
Abstract
The RNA-world theory hypothesizes that early Earth life was based on the RNA molecule. However, the notion that ribose, the sugar in RNA, is unstable still casts a serious doubt over this theory. Recently, it has been found that the silicate-mediated formose reaction facilitates the stabilization of ribose. Using accurate quantum chemical calculations, we determined the relative stability of the silicate complexes of arabinose, lyxose, ribose, and xylose with the intent to determine which would form predominantly from a formose-like reaction. Five stereoisomers were investigated for each complex. The stereoisomers of 2:1 ribose-silicate are the more stable ones, to the extent that the least stable of these is even more stable than the most stable stereoisomer of the other 2:1 sugar-silicate complexes. Thus, thermodynamically, a formose-like reaction in the presence of silicate minerals should preferentially form the silicate complex of ribose over the silicate complex of arabinose, lyxose, and xylose. Key Words: Prebiotic chemistry—Simulation—Silicate-organics interactions—Origin of life—RNA world. Astrobiology 11, 115–121.
1. Introduction
T
In 2004, Ricardo et al. (2004) found that a mixture of formaldehyde, glyceraldehyde, and borate minerals (ulexite, kermite, or colemanite) leads to the formation and stabilization of ribose. One year later, Li et al. (2005) investigated the binding of borate with ribose and other competing sugars, that is, arabinose, lyxose, and xylose. The binding led to 2:1 sugar-borate complexes, with ribose being the sugar that preferentially binds to borate. Those findings indicate that borate minerals might have helped stabilize and select ribose in prebiotic conditions. A similar statement can be made for silicates: in 2004, Lambert et al. (2004) reported that, when silicate and sugars are mixed, ribose is the only sugar that forms a significant amount of sugar-silicate complexes. Recently, Lambert et al. (2010b) also showed that a formose-like reaction in the presence of aqueous sodium silicate leads to the stabilization of ribose. Nonetheless, this recent finding has been brought into question. Kim and Benner (2010) reported that they have performed similar experiments to those reported in Lambert et al. (2010b), yet they did not produce ribose. In turn, Lambert et al. (2010a) responded that the experiments of Kim and Benner (2010) were performed in different conditions, and thus comparisons are confusing.
Compared to experiment, theory has a great advantage when addressing problems related to prebiotic chemistry. While experiments must deal with a complicated matrix of a variety of compounds present in the reaction mixture, theoretical studies are able to examine only the compound of interest. In this way, properly designed calculations can provide results that are free of the errors that arise due to the complexity of the composition of the reaction mixture. In Šponer et al. (2008), we showed the usefulness of theoretical work in prebiotic chemistry by investigating which 2:1 sugar-borate complex will form preferentially from mixing prebiotic precursors in the presence of borate minerals. We assumed that when formaldehyde and glyceraldehyde were mixed in the presence of borate, a single 2:1 sugar-borate complex, where the sugar was arabinose, lyxose, ribose, or xylose, was formed.
Experimentally, mixing formaldehyde, glyceraldehyde, and borate does not produce a unique sugar-borate complex (Ricardo et al., 2004) but a mixture of all of them. This process is complex and does not occur in a single reaction step. However, simplifying it to a single step by splitting it into four, one for each sugar complex, allows for an estimation of which complex should predominantly form from a thermodynamic point of view. There are two ways of estimating this: (1) comparing the change in free energy upon the formation of the sugar-borate complexes, as given by Eq.1; (2) comparing the total energies of the sugar-borate complexes. The change in free energy upon the formation of a 2:1 sugar-borate complex is given by
where G represents the free energy. When ΔG is compared for two different 2:1 sugar-borate complexes, the second and third terms on the right side of Eq. 2 cancel out, which leaves behind the free energies of the corresponding complexes. Thus, comparing ΔG for two different 2:1 sugar-borate complexes is equivalent to comparing their free energies, G. Further, comparing their free energies is nearly equivalent to comparing their total energies, that is, their electronic energies, E. (It is not fully equivalent because, unlike the free energy, the total energy does not contain the zero-point energy or the thermal and entropic corrections). For simplicity, we decided to compare the total energies. The shortcoming of this decision was that we would lose part of the information on the thermodynamics of the reaction, that is, whether the reaction in Eq. 1 is exothermic or endothermic. In Šponer et al. (2008), we assumed that the reaction was exothermic, a valid assumption given the amount of experimental evidence, and then proceeded to compare the total energies. We found that a strong H-bond (between 3-OH of ribose and O of borate) and an energetically favorable contact between 5-CH2OH and the aqueous medium rendered 2:1 ribose-borate the most stable complex. This result indicates that when originating from within a mixture of formaldehyde, glyceraldehyde, and borate, the formation of the borate complex of ribose is thermodynamically more advantageous than the formation of the borate complex of arabinose, lyxose, or xylose.
