
Abstract
The last universal common ancestor of contemporary biology (LUCA) used a precise set of 20 amino acids as a standard alphabet with which to build genetically encoded protein polymers. Considerable evidence indicates that some of these amino acids were present through nonbiological syntheses prior to the origin of life, while the rest evolved as inventions of early metabolism. However, the same evidence indicates that many alternatives were also available, which highlights the question: what factors led biological evolution on our planet to define its standard alphabet? One possibility is that natural selection favored a set of amino acids that exhibits clear, nonrandom properties—a set of especially useful building blocks. However, previous analysis that tested whether the standard alphabet comprises amino acids with unusually high variance in size, charge, and hydrophobicity (properties that govern what protein structures and functions can be constructed) failed to clearly distinguish evolution's choice from a sample of randomly chosen alternatives. Here, we demonstrate unambiguous support for a refined hypothesis: that an optimal set of amino acids would spread evenly across a broad range of values for each fundamental property. Specifically, we show that the standard set of 20 amino acids represents the possible spectra of size, charge, and hydrophobicity more broadly and more evenly than can be explained by chance alone. Key Words: Astrobiology—Evolution—Molecular biology—Modeling studies. Astrobiology 11, 235–240.
1. Introduction
B
Taken together, these findings highlight an important question: Did the incorporation of one specific subset of amino acids into genetic coding owe more to natural selection (e.g., for an optimal set of building blocks) or to pure chance? If chance played the dominant role, then we might anticipate that an independent origin of life would build from a thoroughly different biochemical foundation, which would produce the sort of utterly different evolutionary outcomes suggested by some (e.g., Gould, 1989). If instead natural selection favored particular properties for the amino acid alphabet, then this would imply some level of predictability in the outcome, that is, some expectation for universal biochemistry, with all that this implies for the emerging science of astrobiology (Des Marais et al., 2008).
Previous reasoning on the topic has reached widely differing conclusions (compare, e.g., Weber and Miller, 1981; Pace, 2001; Benner et al., 2004; Cleaves, 2010). The challenge for astrobiologists is, therefore, to define specific, adaptive criteria that distinguish chance from predictability, which would allow researchers to convert carefully reasoned opinions into testable hypotheses (see Freeland and Philip, 2010). Along these lines, some analyses have suggested a role for thermodynamics in determining the order by which amino acids entered the standard alphabet (Higgs and Pudritz, 2009), and perhaps in discriminating chemical isomers (Zhang, 2007). However, this offers only partial insight into the processes that selected a particular subset of amino acids from a broader pool of possibilities. Meanwhile, an explicit attempt to distinguish whether chance or natural selection forms a better explanation for the contents of the standard amino acid alphabet (Lu and Freeland, 2008) produced ambiguous results.
Here, we seek clarity by increasing the sophistication of expectations for an optimal amino acid alphabet. Our hypothesis is that natural selection would have favored a set of building blocks that evenly samples a wide range of sizes, charges, and hydrophobicities. We therefore test whether the standard alphabet of genetically encoded amino acids exhibits greater range and more even sampling for each property than a representative sample of plausible alternative alphabets.
2. Materials and Methods
To test our hypothesis required (i) precise definitions of the pools of amino acids from which to draw plausible alternative sets at random; (ii) quantifications of size, charge, and hydrophobicity for all these amino acids; (iii) a method for quantifying the “coverage” (breadth and evenness of distribution) for a given amino acid set; and (iv) a method for calculating the expectations of a random alphabet of amino acids.
2.1. Defining the pool of plausible amino acids
We followed previous analysis (Lu and Freeland, 2008) in defining two different versions of a pool of plausible candidates for alternative amino acid sets. The first, smaller pool simply reflected the organic chemistry of the Murchison meteorite, as this “offer(s) an invaluable sample for the direct analysis of abiotic chemical evolution prior to the onset of life” (Pizzarello, 2007). However, 16 of the 66 amino acids found within Murchison are classed as non-α-amino acids that contain extra carbon atoms on their “backbone” relative to the α-amino acids from which proteins are built. We accept previous arguments that these elongated backbones obstruct the formation of stable structures (e.g., Weber and Miller, 1981; Cleaves, 2010) and therefore omitted these 16 non-α-amino acids from consideration to leave a pool of 50 prebiotically plausible α-amino acids (comprising 8 that became part of the standard, genetically coded alphabet and 42 that did not).
