They can differentiate, proliferate, and promote angiogenesis to restore tissue oxygenation and nutrient supply, or if the severity or the duration of the hypoxic insult is beyond the limits, they can undergo cell death (Nör and Polverini, 1999).
ECs differentiate from pluripotent stem cells, in which process hypoxia plays a key role, a topic of a recent review (Podkalicka et al., 2020). Briefly, hypoxia has been shown to facilitate EC differentiation of human and mouse mesenchymal stem cells (Han et al., 2010; Prado-Lopez et al., 2010). Further studies showed that hypoxia-induced EC differentiation is HIF-1α-dependent (Tsang et al., 2017). Besides differentiation, hypoxia influences EC growth, cell cycle, and cell death. Tucci et al. showed that in response to hypoxia, EC proliferation slows down but does not arrest (Tucci et al., 1997). Hypoxia triggers alterations in the cell cycle, characterized by a slower transition from the G to the S phase, and an altered progression from S to G2/M, resulting in an increased percentage of ECs in the S phase (Tucci et al., 1997). Stempien-Otero et al. exposed HUVECs to severe hypoxia (<1% O2) for 24 h and 48 h. The results revealed that HUVECs are highly resistant to hypoxia-induced cell death, resulting in only 2% of apoptotic cells after 24 h and 45% of apoptotic cells after 48 h of hypoxia exposure (Stempien-Otero et al., 1999). Hypoxia increased the expression of p53, and overexpression of p53 protein alone was sufficient to initiate apoptosis in HUVECs (Stempien-Otero et al., 1999). A combination of low oxygen levels (3% O2) with low glucose (1 g/L) availability triggers EC pyroptosis characterized by caspase-1 activation (Bellut et al., 2021). In addition, hypoxia may induce EC death through excessive ROS formation. Warren et al. investigated the effect of menadione, a pro-oxidant molecule that increases intracellular •O2− production, on EC viability (Warren et al., 2000). Menadione induced apoptosis in ECs; however, the degree of cell death was remarkably different in capillary ECs with an apoptotic cell rate of ∼5%, compared with ∼45% in large-vessel ECs (Warren et al., 2000).
ECs are capable of undergoing endothelial-to-mesenchymal (EndMT) (Evrard et al., 2016; Piera-Velazquez and Jimenez, 2019) and endothelial-to-osteoblast transitions (Lin et al., 2017; Wylie-Sears et al., 2011). These cellular transdifferentiation processes are characterized by the loss of endothelial markers and the gain of mesenchymal or osteoblast markers, respectively. Zhu et al. showed that hypoxia induces EndMT of pulmonary arteriolar ECs and described the involvement of this process in hypoxia-induced pulmonary vascular remodeling (Zhu et al., 2006). Endothelial-specific lineage tracking revealed the common presence of EndMT-derived fibroblast-like cells in atherosclerotic lesions. Hallmarks of advanced lesions, including hypoxia, oxidative stress, and transforming growth factor-beta signaling, were identified as major triggers of EndMT in the atherosclerotic plaque, and a correlation was described between the extent of EndMT and plaque vulnerability (Evrard et al., 2016). Although we lack information about the influence of hypoxia on endothelial-to-osteoblast transition, accumulating evidence suggests that hypoxia promotes osteogenic differentiation of VSMCs and valve interstitial cells, the underlying mechanism of vascular and valve calcification, respectively (Balogh et al., 2019; Csiki et al., 2023). Further studies are warranted to explore the effect of hypoxia on endothelial-to-osteoblast differentiation.
The effect of hypoxia on EC inflammation
ECs provide the barrier between the blood and tissue and play an essential role in the regulation of inflammatory cell trafficking into the inflamed tissue. Leukocyte infiltration from the bloodstream is a multistep process initiated by the following adhesion molecules: vascular adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and chemoattractants (Luster et al., 2005). The rolling phase is regulated by selectins (E- and P-selectin), slowing the movement of leukocytes at the surface of the endothelium. Then, adherence occurs via the interaction of leukocyte β2-integrin with its ligand, such as ICAM-1 of EC. The last step is the transmigration triggered by the gradient of chemoattractant factors.