In the present study, we carried out a similar analysis for 2:1 arabinose-silicate, 2:1 lyxose-silicate, 2:1 ribose-silicate, and 2:1 xylose-silicate. We started from the experimental observation of Lambert et al. (2010b), which was that a formose-like reaction in the presence of silicate minerals leads to the formation of sugar silicate complexes. We then split this process into four components, as follows:
and determined which 2:1 sugar-silicate complex will be thermodynamically favored by comparing their total energies.
2. Model and Method
The initial structures for the 2:1 sugar-silicate complexes were created by using a combination of the lessons we learned from the studies in Šponer et al. (2008) for the 2:1 sugar-borate complexes and the experimental findings of Lambert et al. (2004) and Kinrade et al. (2003, 2004) for sugar-silicate complexes.
In Šponer et al. (2008), the models for the 2:1 sugar-borate complexes were constructed by evaluating stereoelectronic and solvation effects. The furanose ring conformation was taken from the model with the highest hyperconjugative stabilization; a hydroxyl group in the vicinity of the borate moiety was allowed to form an internal H-bond with the borate oxygen; H-bonds either between free hydroxyls or between a hydroxyl group and an endocyclic oxygen of the sugar were not allowed. Imposing these criteria led to a series of structural characteristics whose main features can be summarized as follows:
The 5-CH2OH group is gauche to O4 and trans to C3; a OH group cis to the borate ion forms a H-bond with the borate O; OH groups anti to the borate ion are fully exposed to solvent (see Fig. 1 for the numbering scheme used to identify the atoms in the 2:1 sugar-borate complexes). In Lambert et al. (2004), it was found that, in the 2:1 sugar-silicate complexes, the silicate and sugar are bound through the atoms C1 and C2. This fact, together with the findings of Kinrade et al. (2003, 2004) for simpler complexes, led Lambert et al. (2004) to propose that each 2:1 sugar-silicate complex has nine different stereoisomers. The stereoisomers are denoted as C(D;E)G, C(D;E)H, and C(D;E)I. In this notation, C, D, and E describe the binding of C1 and C2 to the oxygen atoms of Si(O4)OH in terms of axial and equatorial; G, H, and I describe the orientation of the sugar's ring with respect to the OH group of Si (see Fig. 2). Symmetry operations revealed that, out of these nine stereoisomers, only five are unique, that is, CG, CI, DG, DI, and EH. These five stereoisomers, with their initial structures built by using the structural characteristics mentioned above, are the ones we considered, and we optimized them by using quantum chemistry techniques.
