
Abstract
Nucleic acids, the storage molecules of genetic information, are composed of repeating polymers of ribonucleotides (in RNA) or deoxyribonucleotides (in DNA), which are themselves composed of a phosphate moiety, a sugar moiety, and a nitrogenous base. The interactions between these components and mineral surfaces are important because there is a tremendous flux of nucleic acids in the environment due to cell death and horizontal gene transfer. The adsorption of mono-, oligo-, and polynucleotides and their components on mineral surfaces may have been important for the origin of life. We have studied here interactions of nucleic acid components with rutile (TiO2), a mineral common in many terrestrial crustal rocks.
Our results suggest roles for several nucleic acid functional groups (including sugar hydroxyl groups, the phosphate group, and extracyclic functional groups on the bases) in binding, in agreement with results obtained from studies of other minerals. In contrast with recent studies of nucleotide adsorption on ZnO, aluminum oxides, and hematite, our results suggest a different preferred orientation for the monomers on rutile surfaces. The conformations of the molecules bound to rutile surfaces appear to favor specific interactions, which in turn may allow identification of the most favorable mineral surfaces for nucleic acid adsorption. Key Words: Nucleic acids—Mineral adsorption—Rutile—Origin of life. Astrobiology 10, 311–323.
1. Introduction
M
The interactions of nucleic acids and their components with specific mineral surfaces are especially interesting. Considerable amounts of free nucleic acids are present in the environment, and this pool of material may serve as a vast “nucleic acid library” for microorganisms via transfection (Reanney et al., 1983; Stewart et al., 1991; Khanna and Stotzky, 1992; Lorenz and Wackernagel, 1994; Ogram et al., 1994). A number of studies have examined the adsorption of DNA on various minerals and soil components (Lorenz and Wackernagel, 1987; Romanowski et al., 1991) and the ability of soil-bound DNA to serve as a transfecting agent (Pietramellara et al., 1997). The adsorption of small nucleic acid components (nucleotides, nucleosides, and their nitrogenous base components) in soils and on various minerals is also of interest from the standpoint of geochemical markers for life (Van Der Velden and Schwartz, 1974) and may have some relevance to the origin of a primordial RNA world. The interactions of polynucleotides, nucleotides, and their derivatives such as phosphorimidazolides have been studied extensively in this regard with minerals such as iron oxides (Holm et al., 1993; Arora et al., 2007), gypsum (Orenberg et al., 1985), apatite (Burton et al., 1969), clays (Goring and Bartholomew, 1952; Odom et al., 1979; Banin et al., 1985; Lawless et al., 1985; Ferris et al., 1989), ZnO (Arora and Kamaluddin, 2007), and pyrite (Hatton and Rickard, 2008). Mineral surfaces can also facilitate the oligomerization of activated nucleotides (Gibbs et al., 1980; Ertem, 2004), and adsorption may have protected nascent nucleic acids from degradation in the environment (Franchi et al., 1999; Ciaravella et al., 2004; Scappini et al., 2004; Biondi et al., 2007). While a considerable amount of research has been conducted regarding the interactions of mono-, oligo-, and polynucleotides with mineral surfaces, in particular with clays, relatively less is known regarding the interactions of the component molecules, including ribo- and deoxyribonucleosides and nucleobases. Such studies could lend considerable insight into the nature of the molecular-level interactions of these species.
Metal oxides, while not as abundant as silicates on Earth or Mars, can be especially strong adsorbants of organic compounds. We have investigated interactions of nucleic acid bases, ribo- and deoxyribonucleosides, and nucleotides with rutile (one of the naturally occurring polymorphs of TiO2) to obtain information regarding the molecular mechanisms of adsorption. Rutile is one of the most stable detrital minerals in sedimentary deposits (Morad and Aldahan, 1986; Zack et al., 2004), is common in igneous minerals, and may be common on Mars (Quinn and Zent, 1999). In addition, it has well-characterized surface properties and has been investigated extensively in other studies of molecular interactions (Sverjensky et al., 2008).
Recent investigations of interactions of ribonucleotides with ZnO and hematite (Arora and Kamaluddin, 2007; Arora et al., 2007) have reached conclusions somewhat at odds with previous adsorption studies, and this study thus offers another metal oxide surface for comparison.
2. Materials and Methods
Reagents were purchased from Sigma-Aldrich unless otherwise noted. Ultraviolet measurements were carried out with 1 cm path-length quartz cuvettes and an HP 8452A diode array spectrophotometer. The TiO2 used had a surface area of 2.485 ± 0.009 m2/g as measured by multipoint N2 BET isotherm (Micromeritics Analytical Services, Norcross, GA). Adsorption isotherm experiments were carried out by mixing 50 mg of TiO2 with 1.5 ml of a solution containing various concentrations (5 × 10−6 to 2.5 × 10−4 M) of a nucleic acid component that also contained 0.1 M NaCl to maintain a constant ionic strength in sterile 1.5 ml snap-cap Eppendorf tubes. These were rotated for 24 h on a Labnet Labroller II set at 20 rpm at room temperature (∼20°C). pH-adsorption studies were carried out with 10−4 M nucleic acid derivative, 0.1 M NaCl, and 100 mg TiO2, adjusted to the appropriate pH with aqueous NaOH or HCl in a final volume of 15 ml in sterile screw-top Falcon tubes. pH was measured before and after adsorption with use of an Orion Model 210A pH meter and a Corning Semi-Micro Combo electrode. In some cases, there was a significant change in pH from the initial to the final value, perhaps due to buffering by the dissolved nucleic acid derivative. In all cases, the final equilibrated value is reported.
The time to steady-state adsorption was not measured for these reactions; however, no change in adsorption was observed between reactions rotated for 24 h as opposed to 48 h. Thus, it was assumed that equilibrium had been reached at 24 h. Equilibrated solutions were centrifuged for 15 min with a Fisher Scientific AccuSpin 400 at 4000 rpm (corresponding to a relative centrifugal force of 1073 × g) to pellet suspended mineral particles. Aliquots of the supernatant were then assayed by UV/visible spectrophotometry after dilution to 0.01 M HCl concentration by addition of 0.1 M HCl, which allowed for a direct measurement of the concentration by using literature values for the extinction coefficients of the derivatives at known acidic pH values (Dawson et al., 1986). Measurements were blanked against a 0.01 M HCl solution.
Chromatographic analysis was carried out with a Hewlett-Packard 1050A LC system with a GRAE Altima C18 5μ column with an Agilent 35900E interface controlled by Agilent Chemstation software. The mobile phase consisted of 0.1 M pH 4.8 sodium phosphate at a flow rate of 1 ml/min. Elution was monitored at 260 nm. No significant degradation was noted during the timescales investigated.
The pH of the point of zero charge (pHpzc) of the rutile used was determined by potentiometric titration of the surface with a Metrohm Titrando 386. In some experiments, the pH was not controlled, and the final pH of the solutions was found to be very near the measured pHpzc of the rutile (∼4.3). We estimated error in concentration measurements to be on the order of ±3%, and in pH measurements less than ±0.1 pH unit.
Data were fit to Langmuir-type isotherms where possible. It should be pointed out that this assumes certain behaviors, such as rapid reversible equilibration of adsorption, that all surface sites are equivalent, that the surface sites are saturable, and that adsorption is not cooperative (Davis and Hayes, 1986; Drever, 1988). Although the data in general could be well fit to Langmuir isotherms, this provides no evidence as to the actual mechanism of adsorption.
Error bars were calculated assuming a 3% error in UV absorbance measurements and a 1% error in weighing of the mineral powder.
3. Results
The compounds tested and the atomic numbering conventions used are shown in Fig. 1.
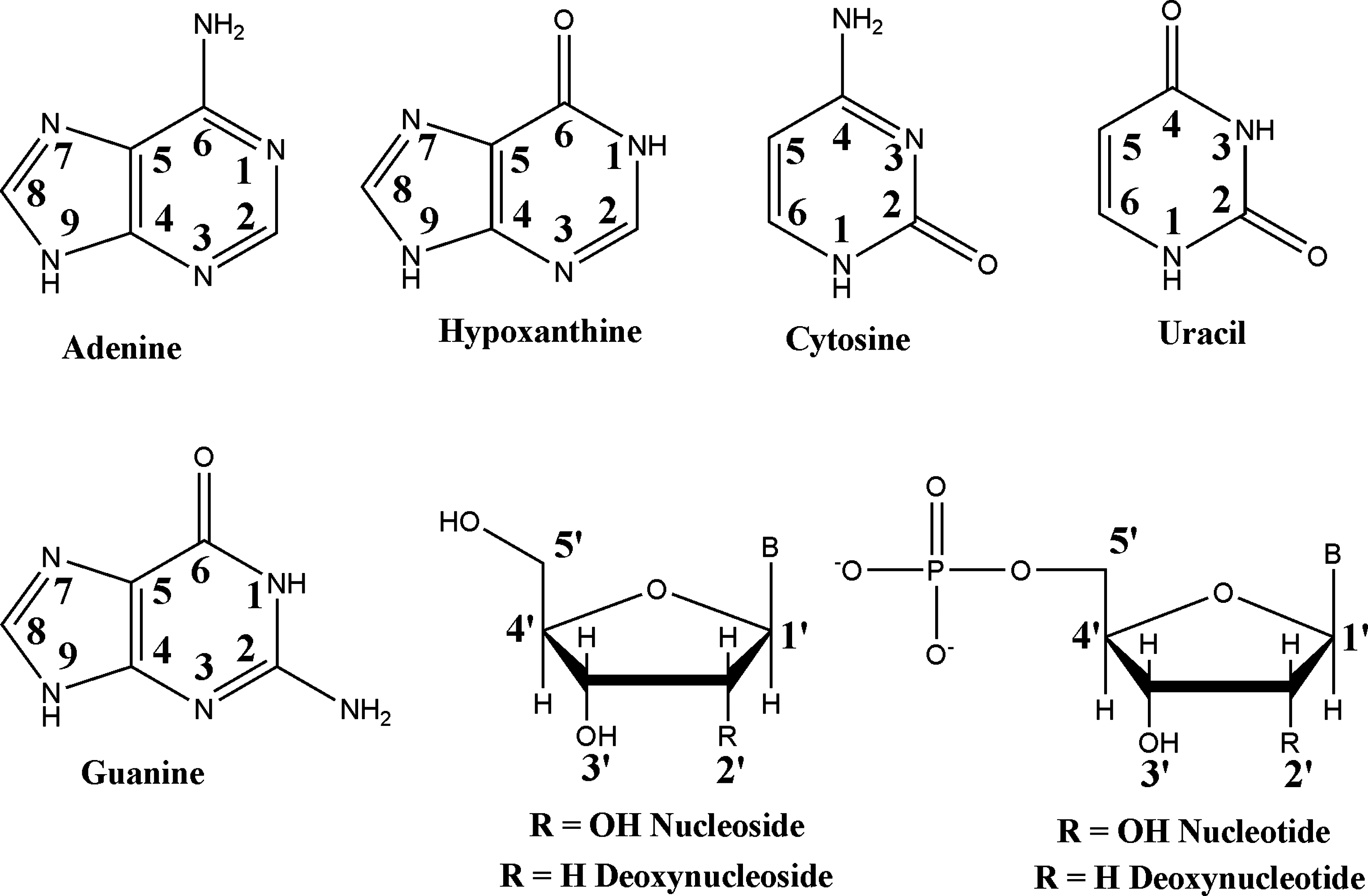
Nucleic acid derivatives tested in this study. B = adenine (A), cytosine (C), uracil (U), or guanine (G). The numbering systems for the nitrogenous bases and sugars are also shown.