Studies showed that hypoxia activates ECs toward a proinflammatory phenotype and increases the adhesiveness of the endothelial surface, leading to enhanced adhesion of neutrophils, monocytes, and lymphocytes. Hypoxia-induced increased leukocyte infiltration plays a pathogenic role in different conditions, including ischemia–reperfusion injury, cancers, and atherosclerosis (Castillo-Rodríguez et al., 2022). Specifically, studies reported that hypoxia increases neutrophil adhesion to ECs in a mechanism mediated by CD18/CD11b, ICAM-1, and platelet-activating factor (Arnould et al., 1993; Ginis et al., 1993). Liang et al. showed that hypoxia increases ICAM-1 protein expression and monocyte–endothelial interaction through increased mitochondrial ROS production (Liang et al., 2019). Han et al. demonstrated that intermittent hypoxia increases the production of IL-6 and IL-8 via activating the NF-κB pathway and inhibiting the nuclear factor erythroid 2-related factor 2/HO-1 antioxidant pathway in ECs (Han et al., 2013). A recent study described a detrimental interaction between hypoxia and mannose-binding lectin-associated serine protease-1, leading to increased production of potent neutrophil chemoattractants (growth-regulated protein alpha [GROα] and IL-8), disruption of vascular network integrity, and activation of the NF-κB pathway in HUVECs and human umbilical artery ECs (Demeter et al., 2024). Oxygen/glucose deprivation has been shown to induce NLR family pyrin domain-containing 1 inflammasome activation and the subsequent production of active IL-1β in human brain microvascular ECs (Jung et al., 2023).
Besides being the source of inflammation, ECs are also targets of inflammatory cytokines, which induce endothelial activation. Groten et al. used a multiomics approach and an unbiased cytokine library and determined that tumor necrosis factor alpha (TNF-α), interferon gamma, and IL-1β trigger the largest EC responses characterized by unique proteomic inflammatory signatures (Groten et al., 2023). Numerous studies have indicated that hypoxia is a common microenvironmental feature at sites of inflammation that can profoundly affect immune cell function and cytokine-induced endothelial responses, too (Taylor and Scholz, 2022). In addition to the complex relationship, inflammatory cytokines, particularly IL-1β and TNF-α, induce HIF-1 activation in a nonhypoxic environment through a mechanism that involves ROS and NF-κB activation (Haddad and Harb, 2005). These studies identified HIF not only as an oxygen sensor but also as a mediator of inflammatory responses (Haddad and Harb, 2005).
The effect of hypoxia on EC-dependent regulation of vascular tone
ECs play a significant role in the local regulation of vascular tone through the production and secretion of vasoactive molecules. Two major groups of these vasoactive molecules are the vasodilators, including NO, prostacyclin (PGI2), and endothelial-derived hyperpolarizing factor, and the vasoconstrictors, such as endothelin (ET) and angiotensin-II (Ang II) (Cahill and Redmond, 2016).
Endothelial NO production is catalyzed by eNOS, which oxidizes its substrate L-arginine to L-citrulline and NO (Förstermann and Münzel, 2006). ECs release NO, which binds to a heme moiety of soluble guanylate cyclase (sGC), leading to sGC activation and a subsequent increase in cyclic guanosine monophosphate (cGMP) levels (Derbyshire and Marletta, 2012). cGMP induces vasorelaxation through directly modulating phosphodiesterases, ion-gated channels, or cGMP-dependent protein kinases (Derbyshire and Marletta, 2012).
Hypoxia modulates NO bioavailability in different ways, including regulation of eNOS expression and activity and L-arginine availability. Hypoxia also regulates NO-mediated activation of sGC and intracellular cGMP levels. These mechanisms contribute to the pathogenesis of hypoxia-driven diseases (Janaszak-Jasiecka et al., 2021).