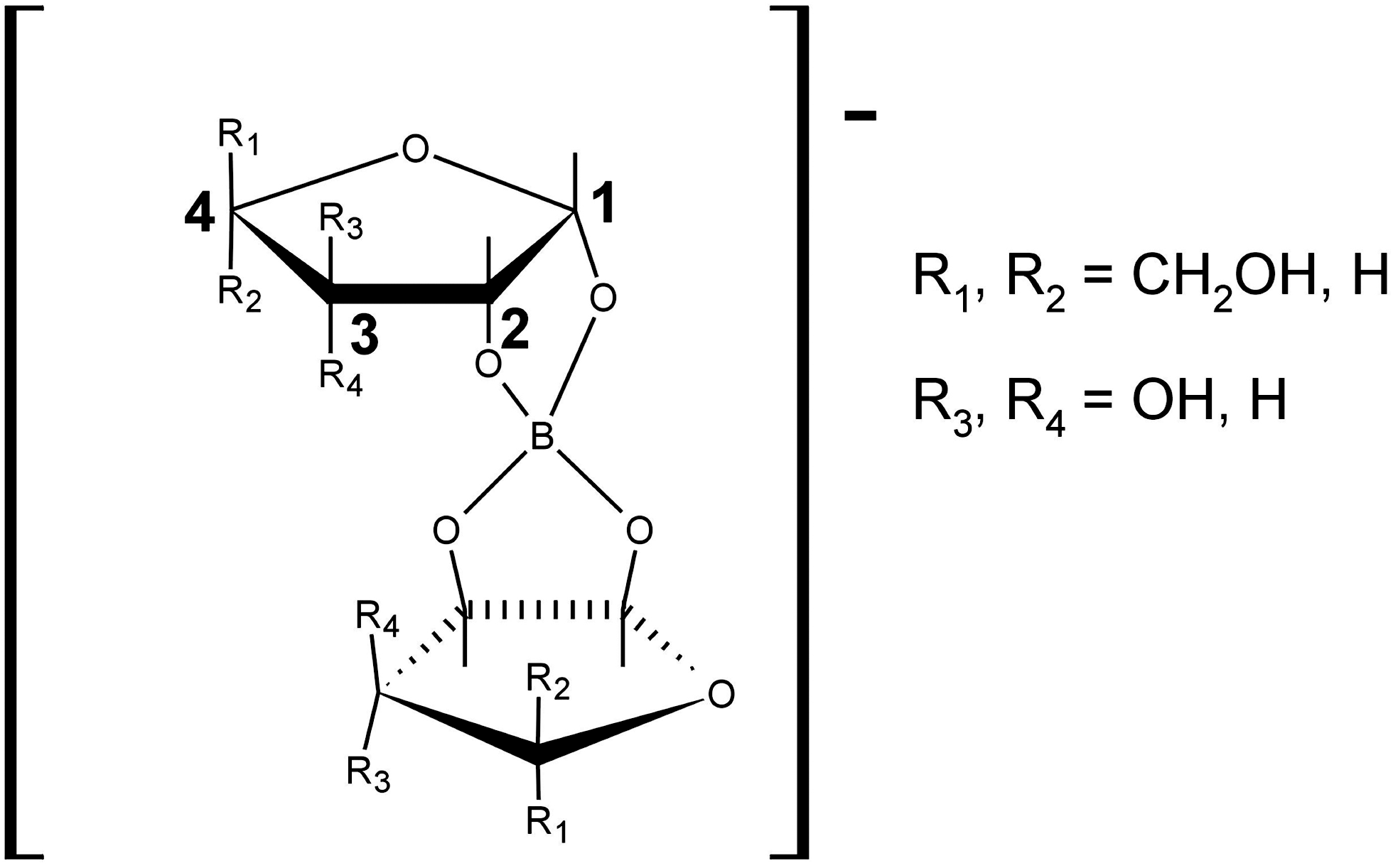
Numbering scheme used to define the atoms in the 2:1 sugar-borate complexes.
