
Abstract
We propose that the paucity of organic compounds in martian soil can be accounted for by efficient photocatalytic decomposition of carboxylated molecules due to the occurrence of the photo-Kolbe reaction at the surface of particulate iron(III) oxides that are abundant in the martian regolith. This photoreaction is initiated by the absorption of UVA light, and it readily occurs even at low temperature. The decarboxylation is observed for miscellaneous organic carboxylates, including the nonvolatile products of kerogen oxidation (that are currently thought to accumulate in the soil) as well as α-amino acids and peptides. Our study indicates that there may be no “safe haven” for these organic compounds on Mars; oxidation by reactive radicals, such as hydroxyl, is concerted with photocatalytic reactions on the oxide particles. Acting together, these two mechanisms result in mineralization of the organic component. The photooxidation of acetate (the terminal product of radical oxidation of the aliphatic component of kerogen) on the iron(III) oxides results in the formation of methane; this reaction may account for seasonably variable production of methane on Mars. The concomitant reduction of Fe(III) in the regolith leads to the formation of highly soluble ferrous ions that contribute to weathering of the soil particles. Key Words: Mars—Methane—Biopolymers—Catalysis—UV radiation. Astrobiology 10, 425–436.
1. Introduction
T
Martian regolith partly consists of submicron metal [mainly, iron(III)] oxide particles (Banin et al., 1993; Rieder et al., 1997; Morris et al., 2000, 2004; Christensen et al., 2004; Herkenhoff et al., 2004; Klingelhöfer et al., 2004, 2005, 2007). There were previous suggestions (Chun et al., 1978; Quinn and Zent, 1999) that the combination of the high flux of UV light through the thin martian atmosphere (Cockell et al., 2000; Patel et al., 2003; Ronto et al., 2003; Schuerger et al., 2003) and metal oxide photocatalysts turned the surface of this planet into a vast photoreactor.
Even if there was no carbon-based life on Mars, the surface of the planet has been bombarded by meteorites, micrometeorites, and comets for billions of years, and the significant fraction of this infalling material consists of carbonaceous chondrites (Benner et al., 2000; Schuerger and Clark, 2008). These include complex organic molecules, mainly in the form of a bitumen-like compound, kerogen. The latter is a cross-linked polymer that consists of polycyclic aromatic hydrocarbons (PAHs) and aliphatic chains (Anders, 1991; Henning and Salama, 1998; Benner et al., 2000). It is expected that ∼25% of the infalling micrometeoritic dust reaches the ground, so the carbon infall is estimated to be 2.4 × 108 g/year (Formisano et al., 2004; Atreya et al., 2007). Through the ages, this bombardment should have enriched the martian soil with organic carbon. Contrary to these expectations, the Viking probes sent to Mars in 1976 found no traces (<1 ppb) of organic molecules in the upper soil, which initiated the controversy about martian soil chemistry (Oyama et al., 1978; Biemann, 1979, 2007; Huguenin et al., 1979; Hunten, 1979; Oro and Holzer, 1979; Mancinelli, 1989; McDonald et al., 1998; Bullock et al., 1994; Schuerger and Clark, 2008).
In view of the low oxygen concentration in the atmosphere (0.13%) and scarcity of water on Mars, the paucity of organic material implied that the soil itself is oxidizing. The latter is also suggested by the fact that the surface of Mars is red, as it contains ferric oxides (0.2–0.4 Fe3+/Fetotal) (Rieder et al., 1997; Christensen et al., 2004; Klingelhöfer et al., 2004; Morris et al., 2004) in the form of fine (0.1–5 μm) spherules and particles of hematite (α-Fe2O3), goethite (α-FeOOH) (Klingelhöfer et al., 2005, 2007), magnetite (Fe3O4), and jarosite [KFe3(OH)6(SO4)2]. These oxides are mainly present as nanoparticles and the composite aggregates of such nanoparticles. The soil also contains ∼1% of TiO2, ∼10% of Al2O3, and ∼60% SiO2 (Rieder et al., 1997).
How do the ferric ions in these oxides originate on a planet that is devoid of an oxidizing atmosphere? The hypothesized presence of liquid water on Mars in the distant past and (episodically) at present does not offer answers to this question, as the ferric ores (“band iron”) on the ocean-covered early Earth were produced through the oxidation of aqueous Fe2+ when the terrestrial oceans were biogenically oxygenated 1–2 billion years ago. Thus, in addition to the need to explain the degradation of the organic material, it is also necessary to find a mechanism that accounts for the oxidation and extreme weathering and dispersion of the soil particles. The current thinking is that the oxidation is caused by corrosive gases released during episodic, large-scale impacts (e.g., Zolotov and Mironenko, 2007).
Until a few years ago, the leading candidate for the oxidizer in the martian soil was H2O2 (Oro and Holzer, 1979; Stoker and Bullock, 1997). However, recent measurements of atmospheric mixing ratios (Encrenaz et al., 2004; Atreya et al., 2007) suggest otherwise, as the concentration is too low (20–40 ppbv) to explain the Viking probe data (requiring >250 ppm in the soil). This result is not surprising, as H2O2 is photolytically unstable in the thin martian atmosphere, which transmits solar radiation down to 190 nm, with a peak in the UVA region (300–400 nm).
These and subsequent measurements have revealed another mystery: the martian atmosphere contains a substantial (10 ± 5 ppbv) concentration of methane. As the latter is photolytically unstable (with a half-life that can be as short as 200 days under martian conditions), this gas must be constantly generated (Formisano et al., 2004; Atreya et al., 2007; Lefevre and Forget, 2009; Mumma et al., 2009). This methane is released in large plumes, which peak during martian summer (Mumma et al., 2009). So it is not just oxidation; there is also an unknown process that generates the methane. Volcanic venting of the methane is unlikely, whereas hydrogeochemical reactions (such as serpentinization of basalt) require high-temperature hydrolysis in the interior of a geologically inactive planet (Atreya et al., 2007). Even if such high-temperature reactions do occur deep underground, it is difficult to explain the seasonal variations of the release (Krasnopolsky, 2006; Atreya et al., 2007; Mumma et al., 2009). Two recent studies imply that there is another unrecognized process responsible for the rapid removal of the methane, as atmospheric photochemistry alone cannot account for its rapid disappearance (Lefevre and Forget, 2009; Mumma et al., 2009). A logical explanation of the observations with regard to methane is that it is both generated and consumed by the soil itself.
Our goal is to demonstrate a surface photoreaction that can produce methane under realistic martian conditions. This photocatalytic process can complement other abiogenic and biogenic paths to methane generation, such as outgassing from comet and asteroid impacts (Sekine et al., 2003; Kress and McKay, 2004), interplanetary dust particles (Atreya et al., 2007), and subsurface methane clathrates (Duxbury et al., 2004; Chastain and Chevier, 2007; Thomas et al., 2009); subsurface serpentinization of olivine (Lyons et al., 2005; Oze and Sharma, 2005); UV photolysis of water in the presence of carbon monoxide (Bar-Nun and Dimitrov, 2006); geothermal outgassing (Atreya et al., 2007); tentative biological processes (Krasnopolsky et al., 2004; Krasnopolsky, 2006; Atreya et al., 2007); and direct (that is, uncatalyzed) UV photodegradation of organic compounds in the soil (Stoker and Bullock, 1997).