There is some inconsistency in how eNOS expression changes upon hypoxic exposure in different ECs. For example, hypoxia-induced downregulation of eNOS was shown in HUVECs (Fish et al., 2010; McQuillan et al., 1994), human coronary artery ECs (Olszewska-Pazdrak et al., 2009), bovine PAECs (Liao et al., 1995), and human saphenous vein ECs (Takemoto et al., 2002). Parallel with these in vitro findings, decreased eNOS expression was found in the aortas and mesenteric arteries of mice exposed to chronic intermittent hypoxia (Wang et al., 2013) and in the lungs of patients with pulmonary hypertension (Giaid and Saleh, 1995). In contrast to HUVECs, hypoxia upregulates eNOS expression in human umbilical artery ECs (Vega-Tapia et al., 2021). Besides its effect on eNOS expression, hypoxia can also influence NO availability by downregulating eNOS enzyme activity through posttranslational modifications (Ghosh et al., 2016; Murata et al., 2002; Ostergaard et al., 2007). Furthermore, hypoxia can lead to decreased bioavailability of the eNOS substrate L-arginine due to hypoxia-induced upregulation of arginase-II that competes with eNOS for the same substrate (Krotova et al., 2010). Studies reported a change of sGC from the NO-sensitive reduced state to the NO-insensitive oxidized form under hypoxia, leading to impaired cGMP production (Crawley et al., 1992; Tawa et al., 2014).
PGI2 is an effective vasodilator, produced mainly by ECs. A few in vitro studies investigated the effect of hypoxia on PGI2 production. Interestingly, depending on the origin of the ECs, these studies found different effects; hypoxia increased the production of PGI2 in HUVECs (Cook-Johnson et al., 2006) while inhibiting PGI2 synthesis in bovine PAECs (Madden et al., 1986). The role of PGI2 is established in PAH, and currently, PGI2 and its analogs are the most effective therapies for PAH (Ruan et al., 2010).
ET is a potent vasoconstrictor, synthesized and secreted predominantly by vascular ECs, which acts in an autocrine and paracrine manner on its receptors in ECs and VSMCs (Khalil, 2011). Four ET isoforms (ET-1–4) are produced by the cleavage of preproET and big ET by ET-converting enzymes. ET signaling is regulated by two G-protein-coupled receptors, ET receptor A and B (Khalil, 2011). Arterial vasoconstriction is mainly regulated by the ET A receptor on VSMCs, which, upon ET binding, increases intracellular Ca2+ levels and induces signaling pathways of vasoconstriction and VSMC proliferation (Khalil, 2011).
Hypoxia (1% O2) increases ET-1 production and secretion in HUVECs and bovine ECs (Hieda and Gomez-Sanchez, 1990; Kourembanas et al., 1991). Minchenko et al. identified an HIF-1 binding HRE motif in the promoter region of the ET gene that mediates the transcriptional responses to hypoxia in microvascular ECs (Minchenko and Caro, 2000). In addition, a study by Ambalavanan et al. found increased ET-1 expression in the lungs of newborn mouse pups exposed to hypoxia (12% O2, 14 days) and suggested a role of hypoxia-induced ET-1 in pulmonary vascular remodeling (Ambalavanan et al., 2007).