A quick mention should be made of some experimental details before proceeding. TiO2 is known to be photocatalytic for the degradation of organic compounds (Linsebigler et al., 1995; Fu et al., 1996; Araujo et al., 2005; Van der Meulen et al., 2007). Mineral surfaces can also promote the dephosphorylation and other degradative transformations of organic compounds (Burton et al., 1969). This effect was deemed unlikely due to the lack of change in the UV/visible spectrum of the supernatants; nevertheless, reactions were assayed for breakdown products in the supernatant, after UV measurements were taken, by high-performance liquid chromatography analysis. No significant degradation products (<0.1%) were detected during the course of these experiments.
Also, experiments were conducted under atmospheric conditions; consequently, no effort was made to exclude CO2 or O2. The concentrations of ionic carbonate species present at equilibrium with the amount of gas in the headspace in both the 1.5 ml and 15 ml tubes (∼10−4 L) were calculated to be much lower than the concentration of NaCl or added adsorbent (∼1 μM maximum). Below ∼ pH 5, essentially no ionized carbonate species should be present; nevertheless, carbonate species can adsorb below pH 5, and these species could potentially compete for adsorption sites.
3.1. Nitrogenous bases
The first set of experiments tested nitrogenous base adsorption, including the purines hypoxanthine (HX) and adenine (A) and the pyrimidines cytosine (C) and uracil (U). A guanine (G) isotherm was not attempted due to its low solubility at the equilibrium pH value studied in these experiments. Results of these experiments are shown in Fig. 2.
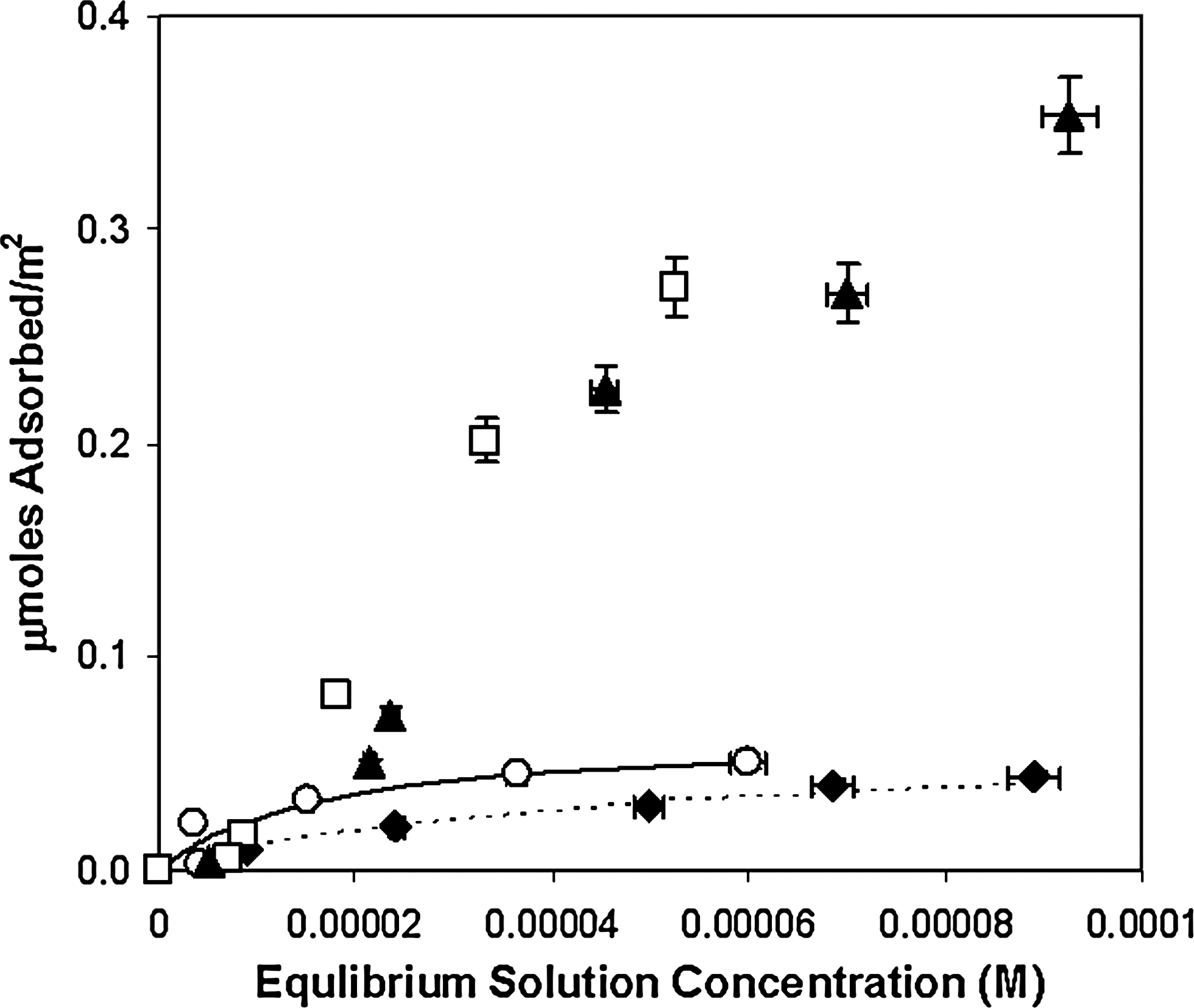
Adsorption of nitrogenous bases per m2 to rutile (33.3 g/L) at ∼ pH 4.3 (final pH) in 0.1 M NaCl. Filled triangles, adenine; filled diamonds, hypoxanthine; open circles, uracil; open squares, cytosine. Uracil and hypoxanthine are shown with Langmuir isotherm curve fits. Adenine and cytosine were not well fit with this model.
The bases with exocyclic amino groups (A and C) generally showed greater binding affinity, perhaps because there are several potential binding modes for these compounds that maximize potential surface interactions as shown in Fig. 3. These compounds could form inner- or outer-sphere interactions with surface functional groups, including
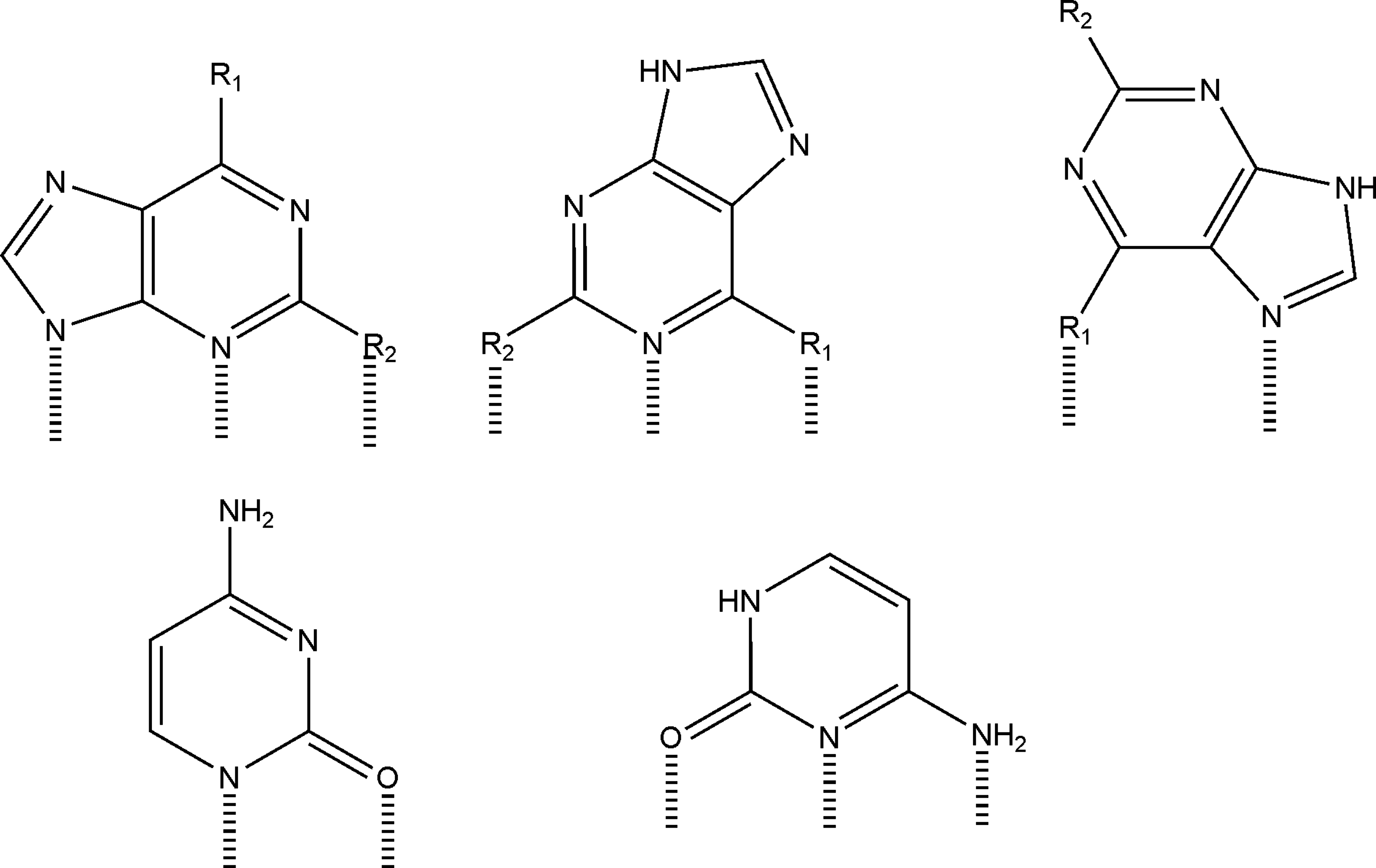
Potential binding interactions of purines (top row, A: R1 = NH2, R2 = H; G: R1 = OH, R2 = NH2; HX: R1 = OH, R2 = H) and cytosine with the mineral surface. Hypoxanthine and guanine are shown in their enolic form.