Photocatalysis by metal oxides (TiO2, γ-Fe2O3 and δ-FeOOH), with or without the presence of H2O2 (Chun et al., 1978; Mancinelli, 1989; Zent and McKay, 1994; Quinn and Zent, 1999); oxidation by smectite clays and various peroxides (KO2, ZnO2, CaO2, and MnO2); reactive radicals and molecules such as ONOO−,
One possible resolution of the problem of the missing organic molecules on Mars may be that these molecules are not missing. It has been suggested (Benner et al., 2000; Navarro-Gonzalez et al., 2006; Stalport et al., 2009) that the organics are, in fact, still present in the martian soil; the aliphatic component of the kerogen is oxidized to carboxylates, such as acetate (that forms nonvolatile salts with metal ions, including Fe3+), whereas the aromatic hydrocarbons are oxidized to polycarboxylated benzenes, such as mellitic (graphitic) acid, which also form nonvolatile salts. Such carboxylated and polycarboxylated compounds are assumed to be stable to further radical oxidation. (Stepwise oxidation is carried by OH radicals and, possibly, other O-centered radicals generated by UV photodissociation and photoionization of water and ice trapped on the regolith particles.) As such, these carboxylated compounds may serve as a reservoir of the organic carbon that was missed by the Viking probes. It was shown that heating of Fe2+ phthalate, mellitate, and benzene-1,2,4-tricarboxylate did not produce volatile products identifiable by mass spectrometry with the Viking protocols (Benner et al., 2000), although this statement has been disputed (Biemann, 2007). Since the flux of shortwave UV photons with λ < 190 nm that can photodissociate water is low at the surface (the UV environment on Mars is extensively discussed by Cockell et al., 2000; Patel et al., 2003; Schuerger et al., 2003; Moores et al., 2007; among others), this scenario requires elevation of the soil particles to high altitudes by strong winds, where these particles are exposed to shortwave UV radiation or other means for generation of O-centered radicals, such as the electric discharge in dust devils, that might also be producing hydrogen peroxide “snow” (Atreya et al., 2006b; Delory et al., 2006).
2. Photocatalysis
In this study, we examine the possible role of photocatalysis on Mars. Our examination is based on our recent study (Shkrob and Chemerisov, 2009) in which the photoinduced catalytic oxidation of aliphatic and aromatic carboxylated and polycarboxylated compounds, including L-α-amino acids and peptides, were systematically examined with electron paramagnetic resonance (EPR) spectroscopy, a technique that allows for detection and identification of reaction intermediates that have unpaired electrons. As the metal oxides are semiconductors, the photoexcitation of these materials results in charge separation, which yields trapped electrons (e−) and electron deficiencies called holes (h+). The important feature of the photocatalysis is that the photochemistry on the oxide surfaces does not depend on the photon wavelength. Above the optical gap, the photons are strongly absorbed by the oxide material rather than the organic substrate at the surface, and the excess photon energy is dissipated in <100 fs, as the photoinduced charges thermalize, migrate, and descend into internal and surface traps (see, e.g., Linsebigler et al., 1996; Shkrob and Sauer, 2004). For this reason, the photochemistry of organic molecules adsorbed on the oxide particles does not change as long as the photon energy exceeds the optical band gap of the semiconducting oxide material [which is ∼3 eV for the anatase and ∼2.5 eV for iron(III) oxides], whereas the photochemical yield scales with the photon absorption cross section of the material rather than that for the adsorbate. For this reason, we chose the photon energy of 3.5 eV (355 nm), which is well above the optical gap energy for these oxide materials (so the light absorption is entirely dominated by the oxides) and falls into the middle of the UVA band corresponding to the maximum in the UV photon flux at the ground level (Ronto et al., 2003; Schuerger and Clark, 2008). This single-light wavelength is fully representative of the photochemistry across the entire region of phototransparency of the martian atmosphere where it overlaps with the absorption spectra of the oxide materials (for the latter, see Fig. 1S in Shkrob and Chemerisov, 2009).
The photogenerated free electrons in the semiconducting oxides are trapped either by the crystalline interior (lattice electrons) or structural defects at the interface (surface electrons). Regardless of the location, such centers are the electrons in d orbitals of the transition metal ion. For example, in TiO2, the electron centers are Ti3+ ions; in Fe3+ oxides, these are Fe2+ ions. As the Ti3+ ions are paramagnetic, these can be readily observed with EPR spectroscopy; different crystal fields around the interior and surface centers allow them to be distinguished. While isolated Fe3+ ions are paramagnetic, the spins of these ferric ions in hematite and goethite are paired, so there is no spin resonance from the non-irradiated oxides. The Fe2+ electron centers in the Fe3+ oxides are EPR silent, but paramagnetic Fe3+ ions that are isolated from the neighboring Fe3+ ions by the reduced Fe2+ ions at the surface can be observed with EPR spectroscopy (Fig. 1). The trapped-hole species, by contrast, are unpaired electrons in the low-energy p orbitals of the oxygen atoms (the so-called oxygen hole centers). These form only at the surface; and, as such, are sensitive to the state of the surface, the presence of defects, hydration, and surface modification by adsorbed molecules. Some of these hole centers can be thought of as trapped OH radicals formed in the reactions of energetic holes with water molecules adsorbed at the surface. These OH radicals are released into the bulk and react with the molecules therein. The quantum yield of the OH radicals is low (<10−2), but these OH radicals are very reactive, and the quantum yield increases when H2O2 is present in solution. By contrast, charge transfer from the organic adsorbate to the surface hole centers can be extremely rapid and efficient (with nearly unit quantum yield) even at low temperature (4–100 K), as these hole centers are strong oxidizers. This charge transfer competes with the recombination of the trapped holes and electrons. Following the charge transfer, the resulting unpaired electron species can recombine with the electron trapped on the same oxide particle; in this case, the original molecule is regenerated. Alternatively, a fragmentation reaction near the heteroatom, such as deprotonation (X = N, O), may occur
(
The resulting radical RX· does not have the sufficient electron affinity to recombine with the trapped electrons (as this charge transfer reaction needs to be strongly exergonic to proceed). In this case, the fragment radical can desorb, undergo further chemical transformations, or both. Another fragmentation is the photo-Kolbe reaction involving the carboxylate anions at the oxide surface
This photoreaction involves (poly)carboxylated molecules that are chemisorbed as μ-O2 complex at the oxide surface. The energetics of Reaction 2 are mainly determined by the stability of the intermediate species. When the residue R in Equation 2 is an aliphatic group, Reaction 2 is strongly exergonic (0.5–1 eV); but, for aromatic carboxylates, the outcome is structure specific. For monocarboxylated benzenes, such as benzoic acid, the reaction is endergonic, and fragmentation does not occur. For di- and polycarboxylated acids, including mellitic acid, the reaction is exergonic and leads to the elimination of CO2. Below, we demonstrate that photoreactions 1 and 2 occur on the surfaces of hematite and goethite particles.
3. Experimental
We outline only general aspects of the experimental approach, as the full details of the method have been given in a previous publication (Shkrob and Chemerisov, 2009).
Anatase nanoparticles were synthesized by hydrolysis of TiCl4 and stabilized in pH = 3 solution (Shkrob and Sauer, 2004). Hematite nanoparticles were prepared by hydrothermal oxidation of FeCl3 (Chen et al., 2002). These nanoparticles (30–50 nm) were stabilized at pH = 4. Goethite particles were synthesized by iron(III) nitrate hydrolysis at ∼320 K (50°C) for three days at pH = 2 and stabilized at pH = 6 (Varanda et al., 2002). The particles ranged in size from individual particles of 50 nm to the agglomerates of 0.2–1 μm, as evidenced by light-scattering measurements and electron microscopy. All other chemicals were obtained from Sigma-Aldrich and used without further purification.
The aqueous solutions/suspensions were irradiated with 6 ns laser pulses from the third harmonic of a Nd:YAG laser. N2-purged aqueous solutions containing the adsorbate were flash frozen at 77 K in Suprasil tubes and subsequently irradiated with 355 nm laser light (15 mJ/pulse, 100–1000 pulses). We stress that the organic molecules that we studied do not absorb at this wavelength, so the UVA light was absorbed by the oxide particles (Shkrob and Chemerisov, 2009). The irradiated samples were transferred into a cryostat of a continuous-wave EPR spectrometer operating at 9.45 GHz, and the radicals and spin centers were detected in situ at 50–200 K (representative spectra obtained at different temperatures are shown in Figs. 2b and 3b of Shkrob and Chemerisov, 2009). First-derivative EPR spectra were taken at several temperatures, microwave power levels (typically, 0.2 mW), and field modulation amplitudes (typically, 2–5 G); the field modulation frequency was 100 kHz. The UV irradiation temperature of 77 K was chosen for convenience, as it allowed irradiation of samples immersed in liquid nitrogen as opposed to a He flow cryostat. In the temperature interval between 25 and 90 K, the relative yield of radical products of the photooxidation does not depend on sample temperature, and the radicals are extremely stable; above 90 K, the primary radicals become mobile and slowly convert to secondary radicals. To observe the latter process, the samples irradiated at low temperature (either in situ or in an external cryostat) were transferred into an EPR cavity, and then the temperature was slowly increased in steps of 5 K until the radicals decayed. In situ irradiation at temperature > 100 K yields mainly or exclusively secondary radicals.