The effect of hypoxia on EC barrier function
Endothelial barrier permeability differs in organs depending on the physiological demands of the particular tissue. The permeability of the endothelial barrier is regulated by cell–cell and cell–extracellular matrix interactions and influenced by multiple mediators (Komarova et al., 2017; Mehta et al., 2014). Intercellular junctions, including adherent junctions, tight junctions, and gap junctions, are integral parts of the endothelial barriers and are required to maintain the integrity of the vessel wall (Claesson-Welsh et al., 2021). Adherens junctions are located at cell-to-cell contacts, mediating cell adhesion and transferring intracellular signals. VE-cadherin is the major component of endothelial adherens junctions and is specific to ECs. Tight junctions regulate the tightness of the barrier, especially in small arterioles. Interactions between claudins, occludin, and junctional adhesion molecules create tight junctions. Zonula occludens protein 1 (ZO-1) associates with VE-cadherin to increase barrier stability. Gap junctions are intercellular pores with approximately 2 nm pore size, principally formed by transmembrane connexins between ECs (Kumar and Gilula, 1996). Contractile machinery and the actin dynamics of ECs, controlled by Rho GTPases, regulate endothelial barrier integrity, too (Aslam et al., 2013).
Accumulating evidence suggests that hypoxia decreases the integrity of the endothelial barrier system. For example, Funamoto et al. showed that hypoxia (3% O2, 6 h) increased HUVEC monolayer permeability by 11-fold for 70 kDa and fourfold for 10 kDa dextrans, resulting from the loss of VE-cadherin from cellular junctions under hypoxia (Funamoto et al., 2017). Ogawa et al. showed that hypoxia-induced hyperpermeability is due to decreased formation of cyclic adenosine monophosphate (cAMP) under hypoxic conditions (Ogawa et al., 1992). cAMP regulates the endothelial barrier by inhibiting the contractile machinery, mainly by activating myosin light-chain phosphatase via inhibiting RhoA/Rock (Aslam et al., 2013). Makarenko et al. showed that intermittent hypoxia induces endothelial barrier dysfunction via ROS-dependent MAPK signaling pathway activation and translocation of junction proteins, such as VE-cadherin and ZO-1, in lung microvascular ECs (Makarenko et al., 2014).
Hypoxia-induced EC responses in the regulation of coagulation
Thrombosis causes 1 in 4 deaths worldwide, displaying a major public health issue (Wendelboe and Raskob, 2016). Increasing evidence suggests the involvement of hypoxia in the pathomechanism of thrombus formation. The von Willebrand factor (vWF) triggers platelet adhesion to subendothelial collagen at sites of vascular injury. ECs store vWF in Weibel–Palade bodies (WPBs), and secretion of WPBs is an important regulatory step in controlling hemostasis. Brill et al. demonstrated that hypoxia/reoxygenation triggers the secretion of WPBs, thereby promoting deep vein thrombosis in mice (Brill et al., 2013). Besides triggering WBP secretion, hypoxia upregulates vWF expression in lung microvascular ECs, leading to increased platelet binding (Mojiri et al., 2013). Hypoxia induces upregulation of factor Xa and promotes thrombosis in bovine ECs and HUVECs (Ogawa et al., 1990; Stavik et al., 2016). Hypoxia and hypoxia mimetics (CoCl2 and DMOG) downregulate the production of the tissue factor pathway inhibitor in ECs, in an HIF-2α-dependent manner (Stavik et al., 2016). Upregulation of the HIF pathway in human PAECs is common in pulmonary vascular diseases characterized by pulmonary thromboembolic remodeling, however, platelet–endothelial interactions were not well-characterized under hypoxic conditions. Alba et al. described a novel mediator, neural precursor cell expressed developmentally downregulated protein 9 (NEDD9), that is expressed on the surface of pulmonary endothelium and interacts with P-selectin to mediate platelet–endothelial adhesion (Alba et al., 2021). NEDD9 expression is upregulated under hypoxia in an HIF-dependent manner, and the anti-NEDD9 antibody neutralizes the NEDD9-P-selectin complex and prevents platelet–endothelial adhesion under hypoxia in human PAECs and inhibits platelet aggregation in vivo (Alba et al., 2021). Pidgeon et al. found that hypoxia-induced increased cyclooxygenase-2 (COX-2) expression and COX-2-induced platelet activation play a pathogenic role in intravascular thrombosis after hypoxia-induced pulmonary hypertension (Pidgeon et al., 2004). Interestingly, in tumors, Evans et al. reported that HIF-1 activation promotes adhesion and clotting in tumor cells, but in tumor ECs, HIF-1 prevented microthrombus formation (Evans et al., 2016).