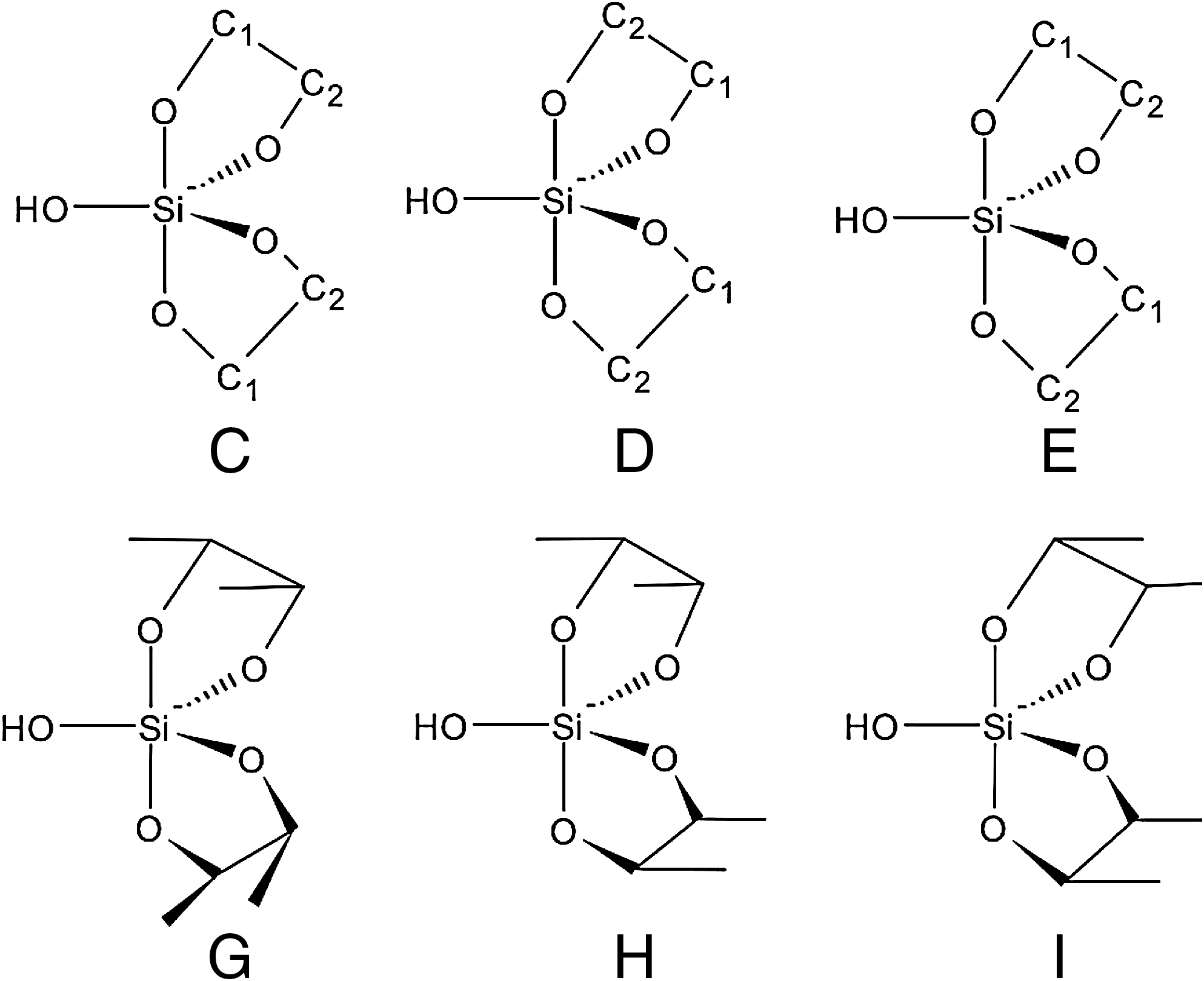
Stereoisomers of each 2:1 sugar-silicate complex. In the C(D) structure C1(C2) is axially and C2(C1) is equatorially connected to the silicate, respectively. In the E structure, one C1 connects to an axial while the other connects to an equatorial silicate-oxygen; the same is true for C2. For C, D, E there are three possible orientations of the furanose ring. The orientations are described by the structures denoted as G, H, and I. In G(I), the furanose rings are syn (anti) to the OH group of Si. In H, one furanose ring is anti, and the other is syn. There are nine stereoisomers for each 2:1 sugar-silicate complex. Symmetry operations reveal that only five of these are unique, i.e., CG, CI, DG, DI, and EH.
All the optimizations were carried out at the B3LYP level of theory (Becke, 1988, 1993; Lee et al., 1988) with the NWChem (Kendall et al., 2000; Bylaska et al., 2006) and GAMESS suite of programs (Schmidt et al., 1993). B3LYP has been used before for studying carbohydrates, and it has been proven to be an accurate technique for these molecules. [See, e.g., Csonka et al. (1996), Ma et al. (1998), Guler et al. (2002), and Lii et al. (1999)]. In particular, it has been shown that B3LYP is very accurate for predicting the energetic order of carbohydrates. However, care must be taken. In fact, H-bonds are known to be the biggest obstacle for determining accurately the right energetic ordering of carbohydrates (see, e.g., Lii et al. (1999)]. This obstacle is avoided by performing single-point calculations with the use of a large basis set, which include diffuse functions. In our calculations, we accounted correctly for the H-bonds by computing the energetic order with diffuse functions.