The comparable adsorption of C and A, and their much higher adsorption relative to HX and U, suggest the involvement of electrostatic interactions (C and A both exist significantly as cations under the pH conditions studied). Van der Waals and hydrogen-bonding interactions may also contribute to some extent (Kochetkov and Budovskii, 1972; Saenger, 1988). In addition, the bases are known to coordinate metal ions via various combinations of the ring nitrogen atoms and the exocyclic groups (Marzilli, 1977).
Adsorption does not correlate well with solubility. Measured solubilities in water at 25°C are as follows: cytosine, 69 mM (Budavari, 1996); uracil, 31.9 mM (Budavari, 1996); adenine, 2 mM (Budavari, 1996) and 8.7 mM (DeVoe and Wasik, 1984); and hypoxanthine, ∼5.3 mM (Budavari, 1996). In contrast, hydrophobicity increases with C < HX < U < A (Shih et al., 1998). These differences suggest that binding is mediated by specific interactions with the surface rather than via hydrophobic effects. There may also be cooperative association with already adsorbed molecules, which may contribute to adsorption.
To test some of these possibilities, we measured molecular adsorption on rutile as a function of pH. Results are shown in Fig. 4.
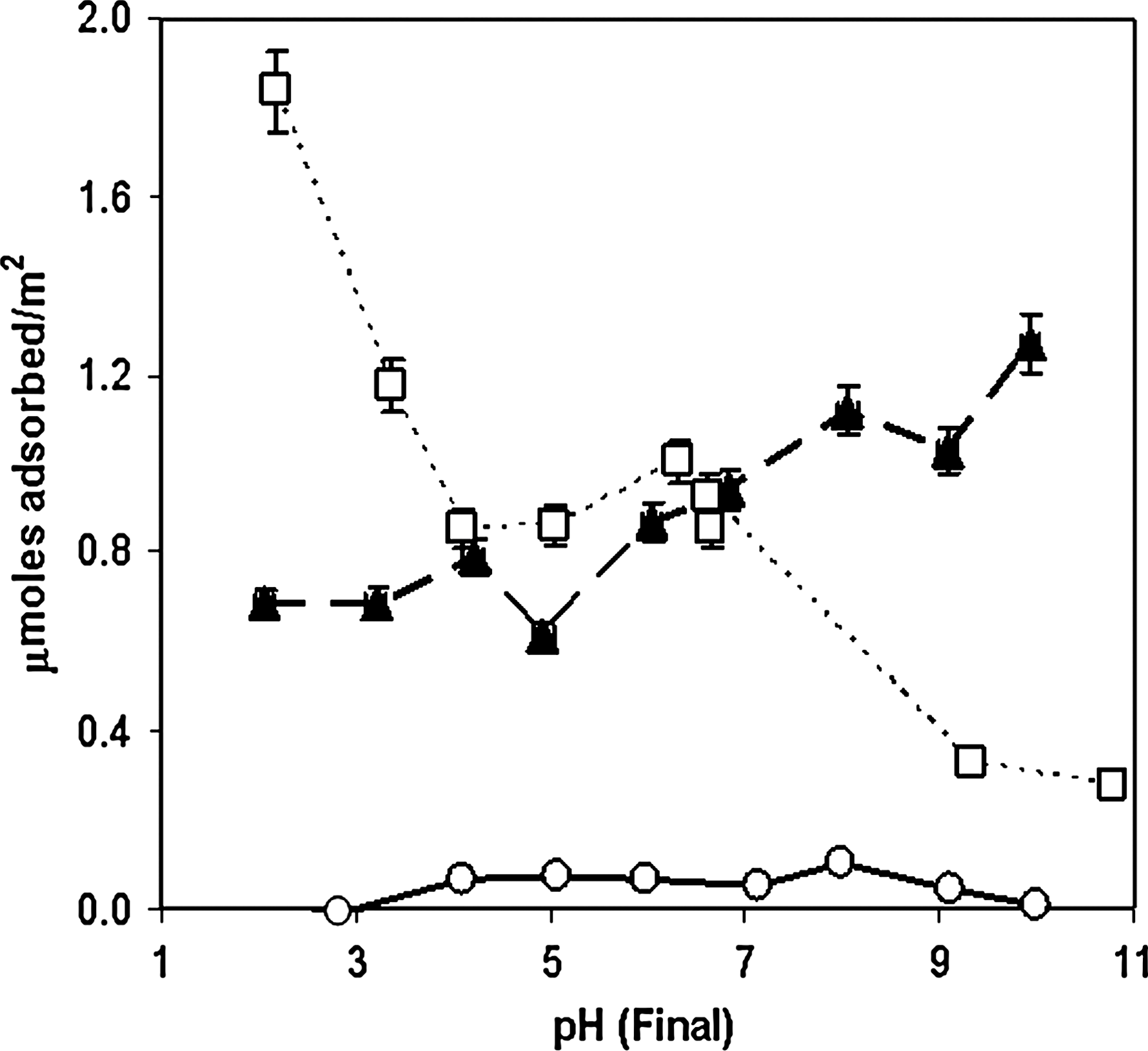
Adsorption vs. pH for guanine, adenine, cytosine, and uracil (10−4 M initial concentration) on rutile (6.67 g/L) in 0.1 M NaCl. Filled triangles, adenine; open squares, cytosine; open circles, uracil. Connecting lines are merely intended as guides for the eye.
As can be seen, uracil adsorption was negligible at most pH values. Although both adenine and cytosine developed positive charges below ∼ pH 5, at which point the surface also developed a net positive charge suggesting adsorption should decrease due to charge repulsion, it appeared that cytosine adsorption increased with decreasing pH. This could be due to cooperative adsorption between cytosine-cytosine+-type base pairs, which may form at lower pH (Cruse et al., 1983). The apparent discrepancies between the amount of cytosine and adenine adsorbed in Fig. 2 could be due to the relative surface loading in the two sets of experiments, which has been documented for the adsorption of glutamate on rutile (Sverjensky et al., 2008).
The nucleobases have several potential points of protonation, including ring nitrogen atoms and functional groups, and may develop charges depending on solution pH (Fig. 5) (Dawson et al., 1986). Cytosine exists largely as a cation below ∼ pH 4.6, and this charged state may influence its adsorption at low pH values. The solubilities of the bases also change as a function of solution pH, particularly in the case of guanine where significant self-pairing may be inhibited at high and low pH (Saenger, 1988). In addition to protonation and deprotonation, the bases exhibited changes in keto-enol tautomerization equilibrium, which are also pH dependent (Kochetkov and Budovskii, 1972). The tautomeric and ionic forms of the bases may have been influenced by the environment of the Stern layer, which may differ from that of the bulk aqueous solution (Ferris et al., 1989).
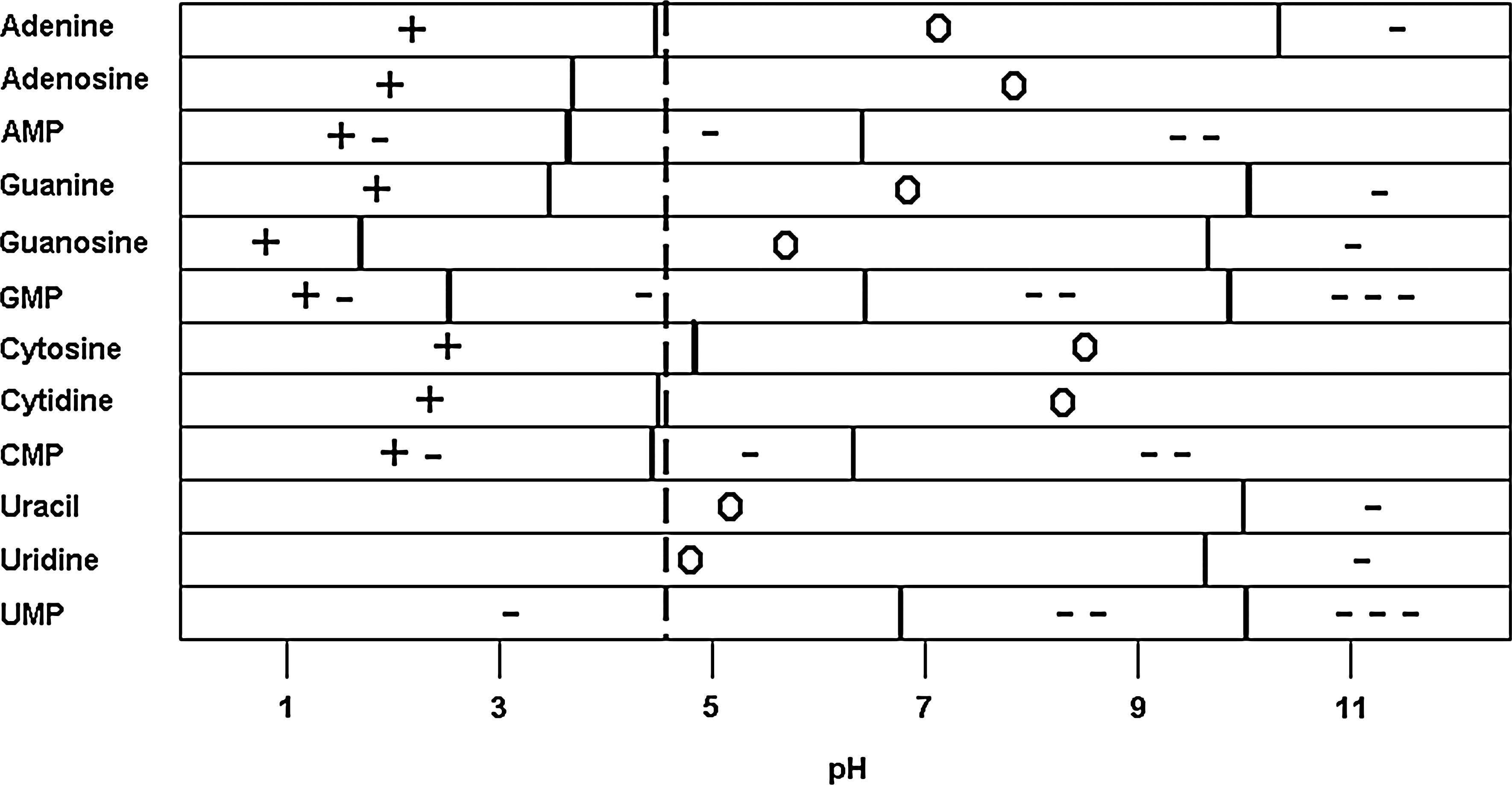
pK a values (horizontal bars) and charges of the compounds tested as a function of pH. The values for 2′-deoxynucleosides and 2′-deoxynucleotides are quite similar to those of the ribose derivatives and thus are not shown for the sake of the clarity of the figure. For reference to many of the adsorption experiments shown, a vertical dashed line has been added at pH 4.3, corresponding to rutile's point of zero charge.