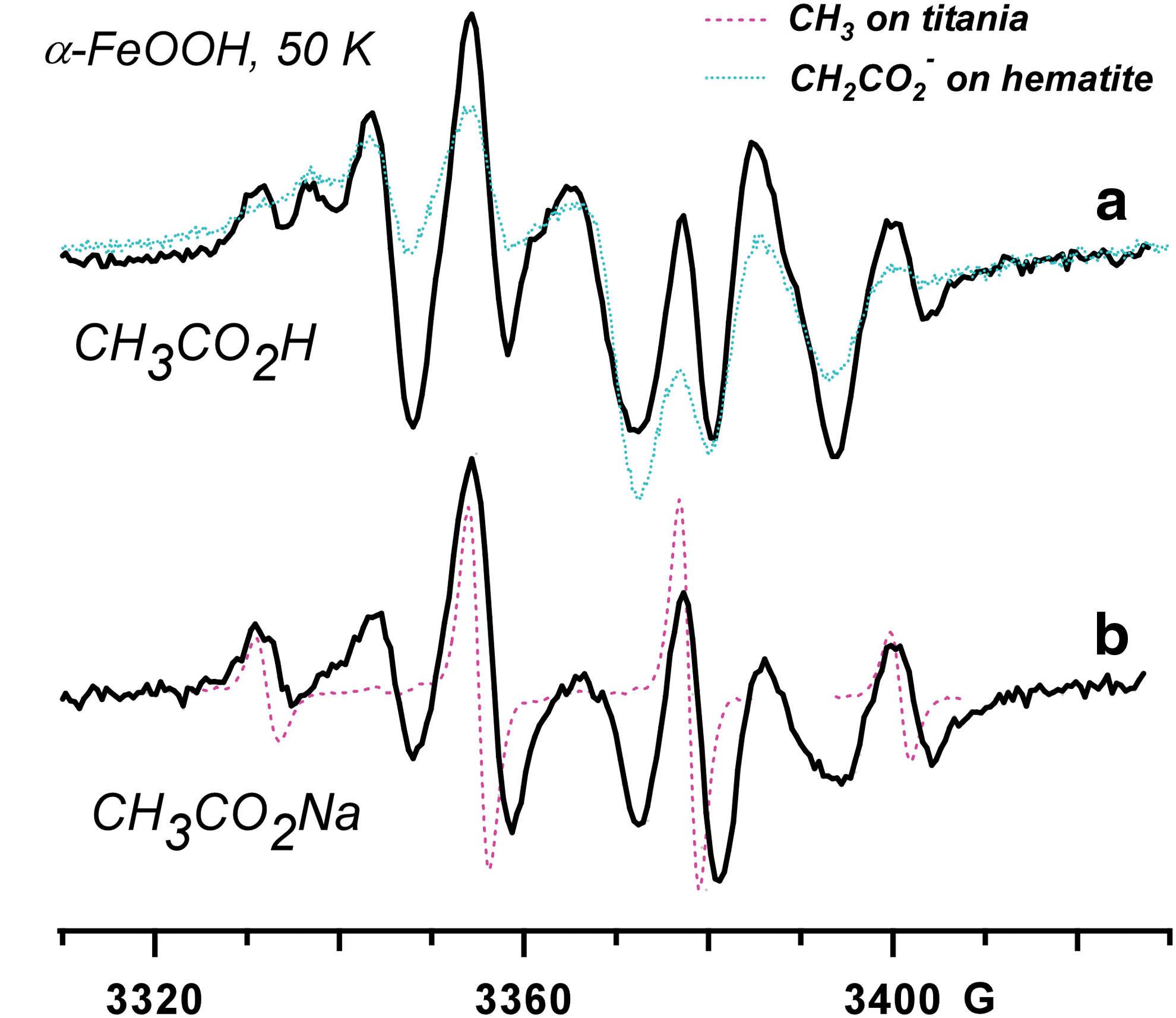
Inset: EPR spectra observed in 355 nm photolysis of 1 M acetic acid (solid line) and 0.1 M sodium acetate (dotted line) on hematite nanoparticles (aqueous solutions). The EPR spectrum from the methyl radical (dashed line) is superimposed on the trace, and the arrows indicate the resonance lines from carboxymethyl,
EPR spectra from photoirradiated (
The standard observation temperature of 50 K was chosen for convenience, too, as the photogenerated radicals are stable for many hours at this temperature; yet it is sufficient for rotational averaging of hyperfine tensors, which greatly simplifies identification of EPR signatures for complex organic radicals. The number of laser pulses used in the photolysis was determined by EPR sensitivity. There was no evidence for secondary product photolysis, as the radical yield increased with the UV dose without changing the EPR spectrum. This behavior is fully expected in that the photons are absorbed by the oxide material rather than the organic adsorbates. The latter yielded no organic radicals in photolysis with 355 nm light, as these molecules do not absorb UVA light directly (with the exception of carboxylated PAH derivatives that yield radicals at the peak laser power due to biphotonic processes resulting in the ionization of these molecules). Such direct biphotonic reactions are suppressed in the oxide solutions and suspensions due to the strong 1-photon absorption of the oxides (the optical density across the samples was 0.2–1), and the general appearance of the EPR spectra does not change as a function of the photon flux for such samples.