EC-Dependent Contribution of Hypoxia to the Pathogenesis of Atherosclerosis
Atherosclerosis, the main underlying cause of cardiovascular disease, is the leading cause of mortality and morbidity worldwide (Libby, 2021). Atherosclerosis is a chronic lipid-driven inflammatory disease in which dysfunctional ECs play critical roles in the initiation and progression phases of the disease (Gimbrone and García-Cardeña, 2016).
Atherogenesis is started by endothelial activation in lesion-prone regions of the arterial vasculature. Diverse stimuli trigger EC activation, including proinflammatory cytokines, oxidized lipoproteins, advanced glycation end-products, and disturbed blood flow (Gimbrone and García-Cardeña, 2016). Activated ECs facilitate lipoprotein transcytosis and increase surface expression of adhesion molecules, leading to the accumulation of low-density lipoprotein (LDL) and remnant lipoprotein particles and inflammatory cells in the subendothelial space (Gimbrone and García-Cardeña, 2016). During the progression of atherosclerosis, ECs undergo EndMT to give rise to plaque-associated fibroblast-like cells whose presence is associated with an unstable plaque phenotype (Evrard et al., 2016). In addition, ECs are involved in plaque neovascularization, contributing to the growth and destabilization of atherosclerotic lesions (Virmani et al., 2005).
Studies on human atherosclerotic lesions revealed the presence of HIF-1α expression in the nuclei of ECs covering the plaque (Akhtar et al., 2015). Similarly, nuclear HIF-1α expression was detected in ECs adjacent to the plaques in apolipoprotein E (ApoE)-deficient mice on a high-fat diet but not in ECs from mice fed a normal diet (Akhtar et al., 2015). Moreover, a recent study found increased expression of HIF-1α and glycolytic enzymes at atheroprone sites in porcine and mouse aorta (Feng et al., 2017). These results suggest that HIF-1 pathway activation occurs upon and even before plaque development. However, oxygen availability through the bloodstream is not limited to ECs at these stages of the disease. Therefore, nonhypoxic HIF-1 activation was proposed as a mechanism behind increased nuclear HIF-1α expression in ECs. Diverse atherogenic stimuli can trigger HIF-1α activation in ECs, including Ang II, high glucose, and mechanical low shear stress (Feng et al., 2017; Li et al., 2022; Luo et al., 2015) (Fig. 6).

In addition, previous studies investigated the effect of systemic hypoxia on atherosclerosis and found that hypoxia promotes atheroprogression and identified several EC-related contributing mechanisms (Song et al., 2015). For example, chronic intermittent hypoxia intensifies high-fat diet-induced atherosclerosis in ApoE-deficient mice through a mechanism involving endothelial NF-κB activation-dependent upregulation of E-selectin and VCAM-1 expression (Song et al., 2018). Harki et al. showed that intermittent hypoxia treatment impairs VE-cadherin integrity, increases intima-media thickness in the aorta of C57Bl/6 mice, and promotes lesion formation in ApoE-deficient mice fed with a standard diet (Harki et al., 2022). In vitro, intermittent hypoxia triggers VE-cadherin cleavage and facilitates the transendothelial passage of LDL and monocytes in human aortic ECs (Harki et al., 2022). Obstructive sleep apnea (OSA) is a frequent disorder that causes chronic intermittent hypoxia and is recognized as an independent risk factor for atherosclerotic cardiovascular disease. A recent study found a strong association between increased EC activation markers (EC-specific molecule-1, VCAM-1, P-selectin, and L-selectin) and carotid intima-media thickness, a measure of subclinical atherosclerosis in OSA patients (Sun et al., 2022).