3.2. Ribonucleosides
The adsorption of the ribonucleosides was then assayed as shown in Fig. 6. The addition of the sugar moiety caused a significant increase in the binding affinity of these compounds, but there were also significant differences between the various species, depending on the base moiety.

Adsorption of nucleosides to rutile (33.3 g/L) surfaces at pH ∼ 4.3 in 0.1 M NaCl. Filled diamonds, guanosine; filled triangles, adenosine; open squares, cytidine; open circles, uridine. Curves are best fits to the Langmuir equation: solid line, uridine; heavy dotted line, guanosine; light dotted line, cytidine; dashed line, adenosine.
Adsorption of uridine relative to uracil was enhanced by a factor of ∼7 near pH 4.3, while there was relatively little difference between cytosine and adenine derivatives. Points of interaction available to the nucleobases, specifically those involving the pyrimidine N1 position and purine N9 position, were suppressed by the blocking of these portions of the molecule by the sugar moiety. However, the adsorption of the ribosides may have been enhanced by the interactions involving the ribose hydroxyl groups, and all showed fairly similar isotherms near pH 4.3, which suggests that the sugar moiety governs the interactions with minor, though significant, contributions from the base moiety.
The adsorption of the ribosides as a function of pH was then assayed (Fig. 7).

Adsorption vs. pH for ribosides (10−4 M initial concentration) on rutile (6.67 g/L) in 0.1 M NaCl. Filled diamonds, guanosine; filled triangles, adenosine; open squares, cytidine; open circles, uridine. Connecting lines are merely intended as guides for the eye.
Note that there are significant differences in the adsorption maxima when measurements were made by using 6.67 g/L versus 33.3 g/L TiO2 at similar pH values. The sources of these differences are unclear; but, as noted for the pH adsorption profiles in Fig. 4, some of the discrepancy may be due to differences in surface loading. This effect is especially pronounced with guanine derivatives (see Figs. 8 and 9 below) and may represent the formation of intramolecularly hydrogen-bonded aggregates, which may precipitate from solution, rather than surface adsorption.
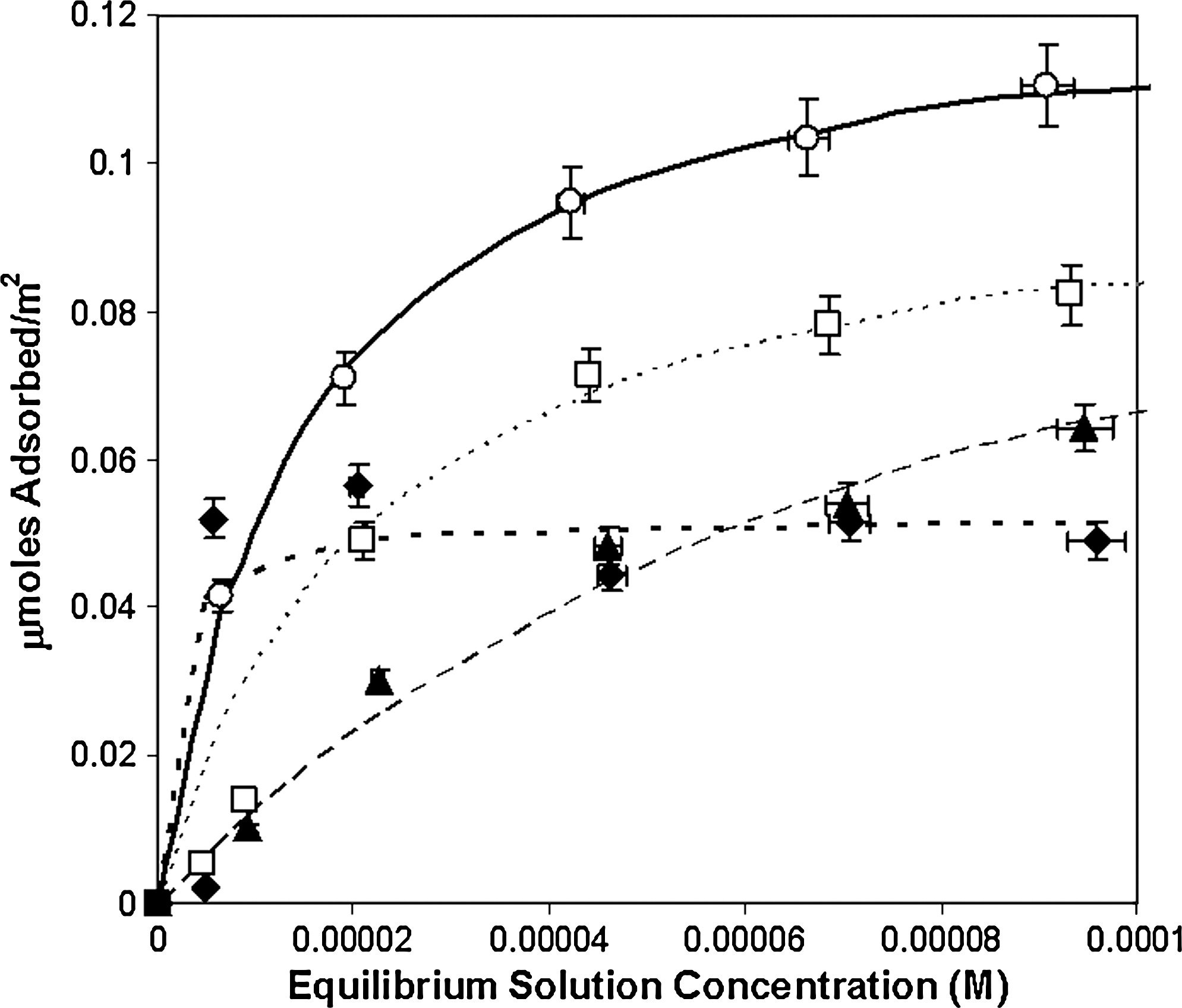
Adsorption isotherms of deoxyribonucleosides to rutile (33.3 g/L) surfaces at pH ∼ 4.3 in 0.1 M NaCl. Filled diamonds, 2′-deoxyguanosine; filled triangles, 2′-deoxyadenosine; open squares, 2′-deoxycytidine; open circles, 2′-deoxyuridine. Curves are best fits to the Langmuir equation: solid line, uridine; heavy dotted line, guanosine; light dotted line, cytidine; dashed line, adenosine.
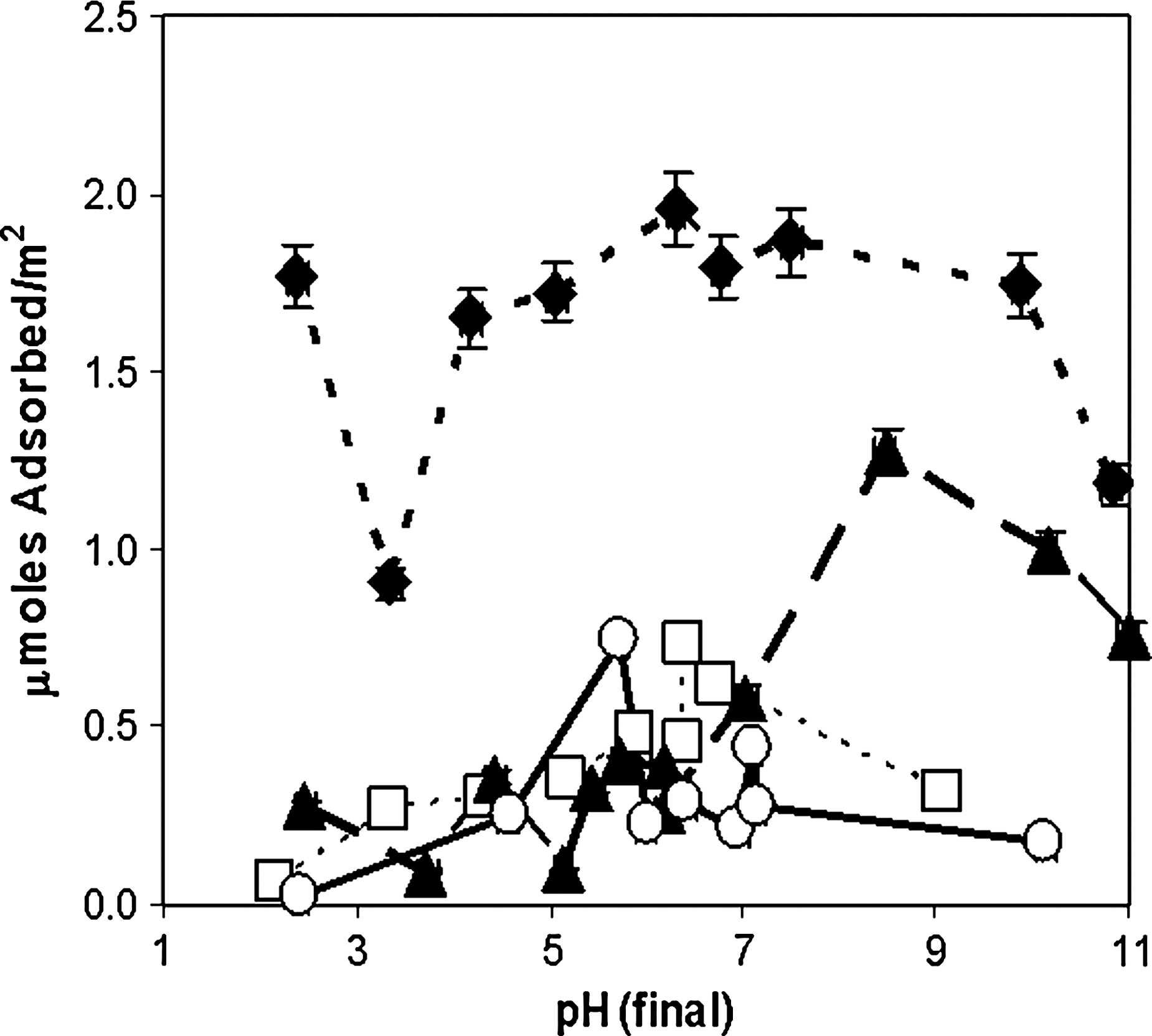
Adsorption vs. pH for deoxyriboside (10−4 M initial concentration) to rutile (6.67 g/L) in 0.1 M NaCl. Filled diamonds, 2′-deoxyguanosine; filled triangles, 2′-deoxyadenosine; open squares, 2′-deoxycytidine; open circles, 2′-deoxyuridine. Connecting lines are merely intended as guides for the eye.