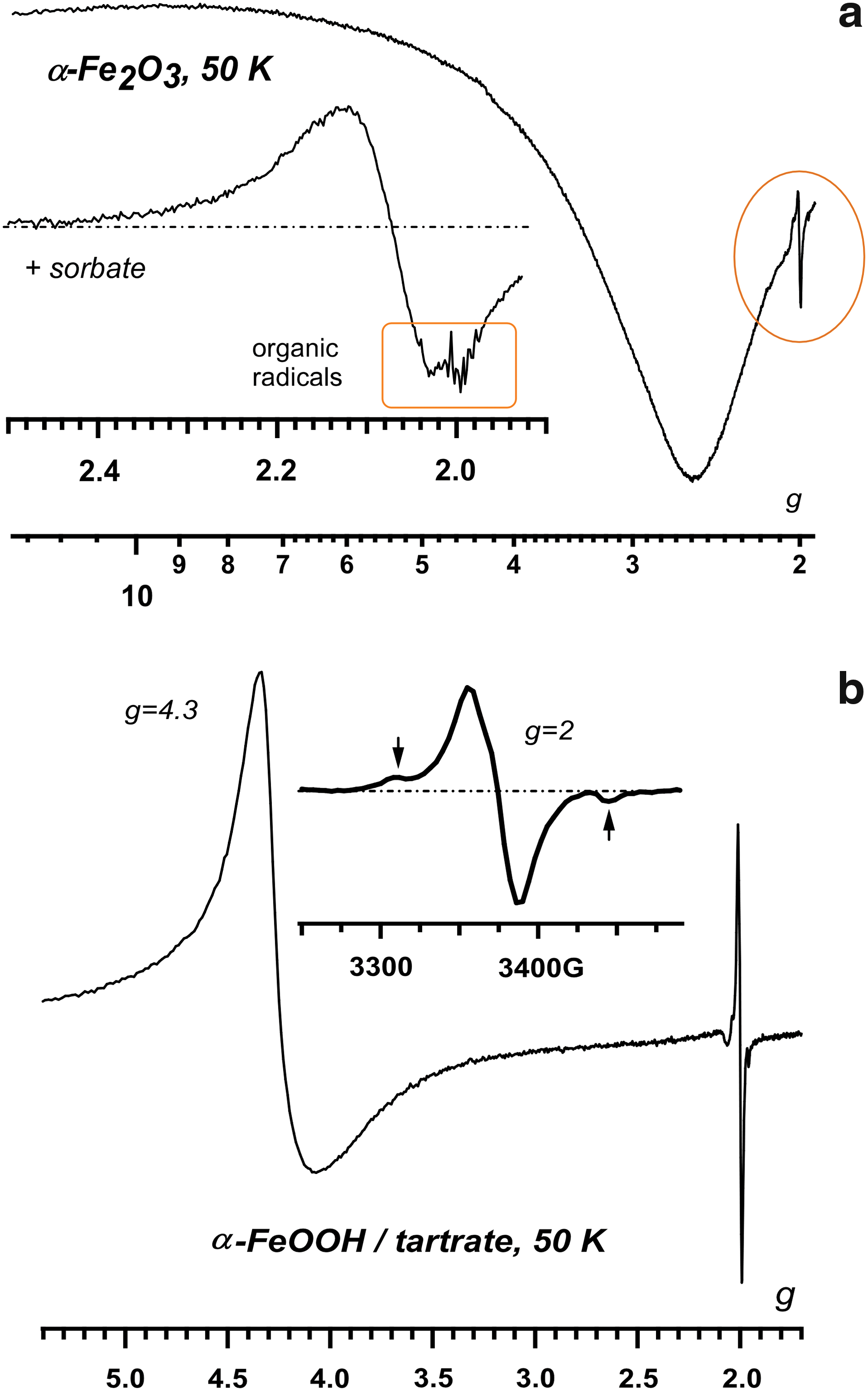
4. Results
Ultraviolet A–irradiated hematite and goethite particles yielded three types of EPR-active species: (i) the broad resonance signal from isolated ferric ions (with the g-factor ∼4.0–4.3) indicative of the magnetically decoupled Fe3+ ions, (ii) a broad signal at g ∼ 2.0 from trapped holes, and (iii) in the presence of the organic adsorbate, the resonance lines from organic radicals (Fig. 1). The latter are often superimposed on the broad and unresolved resonance signal from the hole centers (Fig. 1a). In goethite, there is an additional unassigned doublet, as shown in the inset of Fig. 1b. For some adsorbates, Reactions 1 or 2, or both, were so efficient that the resonance signal from unreacted hole centers is missing from the EPR spectra. In other systems (Fig. 1a), the two resonance signals overlap, which indicates that some of the holes did not react with the adsorbate.
The most important of these adsorbates is the acetate, which is thought to be the main product for the stepwise oxidation of the aliphatic component of the kerogen (Benner et al., 2000). Figure 2 demonstrates the EPR spectrum for UVA-irradiated acetic acid on the hematite; very similar spectra were generated for the acetate. The EPR spectrum is the superposition of the resonance lines from the methyl (·CH3) and carboxymethyl
Similar photo-Kolbe reactions occur for other carboxylic, hydroxycarboxylic, and polycarboxylic compounds (Fig. 4). In all cases studied (Shkrob and Chemerisov, 2009), Reaction 2 resulted in the formation of the corresponding C-centered radical and CO2 molecule. The appearance of these radicals is the same on all three oxides, except for the broader resonance lines observed on the iron(III) oxides due to a more hindered rotation.
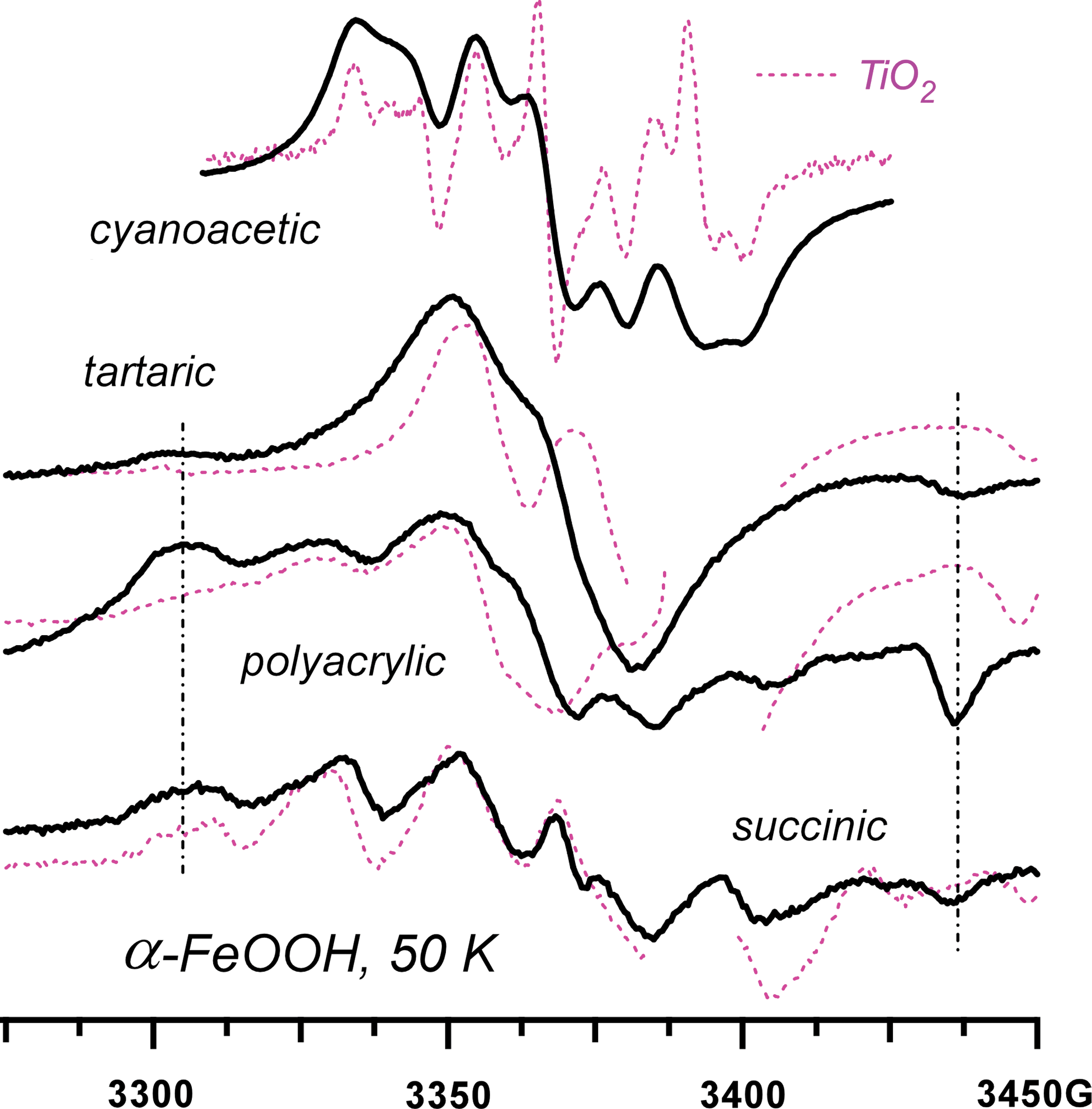
EPR spectra from the radicals generated via photoinduced decarboxylation of cyanoacetic, tartaric, polyacrylic, and succinic acids in the aqueous suspensions of goethite microparticles (solid lines) and solutions of anatase nanoparticles (dashed lines). The vertical dash-dot lines indicate the 120 G doublet shown in Fig. 1b. In the dashed traces, the resonance lines from the trapped-electron centers are not shown. Color images available online at