Our second, enlarged pool of plausible candidates reflects extensive support for the idea that the 12 standard amino acids not found within the Murchison meteorite emerged during early evolution as metabolic modifications of their prebiotic counterparts, via pathways still observed in modern organisms (e.g., see Freeland, 2009; Higgs and Pudritz, 2009; Cleaves, 2010). However, these metabolic pathways produce 14 additional amino acids as intermediate steps; thus, if they truly represent the routes by which the standard amino acid alphabet grew, then all (12 + 14 = ) 26 amino acids must be considered plausible candidates for incorporation into genetic coding. We therefore defined a second, enlarged pool of 76 plausible amino acids, complementing the 50 described above with the 12 standard amino acids that are not found within Murchison and the 14 metabolic intermediates found on the pathways by which this second group are biosynthesized.
These definitions of the pool of plausible amino acid candidates available for inclusion into genetic coding are illustrated in Fig. 1.
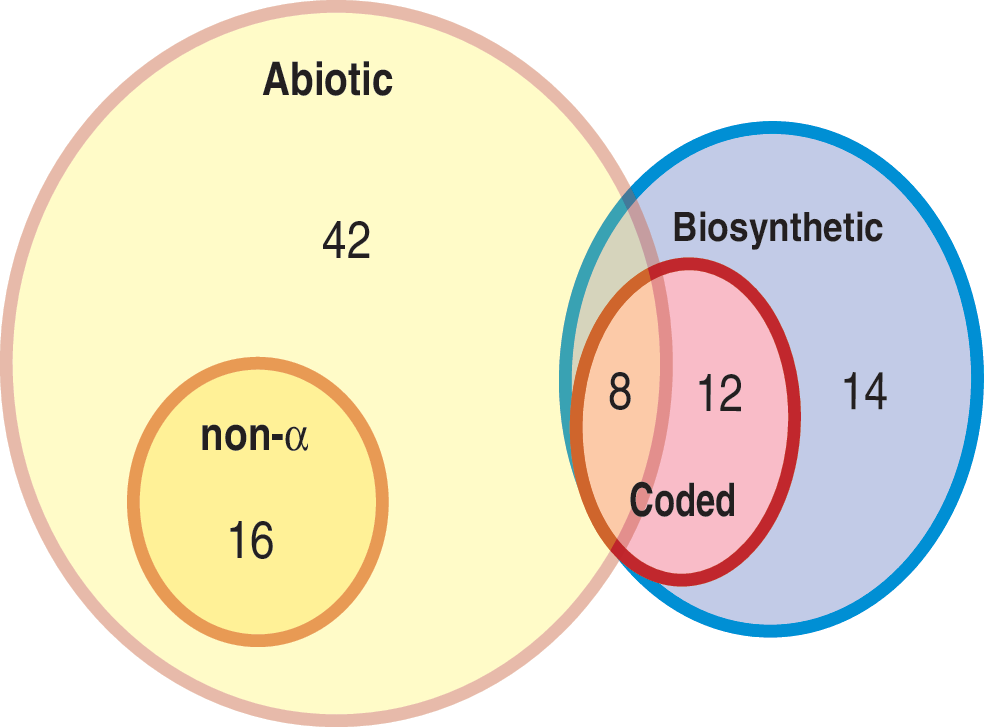
Venn diagram showing the number of amino acids represented in different categories of chemical space. Abiotic refers to the 66 amino acids reported in the Murchison meteorite (including 8 members of the standard alphabet). Non-α refers to the 16 amino acids reported from the Murchison meteorite that have longer carbon “backbones” than those used in genetic coding. Coded refers to the 20 amino acids used within the standard genetic code. Biosynthetic refers to the additional 12 members of the standard alphabet and a further 14 amino acids that are produced as intermediates in their production. Color images available online at
2.2. Quantifying size charge and hydrophobicity of the amino acids
We followed previous analysis (Lu and Freeland, 2008) in quantifying size and hydrophobicity for all amino acids, using the molecular descriptors pI, van der Waals volume, and logP, respectively. As discussed in Lu and Freeland (2006), this choice reflects careful research for precise quantifications that meaningfully represent general amino acid properties known to play an important role in determining what protein structures and functions can be constructed.