3.3. Deoxyribosides
Isotherms for the adsorption of deoxyribosides are shown in Fig. 8. We observed a significant decrease in adsorption compared with the ribosides, from one-third to one-sixth as great at 10−4 M equilibrium solution concentration, which suggests that both the 2′ and 3′-OH groups of the ribose moieties are involved in binding for nucleosides. Small, but significant, differences occurred between the deoxynucleosides, which again suggests that the sugar moiety is not the only interacting part of these molecules. The enhancement of deoxyuridine binding may be explained by the possibility that the C2 ketone of uridine forms a stronger bond with the rutile surface than does the C2 amino group of cytidine. The enhancement of binding of the other deoxynucleosides over deoxyadenosine suggests that the functional group at the C2 position of the base has a significant effect on adsorption.
The deoxyribosides, like the ribosides, showed complex adsorption behavior as a function of pH (Fig. 9).
The purine deoxyribosides in general adsorbed more strongly than the pyrimidine deoxyribosides, and there was little difference between deoxyadenosine, deoxycytidine, and deoxyuridine between ∼ pH 2 and ∼ pH 7 and little difference between the pyrimidine deoxyribosides at any pH, which suggests a similar mode of binding. In addition, in almost all cases, the removal of the 2′ OH group of ribose resulted in considerably weaker adsorption. The significantly higher adsorption of guanine derivatives in both cases suggests either the formation of intramolecularly bonded complexes, insolubility, or a role for specific functional group interactions with the surface.
3.4. Ribonucleotides and deoxyribonucleotides
The ribonucleotides and deoxyribonucleotides showed a marked increase in binding affinity compared to their cognate nucleo- and deoxynucleosides (Figs. 10 and 11).
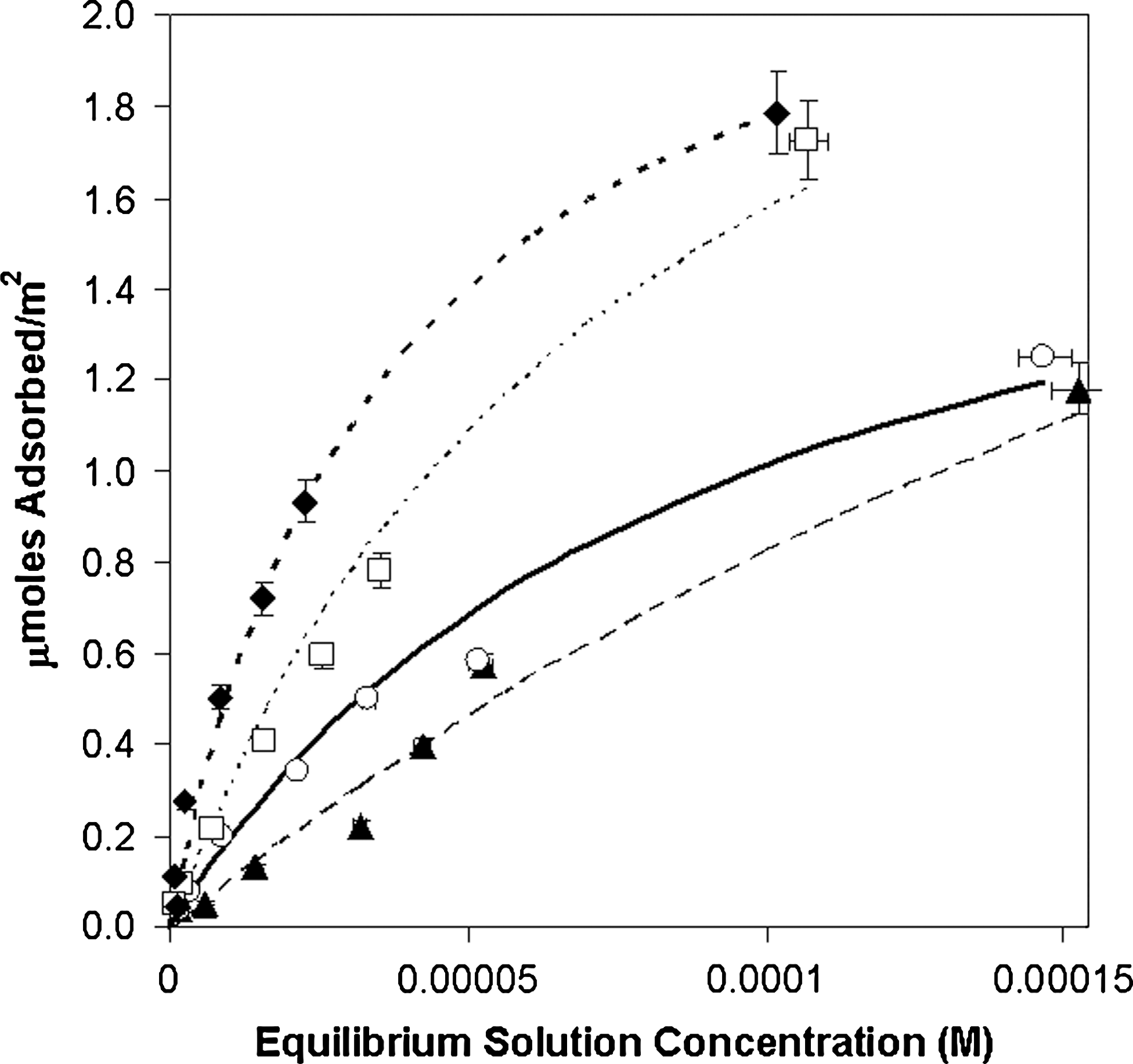
Adsorption isotherms of 5′-ribonucleotides to rutile (33.3 g/L) at pH ∼ 4.3 in 0.1 M NaCl. Filled diamonds, guanosine-5′-monophosphate (GMP); filled triangles, adenosine-5′-monophosphate (AMP); open squares, cytidine-5′-monophosphate (CMP); open circles, uridine-5′-monophosphate (UMP). Curves are best fits to the Langmuir equation: solid line, UMP; heavy dotted line, GMP; light dotted line, CMP; dashed line, AMP.
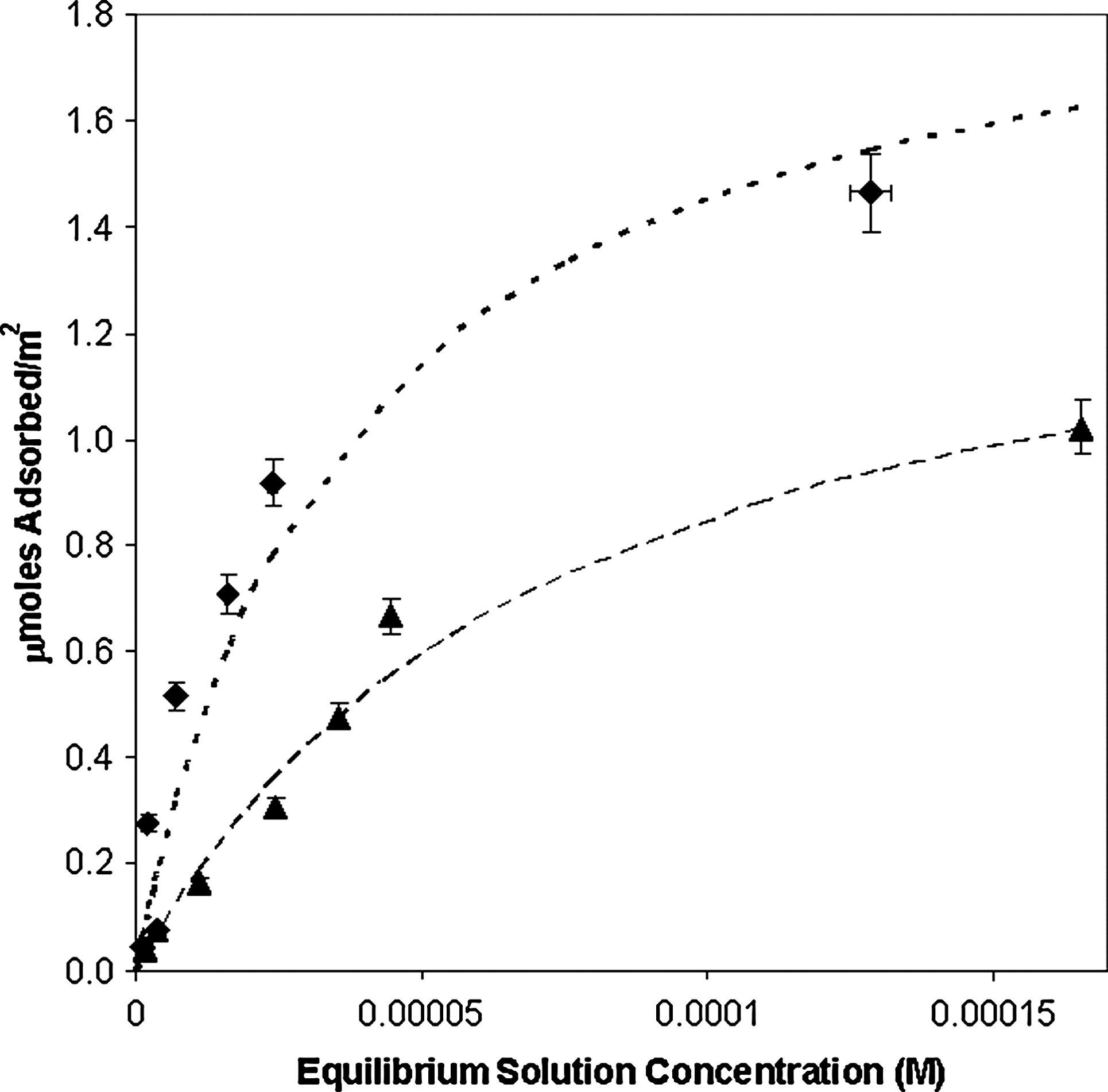
Adsorption isotherms for 2′-deoxyadenosine-5′-monophosphate and 2′-deoxyguanosine-5′-monophosphate on rutile (33.3 g/L) in 0.1 M NaCl at pH ∼ 4.3. Filled diamonds, 2′-deoxyguanosine-5′-monophosphate; filled triangles, 2′-deoxyadenosine-5′-monophosphate. Curves are best fits to the Langmuir equation: heavy dotted line, dGMP; dashed line, dAMP.
The addition of the phosphate group caused a 3- to 6-fold increase in adsorption of the ribotides versus the ribosides. The order of affinity appeared to be G > C > U > A, which again suggests an interaction between the base moiety and the surface. It should be noted, however, that these differences could also be due to cooperative intermolecular interactions such as hydrogen bonding between adjacent molecules.
The phosphate group had the largest influence on adsorption at pH 4.3; however, the ribotides still showed greater affinity than deoxyribotides, which suggests that the 2′-OH group plays a role in adsorption, and the influence of the base (G > A) is still apparent.
The pH adsorption behavior for the ribotides is shown in Fig. 12.