The initial structures of the 2:1 sugar-silicate stereoisomers were optimized with the 6-31G(d) basis set. We did not use diffuse functions during the optimization because it has been found that they are not crucial at this stage; Lii et al. (1999) studied the conformers of D-hexose and found that the energetic ordering is not affected by the use of diffuse functions during the optimization stage. Once the 2:1 sugar-silicate complexes were optimized, we computed the total energies, using a larger basis set that included diffuse functions, that is, 6-311++G(2d,2p). All the calculations were done in aqueous solution (ɛ = 78.4), which was approximated with a conductor-like model (Barone and Cossi, 1998; Cossi et al., 2003), CPCM, and a van der Waals radii scaled by 1.2 for the organic components of the complexes. This scale factor has been used before for organic molecules and proved to be accurate [see, e.g., Barone and Cossi (1998), Tomasi et al. (2005), and Cossi et al. (1996)]. For Si, the scale factor is not critical, because in these complexes surrounding O atoms screen the contact between Si and the solvent.
It should be noted that in Šponer et al. (2008) we used a similar approach for studying the 2:1 sugar-borate complexes, and we found it to be optimally tuned for studying these types of systems. In that work, it was crucial to account correctly for the observation that the role of solvent water molecules in carbohydrate solutions is to disrupt the internal H-bonds wherever it is possible (Kirschner and Woods, 2001). We found that the CPCM continuum solvent method is entirely sufficient for this purpose. Thus, we found that if in a 2:1 carbohydrate-borate complex a H-bond is formed with an anionic oxygen, then the contact is preserved; otherwise it is disrupted. The CPCM model should be able to describe the solvent effect on H-bonding for the carbohydrate-silicate complexes as well.
After optimization, some of the 2:1 sugar-silicate stereoisomers have a furanose ring conformation which differs from that of the 2:1 sugar-borate complexes. Nevertheless, these conformational differences are very minor and can be associated with the steric hindrances brought about by internal H-bond formation. It should be noted that similar conformational changes are also frequently observed in natural RNA nucleotides, where internal H-bond formation between the 2′-OH of ribose and the N3 endocyclic nitrogenous site of the nucleobase leads to the change of the canonical C3′ endo conformation to C2′ endo (Šponer et al., 2005).
The NBO 5.0 program (Glendening et al., 2001) and the B3LYP/6-311++G(2d,2p) wavefunctions have been used for natural bonding orbital (NBO) and natural steric analysis (Badenhoop and Weinhold, 1997,1999).
3. Results
The total energies (without correction for ZPE) of the optimized 2:1 sugar-silicate complexes are summarized and compared in columns 3 and 4 of Table 1. For the silicate complexes of arabinose, lyxose, ribose, and xylose, the most stable stereoisomers are DI, CI, EH, and CI, respectively. Among these, EH-ribose is the most stable, followed by CI-lyxose at 2.72 kcal/mol, DI-arabinose at 3.57, and CI-xylose at 3.80 (Fig. 3 shows the optimized structure of these stereoisomers). In trying to understand this stability trend, it is instructive to refer to Chapelle and Verchere's work on sugar-borate complexes.