It has been suggested (Benner et al., 2000; Navarro-Gonzalez et al., 2006; Stalport et al., 2009) that stepwise oxidation of the aromatic component of kerogen results in the formation of benezenecarboxylates, such as the bases of mellitic and phthalic acids. The photooxidation of PAH molecules on TiO2 and α-Fe2O3 were considered as possible pathways to such products. However, the occurrence of Reactions 1 and 2 indicates that such products are not the terminal products. Figure 5 demonstrates the EPR spectra obtained in photolysis of salicylate and benzene-1,2,4,5-tetracarboxylate on hematite and anatase particles [more examples, including the mellitate and the phthalate, are given elsewhere (Shkrob and Chemerisov, 2009)]. It follows from these spectra that the hydroxylated carboxybenzenes deprotonate via Reaction 1, yielding the corresponding phenoxyl radicals, whereas the polycarboxylated benzenes decarboxylate via Reaction 2, yielding the corresponding phenyl radicals. The only benzenecarboxylic acid that did not undergo Reactions 1 and 2 was benzoate,
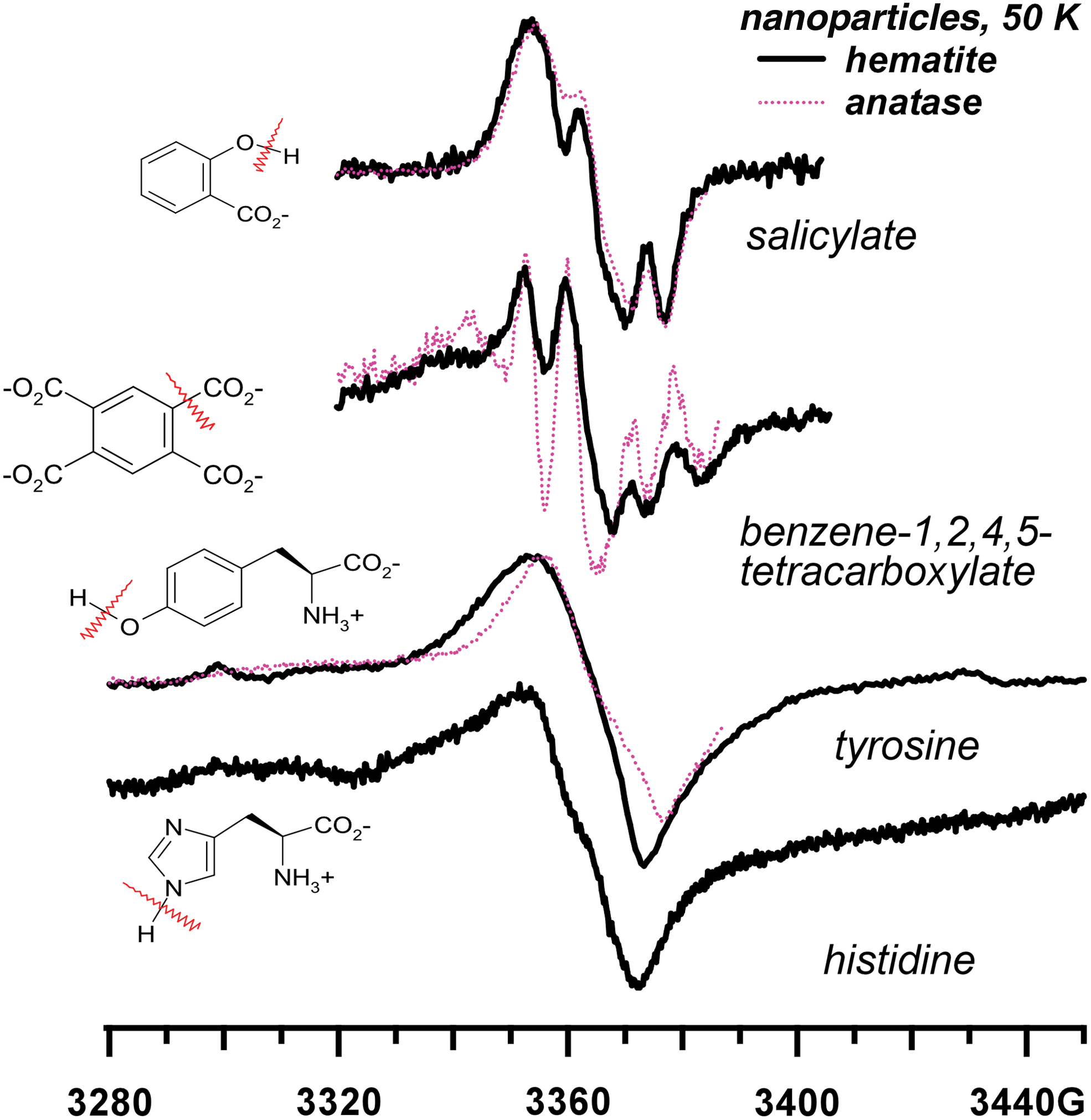
EPR spectra from UVA photoirradiated salicylic, benzene-1,2,4,5-tetracarboxylic acids, and L-α-tyrosine and L-α-histidine. Solid lines are for aqueous solutions of hematite nanoparticles, and dotted lines are for aqueous solutions of TiO2 nanoparticles. The chemical structures of the parent molecules and the fragmentation patterns are shown in the insets. Color images available online at
Reactions 1 and 2 are equally facile for the production of L-α-amino acids and peptides. The photoirradiation of aromatic L-α-amino acids, histidine, and tyrosine on the iron(III) oxides yields the corresponding deprotonated radicals via Reaction 1 (Fig. 5). The irradiation of aliphatic L-α-amino acids generally results in the elimination of CO2 from the carboxyl group in the α-position, by which these acids are chemisorbed (Fig. 6). Only in exceptional cases, when the amino acid includes clustered hydroxyl groups (tricine in Fig. 6) or free amino and thiol groups (lysine and cysteine in Fig. 7a), did we observe the competing Reaction 1. The decarboxylation also occurs for β-amino acids, such as β-alanine (Figs. 7b and 8); dipeptides, such as GlyGly and AlaAla (Figs. 7b and 9); and short peptides (Fig. 9). The cyclization of the amino acids does not inhibit Reaction 2, as seen for proline (Fig. 8). We have also observed this reaction for monohydroxylated L-α-amino acids, such as serine and threonine and other derivitized L-α-amino acids (such as arginine). In fact, all the natural amino acids with the sole exception of phenylalanine were photolytically unstable undergoing Reactions 1 and 2. The phenylalanine escapes this fragmentation by virtue of having the aromatic group that stabilizes the excess positive charge.