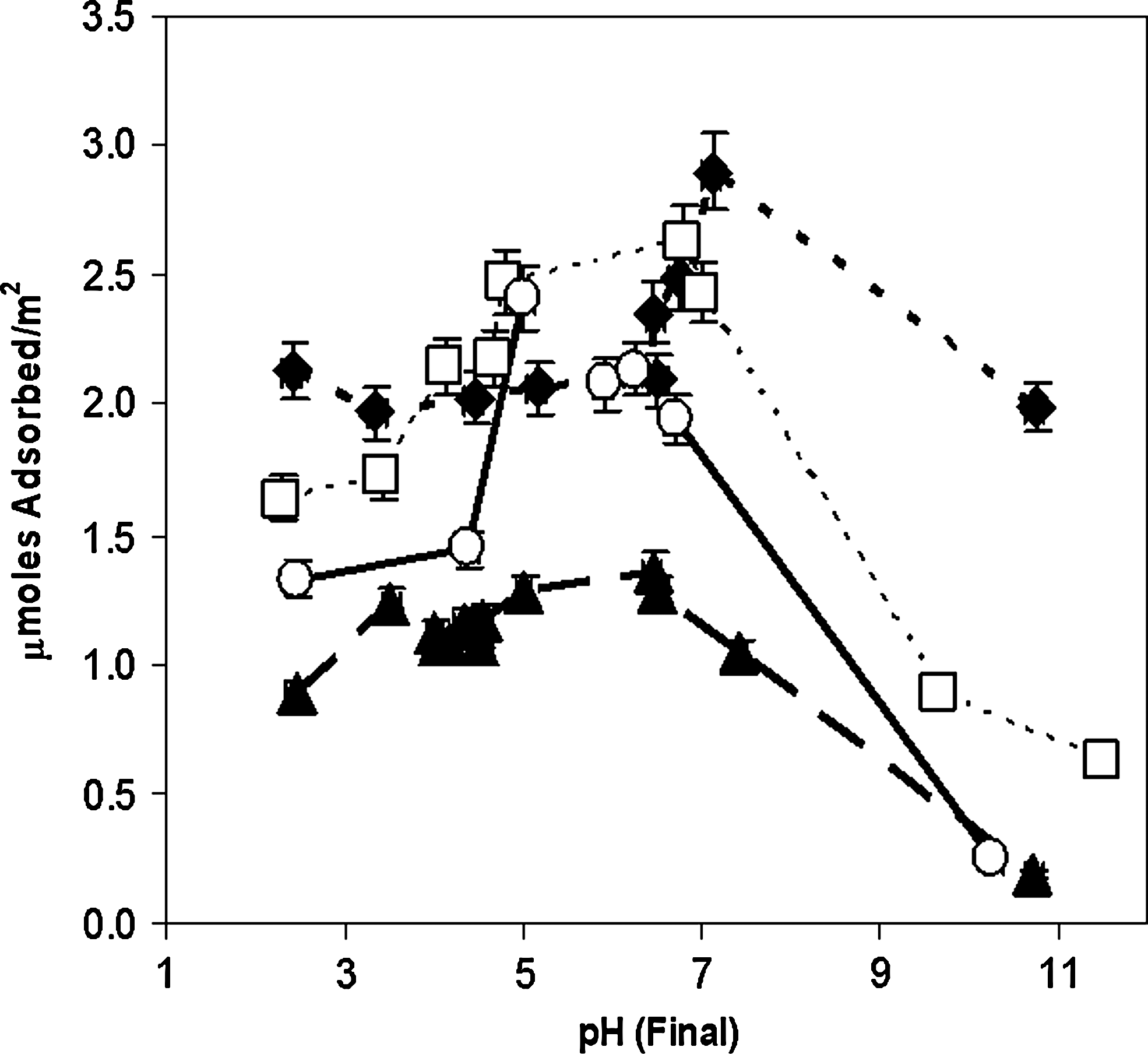
Adsorption vs. pH for ribonucleotides (10−4 M initial concentration) on rutile (6.67 g/L) in 0.1 M NaCl. Filled diamonds, GMP; filled triangles, AMP; open squares, CMP; open circles, UMP. Connecting lines are merely intended as guides for the eye.
The ribonucleotides were adsorbed more strongly than the deoxyribonucleosides, which suggests that the 2′ and 3′ hydroxyl groups and, particularly, the phosphate groups all play a role in binding. An ionic interaction involving the phosphate groups is also suggested by the apparent inflection in adsorption between ∼ pH 5 and 7. The differences in the binding of the individual species may then be attributed to interactions involving the base moieties. The increased similarity between all four species suggests that phosphate group interactions predominate for these species and that the synergism of phosphate group, ribose, and heterocycle C2 functional group interactions are all important, as evidenced by the relatively weaker adsorption of 5′AMP. This suggests a similar orientation of all four species on the surface.
For the sake of direct comparison, the pH adsorption profiles for cytosine derivatives, which present the most internally consistent data of the series investigated, are presented in Fig. 13.
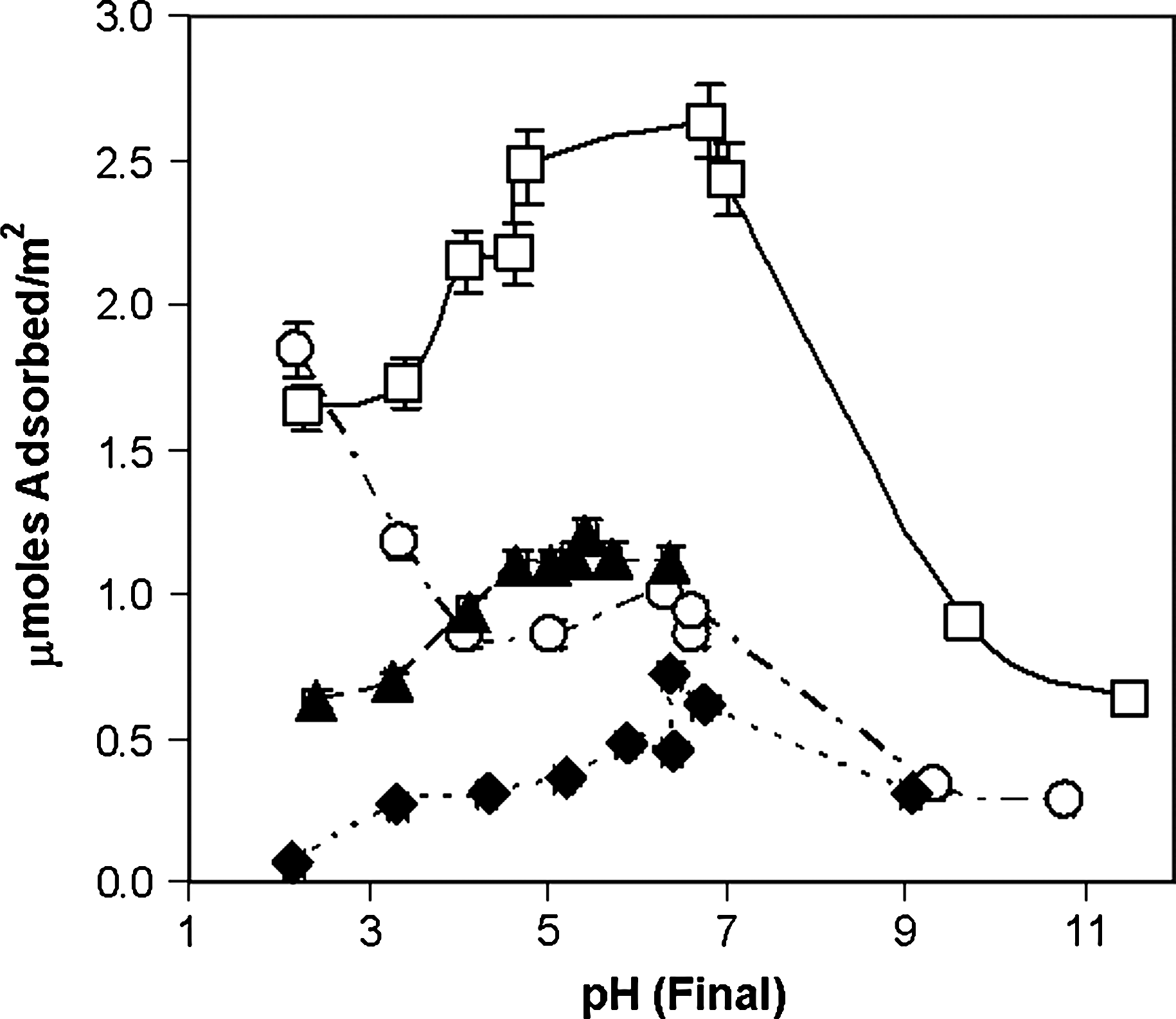
pH adsorption profiles for cytosine derivatives on rutile (6.67 g/L) in 0.1 M NaCl. Open squares, cytidine 5′-monophosphate; filled triangles, cytidine; open circles, cytosine; filled diamonds, 2′-deoxycytidine. Connecting lines are merely intended as guides for the eye.
It is interesting that cytidine-5′-monophosphate (CMP), cytidine, and deoxycytidine have similarly shaped adsorption profiles with respect to pH. The marked increase in binding afforded by the presence of the phosphate group argues for a direct role in adsorption for this group, which is in agreement with a wide range of studies in the literature. The increase in the binding of the riboside over the deoxyriboside argues for the involvement of the 2′-OH group in binding. The difference between the adsorption of cytosine, compared with its derivatives, especially at low pH values, suggests that the free nucleobase may adsorb in a fundamentally different manner.
The relatively low adsorption of the adenine derivatives compared to the other base derivatives (see, e.g., Figs. 8, 11, and 12) suggests that the functional group at the C2 position of the heterocyclic base interacts with the mineral surface. In the case of adenine, this group is a hydrogen atom, and the surface interaction is likely weak. Conformational restrictions of the ribo- and deoxyribonucleosides suggest that the exocyclic C6 substituents of the β-purine nucleoside derivatives and the exocyclic C4 substituents of the β-pyrimidine nucleoside derivatives are unable to interact directly with the surface if the 2′ and 3′ hydroxyl groups of the sugars interact, as suggested by the enhanced binding of the ribose derivatives versus the deoxyribose derivatives, except possibly at step or kink sites on the mineral surface (Fig. 14). This conformational restriction may not be true for α-ribosides and their derivatives (Fig. 14). The prebiotic synthesis of nucleosides from ribose and purines appears to produce both isomers in comparable yield (Fuller et al., 1972). Adsorption to a mineral surface may offer a method for environmental discrimination between these two products. This is an experimentally testable hypothesis.