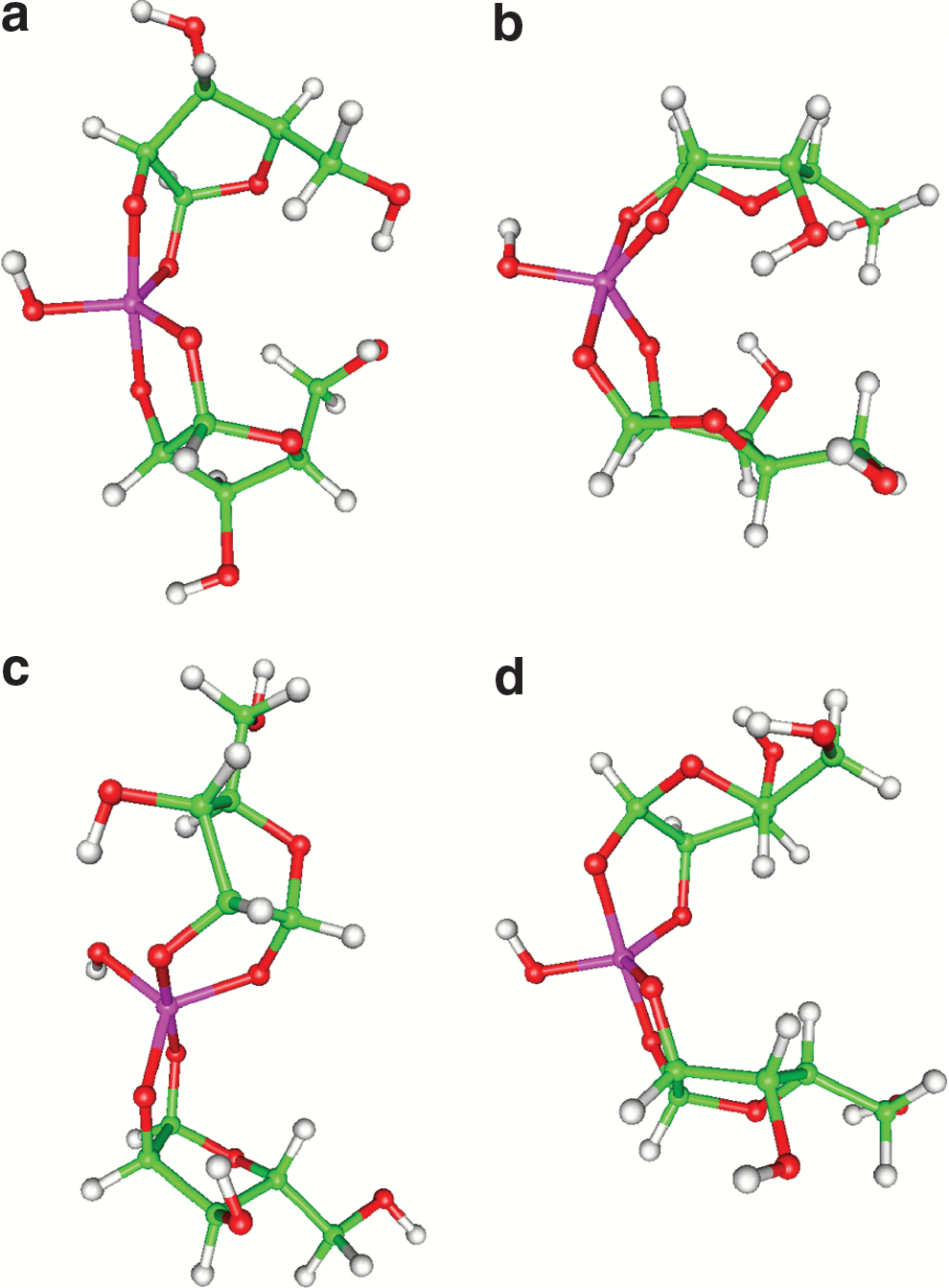
B3LYP/6-31G(d) optimized geometries of (
ΔE, relative energies of the most stable stereoisomers of the 2:1 sugar-silicate complexes with respect to that of ribose; ΔE s, relative energies of the five stereoisomers of a given 2:1 sugar-silicate complex with respect to the most stable one; ΔE l, difference in energy between the least stable stereoisomer of 2:1 ribose-silicate and the most stable stereoisomer of the other 2:1 sugar-silicate complexes. a.u., atomic units.
Chapelle and Verchere (1988) measured the stability constant of the reaction that led to 2:1 sugar-borate complexes from mixing sugar and borate ions. They found that 2:1 ribose-borate has the largest stability constant, followed by the complexes of xylose, lyxose, and arabinose. This trend, they pointed out, seemed to correlate stability with steric effects; the complexes in which the borate ion and 5-CH2OH were trans (2:1 ribose-borate and 2:1 xylose-borate) had larger stability constants than the complexes in which the borate ion and 5-CH2OH were cis (2:1 lyxose-borate and 2:1 arabinose-borate). These authors' subsequent experiments proved that this correlation did not exist. In Šponer et al. (2008), we argued that, besides steric effects, electrostatic repulsions between the borate ion and 5-CH2OH should also be considered. In this way, we were able to qualitatively explain, using both arguments, the energetic trends we found for the 2:1 sugar-borate complexes.