Severe hypoxia induces EndMT in human carotid artery ECs, characterized by increased expression of fibroblast and mesenchymal markers (Evrard et al., 2016). A recent single-cell RNA sequencing study showed that disturbed shear stress promotes EndMT, in which process transcriptional phenotype for hypoxia responses and glycolysis were identified (Chen et al., 2024b). The study provided evidence for the critical role of enolase 1, a hypoxia-regulated key enzyme in glycolysis, in disturbed shear-stress-induced EndMT (Chen et al., 2024b).
Under homeostasis, the vasa vasorum nurtures the outer layers of the vessel wall, while the intima obtains oxygen and nutrients from the lumen. As atherosclerosis progresses, the intima thickens, and oxygen diffusion is impaired, leading to the development of a hypoxic core located 200–300 μm beneath the endothelial lumen. At this stage, hypoxia becomes a critical factor in atheroprogression (Moreno et al., 2006).
Local hypoxia stimulates neovascularization of the plaque from the vasa vasorum through the HIF/VEGF axis (Virmani et al., 2005). Histopathological studies revealed that the newly formed blood vessels that invade the plaque are leaky and immature, leading to intraplaque hemorrhage, erythrocyte lysis, and accumulation of red blood cell membranes and hemoglobin (Hb) (Virmani et al., 2005). The erythrocyte membrane contributes to the accumulation of free cholesterol within the lipid core of fibroatheromas, promoting lesion instability through necrotic core expansion and excessive macrophage infiltration. At the same time, Hb undergoes oxidative modifications and acts in a proinflammatory and pro-oxidant manner toward ECs and macrophages (Bozza and Jeney, 2020; Kolodgie et al., 2003; Silva et al., 2009).
Endothelial-specific HIF-1α deletion in ApoE-deficient mice was associated with decreased lesional macrophage accumulation, reduced atherosclerotic plaque size, and downregulation of miR-19a (Akhtar et al., 2015). This study proposed that endothelial HIF-1α promotes atherosclerosis by triggering miR-19a-mediated CXCL1 expression and monocyte adhesion (Akhtar et al., 2015).
To sum up, these pieces of evidence suggest that endothelial HIF-1 pathway activation occurs and facilitates the progression of atherosclerotic plaques (Fig. 6).
Concluding remarks
Global expression profiling approaches revealed that ECs derived from different vascular beds have distinct and characteristic gene expression profiles. ECs live and function at various oxygen levels, and physiological hypoxia may influence their phenotype and tissue-specific functions. Moreover, almost all major diseases, including diabetes, chronic kidney disease, atherosclerosis, cancers, pulmonary diseases, eye diseases, and neurodegenerative diseases, are associated with pathological hypoxia.
Although ECs primarily operate in low-oxygen environments, our understanding of their behavior under these conditions remains limited. We need to systematically characterize ECs from various vascular beds, using appropriate oxygen concentrations to replicate the physiological and pathological oxygen levels specific to their origin and associated diseases. This approach may improve our understanding of how hypoxia-induced EC phenotype and function alterations contribute to the different pathologies. A deeper understanding of these mechanisms may uncover the therapeutic potential of targeting endothelial hypoxia.
In addition, the number of drugs that target the HIF pathway is increasing. These drugs can cause HIF pathway activation or inhibition. For example, prolyl hydroxylase inhibitors activate the HIF pathway, leading to increased erythropoiesis, an effect used in anemia treatment in chronic kidney disease patients. In contrast, HIF inhibitors are used in patients with different advanced/refractory cancers. Most likely, these drugs influence ECs’ phenotype and function, which warrants further investigations.
Authors’ Contributions
A.T.: Conceptualization, writing—original draft, and writing—review and editing. V.J.: Conceptualization, writing—original draft, writing—review and editing, and funding acquisition.
Footnotes
Author Disclosure Statement
The authors have no relevant financial or nonfinancial interests to disclose.
Funding Information
The Hungarian National Research, Development, and Innovation Office (NKFIH, K146669 to V.J.) funded this work.