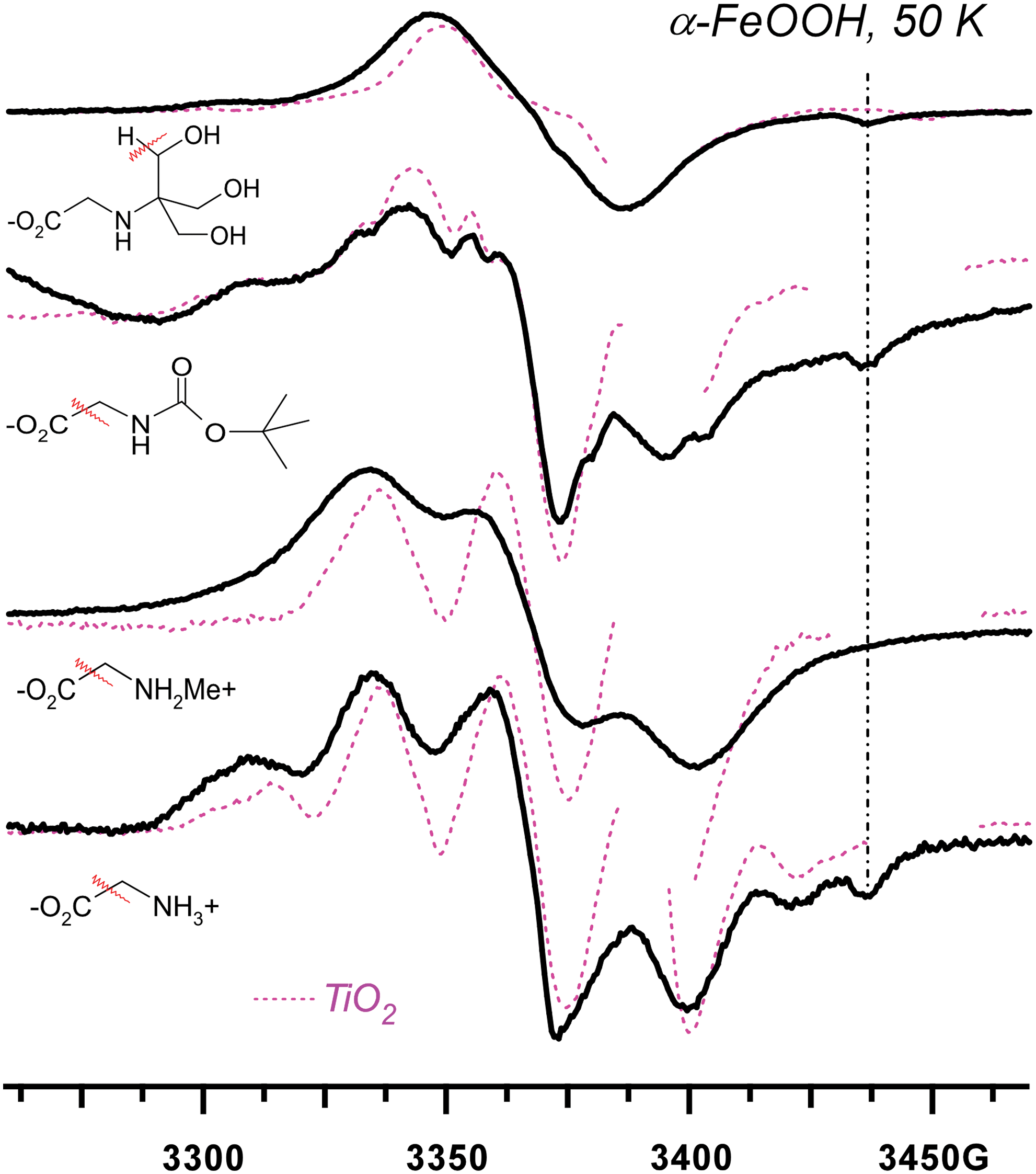
Like Fig. 5, for the following amino acids and their derivatives (from top to bottom): tricine, N-(tert-butoxycarbonyl)glycine, sarcosine, and glycine. Solid lines are for aqueous suspensions of goethite microparticles, and dashed lines are for aqueous solutions of TiO2 nanoparticles. The EPR lines for radicals on the goethite are significantly broader as compared to TiO2 traces; however, the radicals are the same. The dash-dot vertical line indicates the high-field component of the 120 G doublet (cf. Fig. 1b). The chemical structures of the parent molecules and the fragmentation patterns are indicated in the plot. Color images available online at
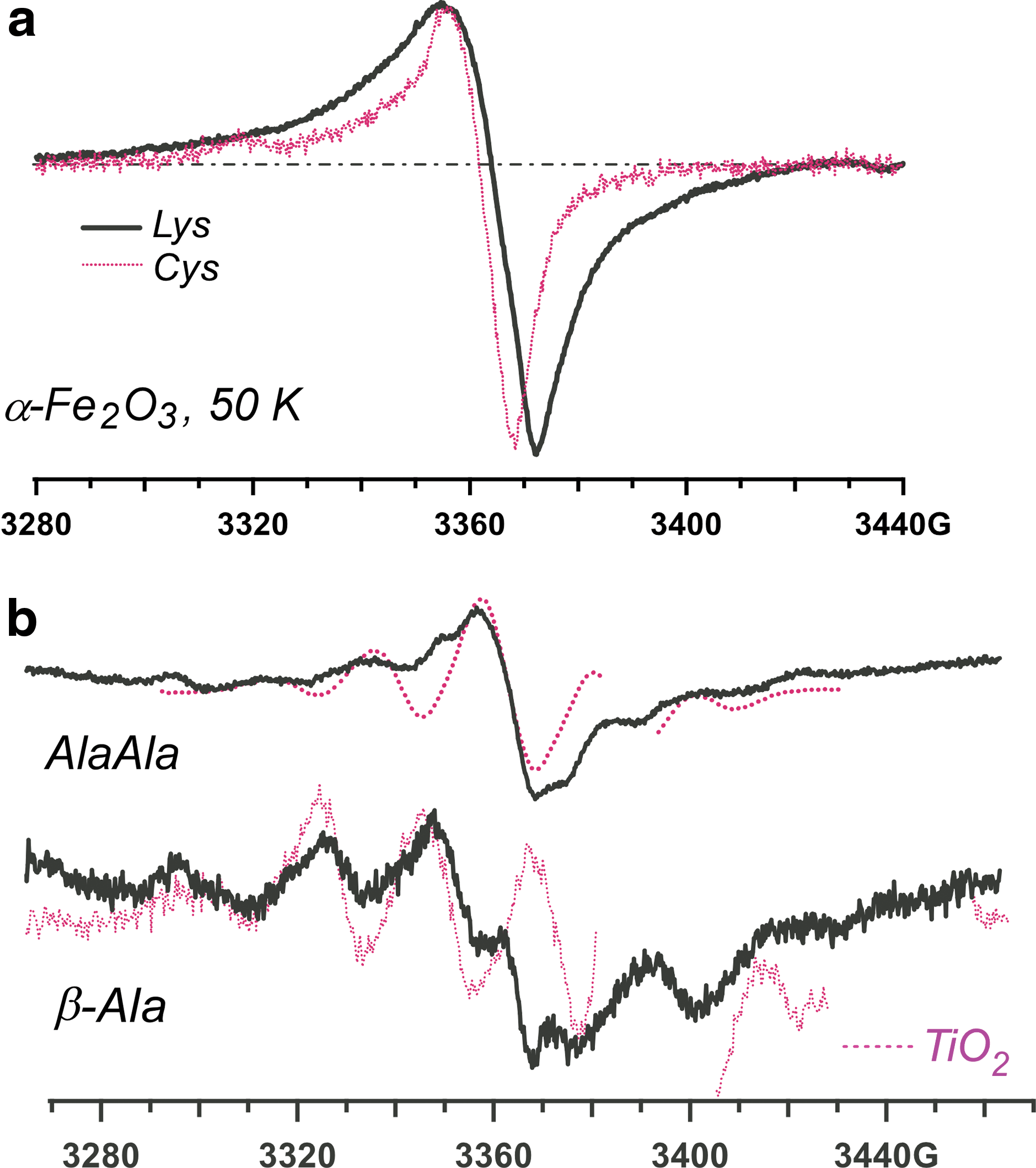
EPR spectra observed in 355 nm photolysis of chemisorbed carboxylated molecules in aqueous solutions of hematite nanoparticles. Panel (
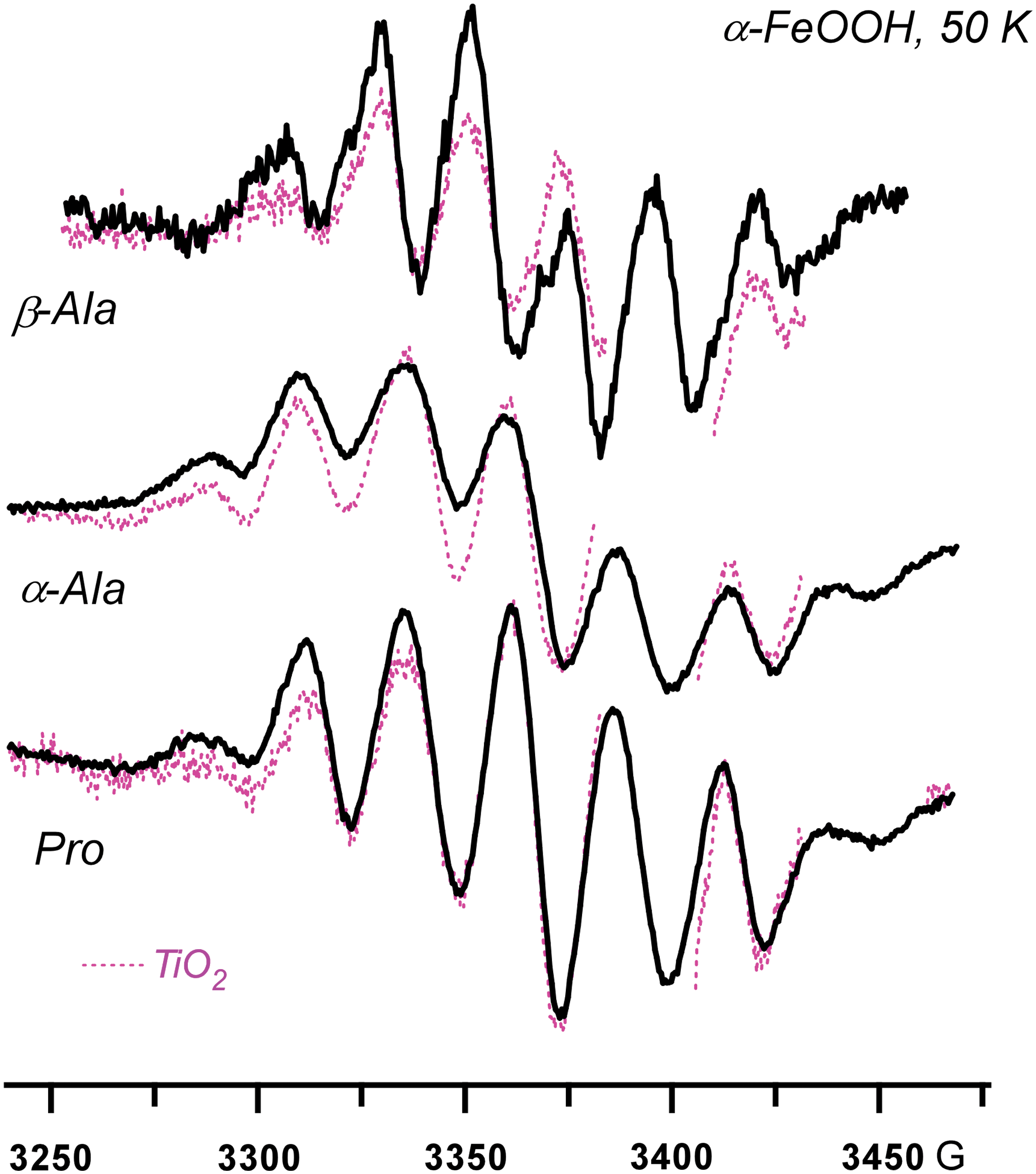
As Fig. 6, for β-alanine, α-alanine, and proline on the goethite. Color images available online at
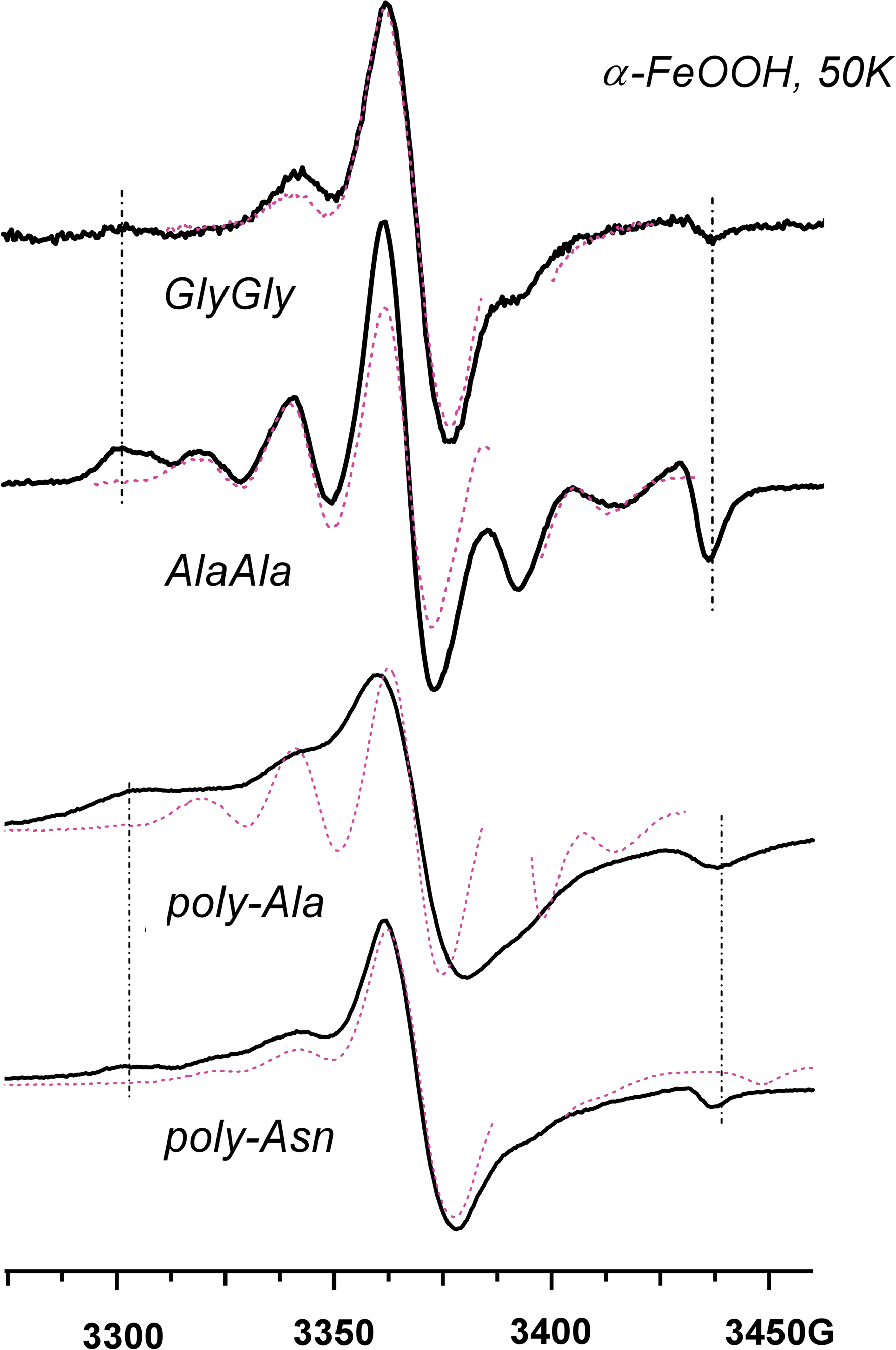
As Fig. 6, for di- and polypeptides on the goethite: (from top to bottom) glycylglicine and AlaAla, poly-L-alanine, and poly-L-asparagine. These radicals are produced via Reaction 2 involving the terminal carboxyl group. Color images available online at
5. Discussion
The photogeneration of the methyl radical from chemisorbed acetate is via Reaction 2. The desorbed methyl radicals abstract hydrogen from the parent compound yielding the secondary carboxymethyl radicals:
As shown above, this reaction can occur even at 70–90 K, provided that the fragmented and H-donating acetate ions are close to each other, which happens at high concentration of the acetate. The observation of the carboxymethyl radicals, therefore, is the proof for the occurrence of low-temperature methane generation via the coupled Reactions 2 and 3. The direct detection of methane (and ∼5% ethane, which is generated via recombination of two methyl radicals) in photoirradiated room-temperature aqueous solutions of anatase, by gas chromatography, has been reported by several authors (e.g., Kraetler and Bard, 1978). For hematite microparticles on clay, photogeneration of the gaseous alkanes from saturated carboxylic acids was observed by using head space analysis (Miyoshi and Yoneyama, 1989). In the presence of organic donors of hydrogen, the formation of the secondary radical and the disappearance of the primary methyl radical by EPR are the direct evidence for the formation of methane.
In the temperature range on Mars (>130 K), Reaction 3 is so facile that only the carboxymethyl radical is observed. (Shkrob and Chemerisov, 2009). Our experiment, therefore, indicates that acetate is photolytically unstable on the iron(III) oxide surface under the UVA illumination: this anion promptly decarboxylates, and the resulting methyl radical abstracts hydrogen from organic molecules, such as another acetate anion.