It would be expected that steric and electrostatic effects are more important for the 2:1 sugar-silicate complexes than for the borate ones, simply because the former are more crowded. In other words, one would expect a trend that states that the less-crowded species are more stable than the more-crowded ones. Yet we do not observe this trend. Indeed, to evaluate the extent of steric crowdedness of the complexes, we computed the steric exchange energy from natural steric analysis (Badenhoop and Weinhold, 1997, 1999). We have observed cases in which more-crowded structures are more stable than less-crowded ones: CI-lyxose (steric exchange energy: 1250.6 kcal/mol) and DI-arabinose (1248.0 kcal/mol) are more stable than CI-xylose (1238.1 kcal/mol). And the situation is not any better if one decides to simplify the comparison by focusing on a particular stereoisomer. For example, DG-xylose (1229.8 kcal/mol) is less stable than DG-lyxose (1253.6 kcal/mol) but more stable than DG-arabinose (1210.7 kcal/mol); and DI-xylose (1248.02 kcal/mol) is as stable as DI-arabinose (1248.04 kcal/mol) and more stable than DI-lyxose (1233.4 kcal/mol). Thus, all that can be said is that, for the 2:1 sugar-silicate complexes, there is no relation between stability and steric and electrostatic effects. *
Further inspection of the data reveals that, because all five stereoisomers of 2:1 ribose-silicate have similar energies (the largest difference being 0.63 kcal/mol, see column 5 Table 1), the least stable stereoisomer of 2:1 ribose-silicate (CI) is more stable than the most stable stereoisomer of the other 2:1 sugar-silicate complexes (see column 6 of Table 1). These results indicate that, from a thermodynamic point of view, a formose-like reaction in the presence of silicate minerals will rather form the silicate complex of ribose than the silicate complex of arabinose, lyxose, or xylose.
4. Discussion
Our theoretical results in Šponer et al. (2008) and those presented here indicate that, thermodynamically, a borate- or silicate-mediated formose-like reaction will favor the formation of the borate complex or silicate complex of ribose, respectively. However, these results cannot be directly compared with the experimental ones. For example, Chapelle and Verchere (1998) measured the stability constant of the borate complexes of arabinose, lyxose, ribose, and xylose when they were produced by mixing the corresponding sugar with borate. Li et al. (2005) measured the binding of these sugars to borate. And Lambert et al. (2004) studied the formation of sugar-silicate complexes that resulted from mixing silicates and sugars (the stability constants were not measured, though). A comparison between those experiments and our studies is not possible, because we have not calculated the stability constants or the binding energies of the sugar-borate/silicate complexes. We have not computed these quantities because, in our opinion, they are not meaningful for the pertinent question, that is, whether the formose reaction in the presence of borate/silicate minerals can lead to the preferential formation of ribose under prebiotic conditions.
When measuring/computing the stability constant or binding energy of a 2:1 sugar-borate/silicate complex, it is inherently assumed that the sugar already exists. That is, the stability constant that is measured/computed is that of the reaction sugar+silicate/borate → sugar-silicate/borate complex. This reaction does not say anything about how the sugar was formed in the first place on early Earth. The formose reaction, however, does provide that information. The formose reaction involves many steps, which start from simple molecules that could have presumably existed on early Earth. After a series of reactions, these simple organic precursors produce the sugar. Thus, computing/measuring the stability constant of the reaction sugar+silicate/borate → sugar-silicate/borate does not provide any information as to how the sugar could have formed on early Earth; that is, it does not provide any information about the formose reaction. This is the crucial information in this problem. For this reason, we do not calculate stability constants but focus instead on explaining how the sugars could have formed from simple organic precursors.