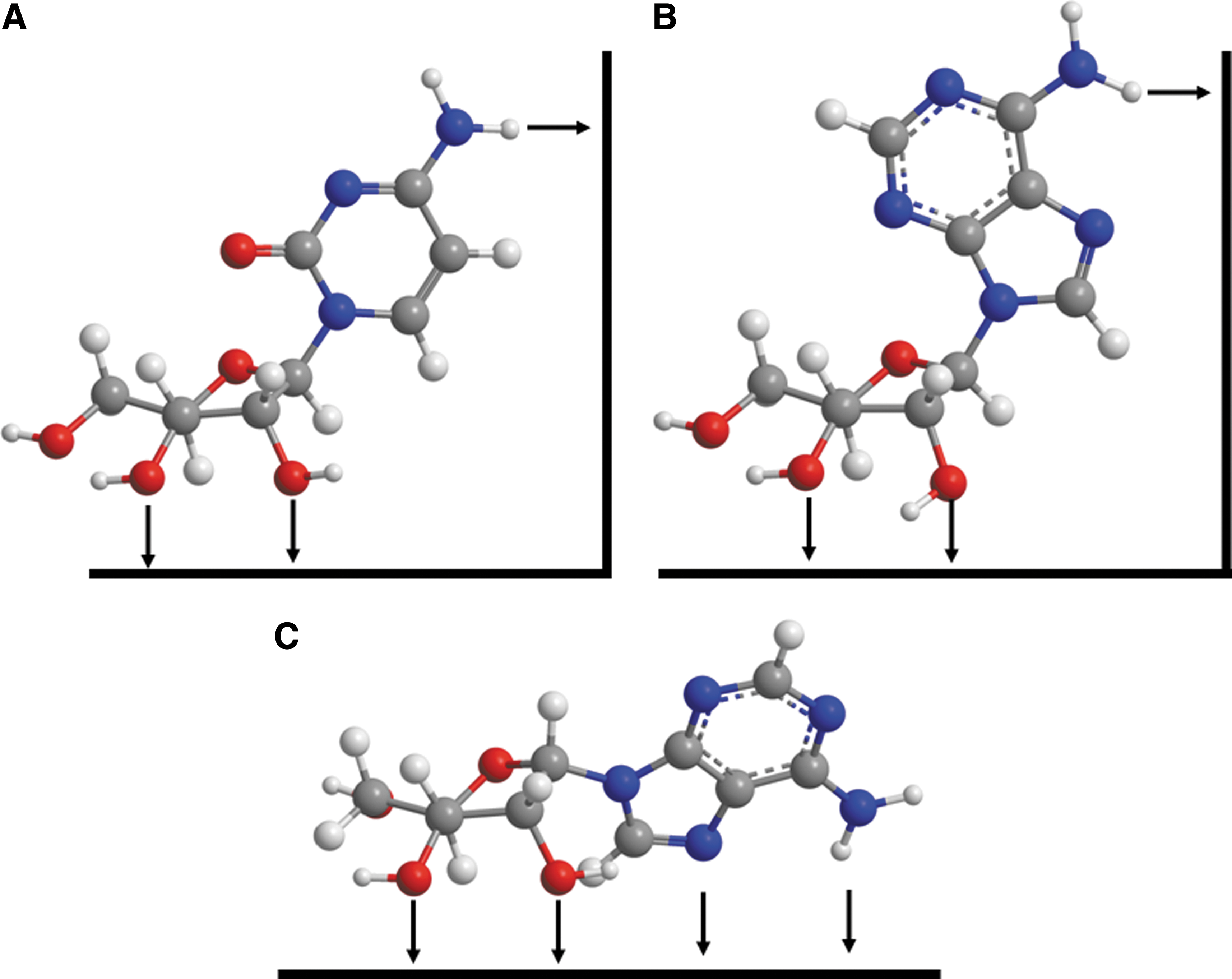
Adsorption of β-D cytidine (
Note that in Fig. 14 A and B, rotation about the nucleobase N1-ribose C1 bond cannot bring the substituents at the pyrimidine C4 position or purine C6 position into proximity with the same surface on which the ribose 2′ and 3′-OH groups interact, although the nucleobases' C2 substituents can interact with that surface. The pyrimidine C4 and purine C6 substituents could interact with stepped or kinked surfaces as shown (right-hand side). In the case of the α-nucleoside shown in C, both the N7 and N6 substituents appear to be able to interact with the surface. It is of course also possible that the molecules adsorb with the heterocyclic rings lying down parallel to the mineral surface.
The binding of the nucleotides in the pH range 4–9 can be explained by binding via hydroxyl groups and the phosphate group as shown in Fig. 15, via hydrogen bond acceptors or electrostatic interactions. The addition of the sugar moiety blocks the N9 and N1 positions of the purine and pyrimidine bases, respectively, so that they are unable to interact with the mineral surface. Rutile may present acceptor groups at an especially favorable spacing for these molecules, and this favorable surface geometry may be true for other metal oxides, many of which display similar surface spacing of oxygen atoms (e.g., Bragg et al., 1965; Downs and Hazen, 2004; Hazen, 2004).

Potential interaction of 5′GMP with the rutile mineral surface. See Fig. 14 caption for additional details. Phosphorous is shown in purple. Color images available online at
This geometry might also facilitate the interaction of adjacent monomers to polymerize on a surface, by positioning the reactive 3′ and 5′ groups for polymerization, and also might allow for the display of the Watson-Crick (WC) hydrogen bonding surfaces to the solution phase. Thus, binding to rutile surfaces might promote template-directed polymerization, as has been noted for activated nucleotides on clays (Ertem et al., 1989), iron oxides (Schwartz and Orgel, 1984), and apatite (Gibbs et al., 1980).
The TiO2 used here had a surface area of ∼2.5 m2/g; thus the isotherms suggest that there is generally less than a monolayer adsorbed even at the highest surface coverages, assuming a surface area of ∼1 nm2 per molecule for a nucleoside derivative (Orenberg et al., 1985). This suggests that there may not be enough surface sites per square meter to adsorb a theoretical monolayer via direct molecule-surface interactions. Surface interactions may be formed through inner-sphere or outer-sphere interactions. It has been suggested that the binding of organic molecules to mineral surfaces occurs principally via minimization of hydrophobic effects and optimization of covalent, ionic, and hydrogen bonding and van der Waals interactions (Stumm, 1992).
4. Discussion
Mineral surfaces can clearly concentrate nucleic acid components from dilute solution, which may have been important for prebiological chemical organization (Lahav and Chang, 1976; Nissenbaum, 1976). We have conducted a literature survey of the adsorption of various nucleic acid components with mineral surfaces for comparison with the results of this study (Fig. 16). It is difficult to compare adsorption isotherms for nucleic acid components between mineral types, because adsorption can vary dramatically as a function of pH, temperature, and background electrolytes, especially divalent and trivalent cations (Lailach et al., 1968), as well as the mineral surface characteristics. Few exhaustive studies have been carried out to allow for such comparisons among these compounds. Several studies have been conducted with ribonucleotides; however, surface area data are often lacking (Lahav and Chang, 1976) and therefore must be crudely estimated. The following discussion must therefore be considered somewhat preliminary.
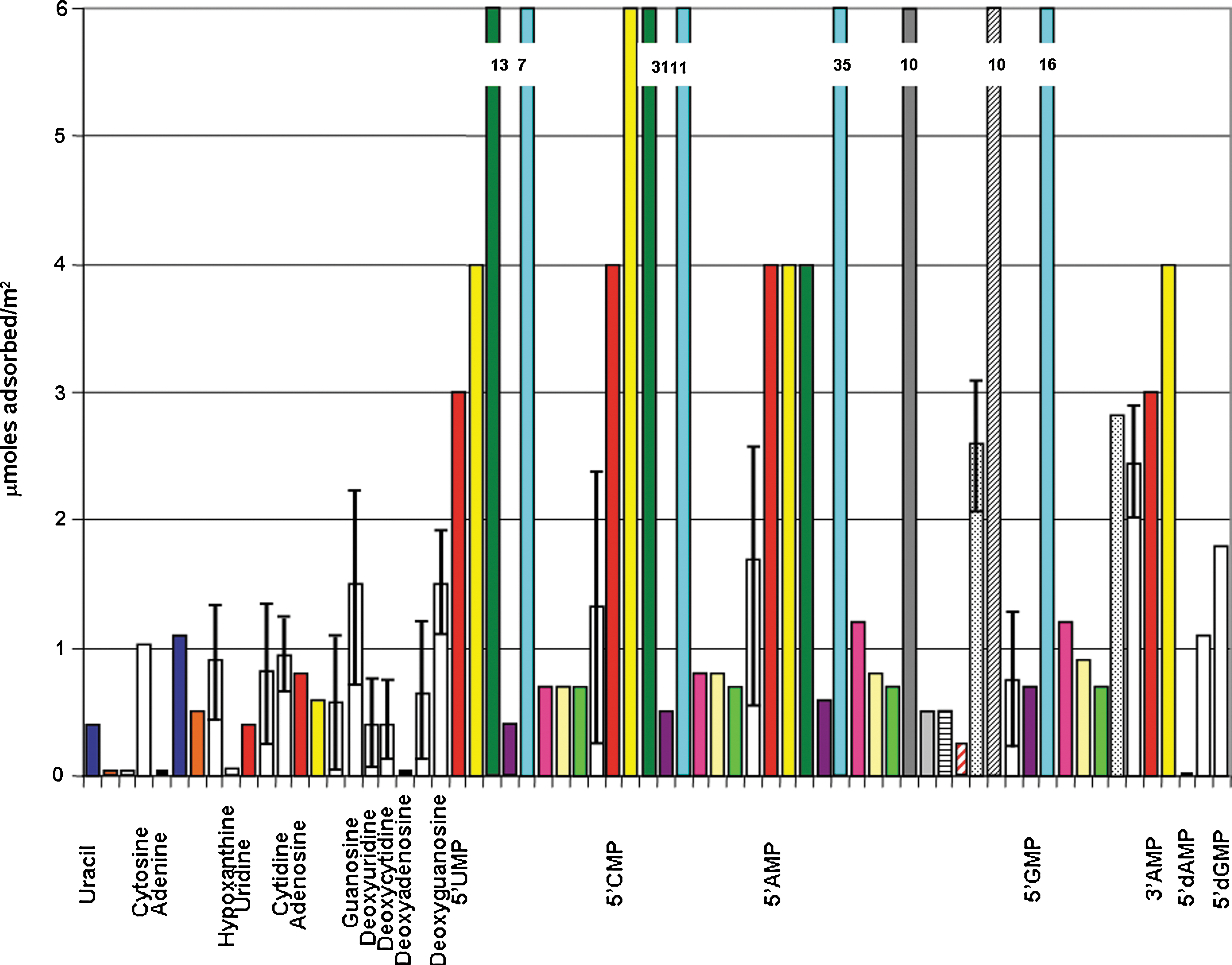
Maximum number of μmol adsorbed m−2 for this study compared with various reported literature values. Legend:
In previous studies of nucleobase adsorption to clays, it was suggested that cation exchange is the principal mechanism of adsorption, which explains well the adsorption of the bases, which are cations at low pH (Lailach and Brindley, 1969; Cortez and Schnitzer, 1981). For nucleosides, this may not be the case, as the adsorption is generally increased relative to the free nucleobases, and this effect is lowered slightly for the deoxyribosides. This difference suggests a contribution of the cationic forms of the adsorbed species in the overlapping region between the pHpzc of the mineral and the pK a of the bases at low to neutral pH, enhanced by the presence of the sugar hydroxyl groups. For nucleotides, the phosphate group may be involved in adsorption via anion exchange processes, and this effect is likely to be greatest near ∼ pH 4.5, close to the second pK a values of the phosphate groups (∼4.5) and the pHpzc of the surface.