Given these facile photo-Kolbe reactions, it is difficult to argue that the compounds derived from kerogen decomposition and/or α-amino acids and peptides can survive for extended periods of time on UV-illuminated oxide surfaces. This interfacial photooxidation alone (without the involvement of the mobile OH and
5.1. Oxidative decomposition of meteoritic kerogen
The formation of methyl and carboxymethyl radicals in the photolysis of acetates on the particulate iron(III) oxides that are ubiquitous on Mars indicates the facility of Reactions 2 and 3 for the chemisorbed acetate. The first step of the reaction, the decarboxylation, occurs at any temperature, as it is strongly exergonic (Shkrob and Chemerisov, 2009); the second step requires thermal activation of methyl migration. Similar H-abstraction reactions have been observed for other C-centered radicals formed via Reaction 2. The H-abstraction reactions can be expected to compete with recombination and disproportionation of these radicals provided that the concentration of the H-donating organic molecules is sufficiently high. For methyl radicals, the products of the photoreaction are methane (H abstraction), ethane (recombination), and CO2. Other carboxylic acids can yield the corresponding alkanes and alkenes (via disproportionation). Thus, the UVA photolysis of aliphatic carboxylates on iron(III) oxides provides a chemical pathway for generating volatile hydrocarbons and CO2 over a wide temperature range, with little chemical activation other than that supplied by the photolysis. Remarkably, the products typically associated with reductive chemistry can also be formed via oxidative chemistry. Such a possibility has so far escaped the attention of planetary scientists; the photoproduction of volatile hydrocarbons was discussed only in the context of direct photolysis of the organics that requires harsh UV light (Stoker and Bullock, 1997).
These observations are important for martian surface chemistry, with its combination of iron(III) oxide particulates, traces of solid and (occasionally) liquid water, and abundant UVA radiation. As explained in the Introduction, the current ideas about the chemical evolution of meteoritic kerogen favor stepwise oxidation of this material by photogenerated O-centered radicals. The aliphatic component of the kerogen is slowly transformed to carboxylic and polycarboxylic acids, whereas the PAH component is transformed to carboxylated benzenes. The contention is that this “oxidative diagenesis” (whether it is driven by photocatalysis, decomposition of H2O2 and other peroxides, solar radiation, or electric discharges) results in the accumulation of carboxylated molecules that are resistant to further oxidation, such as the acetates and mellitates. Nonvolatile salts of these bases gradually accumulate in the soil and serve as the long-term repository of the organic carbon on Mars.
Given our observations, the long-term stability of the aliphatic carboxylates on the UVA light-exposed martian regolith seems unlikely. On the other hand, Reactions 2 and 3 for the acetate provide a rationale for abiogenic methane production on the sunlit soil, possibly accounting for the seasonal variation of methane release (Mumma et al., 2009). There are few known geochemical processes that can explain the continuous, seasonably variable production of methane on a geologically inactive planet (see the Introduction). We suggest that the oxidative decomposition of the kerogen does not terminate in the acetate formation; rather, this process terminates in Reactions 2 and 3, which enrich the martian atmosphere in methane. The fate of this methane in the atmosphere and its possible reactions with the soil particles are beyond the scope of this study.
Likewise, our study indicates that polycarboxylated benzenes would be unlikely terminal products of the stepwise oxidation of the PAH components of the kerogen, as these carboxylates are photolytically unstable on iron(III). Only benzoates and polycarboxylated PAHs are resistant to this photoreaction, but such molecules are open to further oxidation by photocatalytically generated OH radicals yielding hydroxy-substituted polycarboxylic molecules. The latter are further oxidized and subsequently decarboxylated. These considerations indicate that the concerted action of (i) the oxidation by OH, H2O2,
Another consequence of these photoreactions is the degradation of the photocatalyst itself. Since the Fe2+ centers (trapped electrons) in the hematite and goethite are stable, photooxidation (removing the holes from the particle via the formation of mobile organic radicals) results in the gradual reduction of the ferric ions at the surface. The photoinduced dissolution of iron(III) oxides in room-temperature aqueous suspensions containing oxidizable anions (such as the oxalate) is well documented (see Shkrob and Chemerisov, 2009, for review). On Mars, where water is scarce, such dissolution cannot be facile, but even episodic exposure of the regolith particles to liquid water would cause leaching of these photogenerated ferrous ions and lead to chemical erosion of the oxide particles (Zolotov and Mironenko, 2007). The leached ferrous ions can be subsequently oxidized to ferric ions that are reabsorbed by the oxide particles. The simplest chemical path to such oxidation is the well-known Fenton reactions:
where the oxidizers (HO and H2O2) are formed in water/ice UV photolysis (for example, during the periodic elevation of dust particles to the upper atmosphere). These reactions may explain how ferric oxides are formed and maintained on a planet with a reducing atmosphere. The UV light-driven chemical weathering, working in concert with erosion by wind, naturally accounts for the extreme dispersion and erosion of the martian soil, as the particles are constantly cycled through the photoinduced catalytic reduction of Fe3+ to Fe2+ and the Fenton oxidation of the released Fe2+ back to Fe3+.
5.2. Decomposition of the astrobiologically relevant carboxylated compounds
The potential for long-term preservation of the tentative markers for life, such as L-α-amino acids and peptides, is no higher than this potential for the products of kerogen oxidation; the UVA light-induced Reaction 2 on goethite destroys the amino acids
6. Conclusion
We examined the chemical underpinnings for photocatalysis on the surface of iron(III) oxide particles that are common in martian soil. Our study indicates instability of carboxylated organic molecules on oxide surfaces due to the occurrence of CO2 elimination. This photoreaction readily occurs under UVA light illumination and requires no thermal activation. The occurrence of this photodegradation is inconsistent with the hypothesis that chemical evolution of the organic component of the soil results in the accumulation of stable, nonvolatile carboxylated and polycarboxylated molecules in martian soil. In this sense, the import of our studies is negative.
The same photoreaction, however, may account for the seasonably variable martian methane production. The argument originally suggested by Benner and coworkers (2000) is absolutely correct; stepwise oxidation of the aliphatic component of the kerogen necessarily results in the production of carboxylates, such as acetates. However, acetates are not the terminal product. Due to the occurrence of the photo-Kolbe reaction, the terminal products are CH4 and CO2. The aliphatic component of the kerogen is slowly degraded through “oxidative diagenesis,” which results in the formation of acetate. When the chemisorbed acetate on the iron(III) oxide is exposed to UVA light, this anion decarboxylates and releases methyl radicals. The latter abstract hydrogen from organic molecules in the soil and form methane that is subsequently released into the atmosphere. In this scheme, methane production is the last step in a long chain of (photo)oxidative reactions that lead to the mineralization of meteoritic carbon. In this scheme, the methane ultimately originates from the meteoritic organic material delivered on Mars.
The same photoreactions are equally efficient in destroying carboxylated biomolecules. Our examination suggests that both L-α-amino acids and peptides undergo photocatalytic deprotonation and decarboxylation. Once again, there seem to be no products of the oxidation that resist further oxidation. The inescapable conclusion is that the protracted survival of these products is unlikely. A planet that is covered by highly dispersed photocatalyst and generously bathed in UV light is a hostile environment for long-term preservation of organic and bioorganic molecules.
Footnotes
Acknowledgments
Work performed under the auspices of the Office of Science, Division of Chemical Science, US-DOE under contract No. DE-AC02-06CH11357 and NASA's Mars Fundamental Research Program grant No. NNH08AI65I. I.A.S. thanks T. Rajh and N. Dimitrijevic for introduction to the oxide photocatalysis and M. Zolotov, G. Delory, M. Moore, and A. Schuerger for their insights in martian chemistry and geochemistry. We also thank the anonymous reviewers for critical reading of this paper.
Abbreviations
EPR, electron paramagnetic resonance; PAHs, polycyclic aromatic hydrocarbons.