The experiments of Ricardo et al. (2004), Lambert et al. (2010b), and Kim and Benner (2010) involved investigation of the formation of the sugar-borate/silicate complexes that resulted from mixing formaldehyde and glycolaldehyde in the presence of borate/silicate minerals. Their studies could be compared to ours, but only if the relative stability of the 2:1 sugar-borate/silicate complexes would have been measured. This is not the case. Thus, none of the currently available experimental studies of borates/silicates have data that can be directly compared to ours. Furthermore, in our calculations we only considered the thermodynamics aspects of the problem, not the kinetics ones, and kinetics could change the trends we observed here.
In fact, not all the experimental studies are comparable to each other. Concerning the sugar-borate complexes, the findings in Li et al. (2005) are comparable to the findings in Chapelle and Verchere (1988), but neither can be compared to the findings in Ricardo et al. (2004). The situation for sugar-silicate complexes is similar. Some aspects of the work in Lambert et al. (2004) and Kinrade et al. (2003, 2004) can be compared, but neither study can be compared to those in Lambert et al. (2010b) and Kim and Benner (2010). Moreover, the only two experiments that could be directly compared to each other, those of Lambert et al. (2010b) and Kim and Benner (2010), cannot be compared at all, because the conditions used in both experiments were different. In view of all this, our theoretical findings become rather relevant, as they provide a different view of the problem.
5. Conclusions
This view suggests that, thermodynamically, a formose-like reaction in the presence of silicate minerals should preferentially form the silicate complex of ribose over the silicate complex of arabinose, lyxose, and xylose. This result is in disagreement with the findings of Kim and Benner (2010). Yet, it is not in complete agreement with the findings of Lambert et al. (2010b), as this experiment only reports that ribose did form, but not that it formed preferentially over the other sugars. Our statement is more forceful in this respect: a formose-like reaction in the presence of silicate minerals will preferentially form ribose. It is clear, however, that the experimental data on this subject is quite scattered and that much benefit could be obtained if theory and experiment were to focus on this problem together. In this respect, we suggest measuring the amount of 2:1 sugar-borate/silicate produced in a borate/silicate mediated formose-reaction. If this measurement is available, it will provide an opportunity for a direct comparison with our theoretical results. On the other hand, if the experiment suggested above could be performed, and our theoretical predictions were to be found wrong, it would indicate that kinetics play an important role, as our calculations only address the thermodynamics aspects of the problem.
Footnotes
Acknowledgments
This work was supported by the Center for Nanophase Materials Sciences, sponsored by the Division of Scientific User Facilities, U.S. Department of Energy (USDOE) (M.F.C., B.G.S.). S.R.H. was supported by an appointment under the Higher Education Research Experience (HERE) program, administered by the Oak Ridge Institute for Science and Education under contract number DE-AC05-06OR23100 between the USDOE and Oak Ridge Associated Universities. A.V.M. acknowledges support from the DOE, Offices of Basic Energy Science and Advanced Scientific Computing Research as part of the SciDAC program. Authors are also thankful to the computational resources of the UT/ORNL National Institute for Computational Sciences (A.V.M., M.F.C.). J.E.S. and J.S. are thankful for the financial support by the Ministry of Education of the Czech Republic (grant numbers AVOZ50040507, AVOZ50040702, MSM0021622413, and LC06030, MSM6198959216, LC512), by the Grant Agency of the Academy of Sciences of the Czech Republic (grant number IAA400040802) and Grant Agency of the Czech Republic (grant numbers P208/10/2302, 203/09/1476 and 203/09/H046).
Disclosure Statement
No competing financial interests exist.
Abbreviation
NBO, natural bonding orbital.
*
On the other hand, it should be noted that the main orbital delocalizations from NBO analysis are very similar both for silicates and borates. The energetic stabilization brought about by the delocalization of the lone electron pair at the ring oxygen of the furanose ring is ca. −10.3 to −12.0 kcal/mol for silicates, depending on the position of the ring, which agrees very well with the value obtained for borates, −12.7 kcal/mol. Similarly, delocalization of the lone pair of the glycosidic oxygen to the adjacent C-O antibonding orbital of the furanose ring contributes to the stabilization of the silicate complexes by −19.3 to −22.6 kcal/mol, again in good agreement with the corresponding value computed for borates, −20.2 kcal/mol.