It should be noted that the surface area was not directly measured in all the previous studies, and thus the maximal adsorption values could be considerably different than estimated here. Many mineral surfaces thus appear to have a binding capacity for nucleoside monophosphates and other nucleic acid components, which span a range of approximately 0.05–35 μmol/m2. Lahav and Chang (1976) conducted a survey of the adsorption of nucleobases on mineral surfaces and found adsorption constants in this range for a variety of mineral surfaces. It should be noted that there is often considerable disagreement between the measured values for the same molecules on the same surfaces under almost identical conditions, for example, the studies of nucleotides on goethite by Holm et al. (1993) and Arora et al. (2007), whose measured values differ by a factor of ∼7. Possible reasons for these differences include choice of buffer, different particle size, and differing methods of mineral preparation. There is also a considerable difference between different types of surfaces; for example, ZnO appears to have a much greater affinity than goethite for nucleotides. The trend that bases are adsorbed less strongly than nucleosides, which are in turn adsorbed less strongly than nucleotides, does not appear to hold true for all surface types for which this comparison has been attempted. These differences open the possibility that different mineral classes—for example, clays, oxides, and sulfides—present quite different surface characteristics for adsorption, and that different minerals within the same mineral class (e.g., iron oxides, ZnO, and rutile) do likewise. A systematic study of this phenomenon has unfortunately not been conducted.
Another possible source of adsorption variability may be related to initial molecular concentration. For example, Sverjensky et al. (2008) demonstrated that at lower concentrations glutamate tends to adopt a flat adsorption orientation on ferrihydroxide, whereas at high concentrations these molecules stand on end and, thus, pack the surface more closely. Similarly, uracil viewed face-on has a molecular cross section of ∼0.25 nm2, while 5′-GMP's value is ∼0.6 nm2. Assuming a molecular cross section of ∼0.5 nm2, a value of 10 μmol/m2 corresponds to slightly more than a monolayer, which suggests that at values of 10 μmol/m2 the molecules may not be laid down flat but rather adsorbed edge-on or bind cooperatively. Of course, the surface site density for different minerals may also be sparser than what would allow for the adsorption of a complete monolayer.
The general adsorption trend is bases < nucleosides < nucleotides near neutral pH for most minerals, but this sequence depends on pH and mineral type, in part due to the differing surface charge (Parks, 1965) and surface species properties of the different minerals (Stumm, 1992).
Graf and Lagaly (1980) suggested that adenosine-5′-monophosphate (AMP), ADP, and ATP interact with apatite surfaces principally through anion exchange processes, analogous to the way phosphate exchanges with TiO2 surfaces (Flaig-Baumann et al., 1970; Cornejo et al., 1978). Orenberg et al. (1985) suggested that AMP, CMP, and ATP interact with gypsum surfaces via the same mechanism. Lailach et al. (1968) and Ertem et al. (1989) suggested that the purines, cytosine, and their nucleosides and nucleotides are principally adsorbed to clays via cation exchange.
These considerations seem reasonable to a first-order approximation due to the relatively strong adsorption of nucleotides and deoxynucleotides relative to the nucleosides and deoxynucleosides. However, the increasing strength of adsorption of the series A < U < C < G suggests that there may be interactions involving the base moiety as well. Notably, A has only a hydrogen atom at the 2-position of the purine ring, while U and C have an oxygen atom and G has an amino group, which are likely able to interact more strongly with the surface than the hydrogen atom.
The deoxynucleotides dGMP and dAMP generally adsorb slightly less strongly than the ribonucleotides, which suggests that there is a contribution by the 2′OH group of the sugar moiety. This suggests binding as shown in Fig. 15.
In contrast, Arora et al. (2007) and Arora and Kamaluddin (2007) suggested that, in addition to phosphate group interactions, the purine nucleotides interact via the N1 and N7 positions and the pyrimidine nucleotides via the N3 position. Infrared spectra were offered as evidence for this N involvement. Such an orientation would not allow for the exposure of the WC hydrogen bonding surfaces of the base moieties. However, it has been noted that the adsorption of polycytidine on iron oxides has no effect on the template polymerization of 5′ImpG (Schwartz and Orgel, 1984), which suggests that the WC sites of the adsorbed template are not blocked by adsorption to the mineral surface. Also, it would be physically impossible for both the N1 and N7 positions of the purines to interact simultaneously with the mineral surface except perhaps at step or kink sites (Fig. 14). Although it would be feasible for both the phosphate group and either the N1 or N7 groups of β-D-purine nucleotides to interact with a planar surface simultaneously, which might allow the 2′ and/or 3′OH groups of the ribose moiety to be solvated, it would prevent their interaction with the surface, and the ribosides and ribotides appear to interact more strongly than their 2′-deoxy analogues. Cis-diols have been found to chelate various inorganic species (Kolb and Zhu, 2004; Lambert et al., 2004; Shkrob et al., 2004). Lailach and Brindley (1969) noted that pyrimidines could be co-adsorbed with purines onto clay surfaces under conditions in which the pyrimidines alone did not adsorb, which suggests specific, possibly WC-type, interactions. This further suggests that, even for the nucleobases by themselves, the WC surfaces of these molecules are not involved in surface binding. This remains to be demonstrated for monomers on metal oxide surfaces.
Holm et al. (1993) noted the cooperative adsorption of adenosine to polyuridine adsorbed to goethite and akaganeite. Gibbs and coworkers (1980) observed WC hydrogen bonding of polyadenosine to polyuridine bound on apatite, although oligonucleotides may bind differently than monomeric nucleoside monophosphates. Ferris et al. (1989) showed that polyuridine and polycytidine allow interactions with monomers when bound to clays, which suggests that the WC faces are not blocked by adsorption.
It is possible that the bases, nucleosides, and nucleotides all bind to mineral surfaces in different orientations. These differences could be a positive phenomenon for chemical evolution and the origin of the RNA world. Prebiotic syntheses of the purine nucleosides have been shown to result in the equal synthesis of both α- and β-anomers of the nucleosides (Fuller et al., 1972). Based on the data presented here, we cannot determine whether rutile surfaces would preferentially select β- versus α-ribonucleotides; however, there could be a significant difference. Bielski and Tencer (2007) proposed that the regiochemistry of sugars may have been a factor for the selection of ribose among the numerous possible sugar isomers. It may be preferable from the standpoint of prebiotic chemistry for the N1 and N9 positions of the pyrimidine and purine bases, respectively, not to be directly involved in surface adsorption so that they may react to form nucleosides. α-Ribosides could have different interactions with mineral surfaces, which could lower their reactivity to both oligomerization and template-directed polymerization. Joshi et al. (2007) found that montmorillonite could selectively orient mixed isomers of β-
In summary, we found there are profound differences in the adsorption of nucleic acid bases, nucleosides, and nucleotides on rutile surfaces. There are clearly base-, sugar- and phosphate-dependant interactions that mediate adsorption, and these each show pH-dependant behavior, consistent with multiple modes of interaction. The inferred nature of these interactions, at least for the nucleosides and nucleotides, appears to be consistent with the display of the WC hydrogen bonding surfaces of the base moieties, which would allow them to engage in molecular recognition of their complements. It is possible that these differences in adsorption could influence the relative ratios of bases in oligonucleotides synthesized abiotically from activated nucleotides on a surface, if a plausible prebiotic mechanism were available. This effect has been observed both on surfaces and in eutectic solution (Monnard et al., 2003; Ferris, 2006) but would of course depend on the relative amounts of the different precursors available in the environment and the mechanism of activation.
The adsorption isotherms are, with few exceptions, comparable on a μmol adsorbed/m2 and phenomenological basis with previous studies of the adsorption of nucleic acid derivatives with other mineral surfaces, which suggests that nucleic acid component adsorption to mineral surfaces may be somewhat generalizable and is mediated by both ion exchange and weak interactions.
The disparity between these results and those of Arora et al. (2007), and the ultimate resolution of how these compounds adsorb to mineral surfaces, should be resolved by spectroscopic studies or adsorption studies with analogues blocked at the appropriate positions, such as O-methyl ribosides or ribotides and N 3-methyl pyrimidine and N 1/N 7-methyl purine ribosides or ribotides, pyrimidine and purine nucleosides and nucleotides without exocyclic groups on the heterocyclic rings, and dideoxynucleosides and nucleotides.
5. Conclusions
Adsorption experiment results suggest that there is considerable difference in the adsorption of the nucleobases and their derivatives on rutile surfaces, dependant on the molecular architecture of the adsorbing species. The degree of adsorption is comparable to that determined previously for other metal oxide surfaces, gypsum, apatite, and clays; however, the results presented here are consistent with adsorption principally through the phosphate, sugar, and base portions of the molecules in an orientation that leaves the WC surfaces of the bases largely exposed to solution, which could facilitate template-directed oligomerization.
Surface adsorption for even relatively simple species such as oxyanions or metal cations (Davis and Hayes, 1986; Stumm, 1992), or amino acids (Sverjensky et al., 2008) can exhibit complex behavior. It should not be particularly surprising that more complex species, especially compounds such as nucleic acid components that have complex geometries and multiple functional groups and have been selected during biochemical evolution for their self-associative properties, should have considerably more complicated adsorption properties. Nevertheless, there may be experimentally discernible adsorption behavior trends that may have assisted in prebiotic self-organization.
Biological evolution has selected its constituents for use based on a number of criteria. Minerals may have played a role in this selection. Perhaps the most intriguing possibility is that minerals have fostered the selection of certain molecular characteristics that may be reflected in modern biochemistry.
Footnotes
Acknowledgments
We gratefully acknowledge support from the national Science Foundation, NASA Astrobiology Institute, and the Carnegie Institution of Washington. No competing financial interests exist.
Abbreviations
A, adenine; AMP, adenosine-5′-monophosphate; CMP, cytidine-5′-monophosphate; C, cytosine; G, guanine; GMP, guanosine-5′-monophosphate; HX, hypoxanthine; U, uracil; UMP, uridine-5′-monophosphate; WC, Watson-Crick.