2.3. Quantifying “coverage” for a given set of amino acids
The novelty of the study presented here lies in a new method for measuring the adaptive value of a given amino acid alphabet. Previous analysis (Lu and Freeland, 2008) focused on biochemical diversity, arguing that an optimal set of amino acids would be one that exhibits the largest possible statistical variance. Here, we retain the underlying concept but regard its previous measurement as flawed.
In particular, variance measures “diversity” as the sum of squared deviations from arithmetic mean of a group of objects. For the purpose of scoring the adaptive value of a set of amino acids, this has the unfortunate effect of awarding the highest scores to alphabets in which amino acids cluster at the maximum or minimum values of a given property. To take the example of amino acid size, under the previous scoring system, an amino acid alphabet that comprises some very small amino acids and other very large amino acids would score higher than one in which amino acids are evenly distributed across an equally large range of sizes, from smallest to largest (Fig. 2B, 2C).
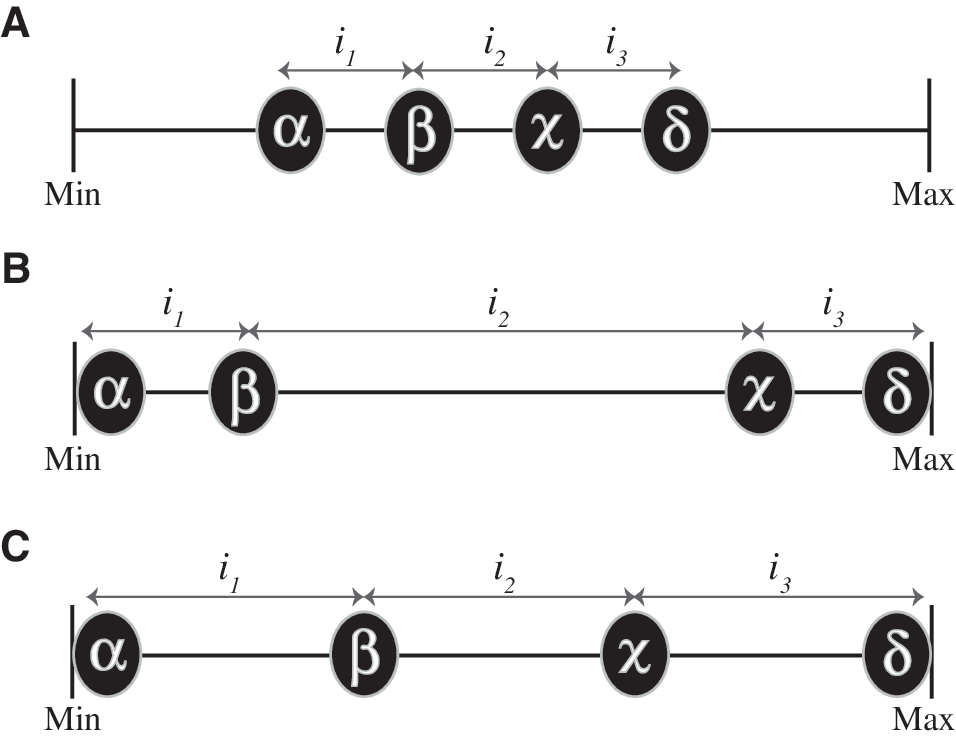
A comparison of methods for scoring the adaptive value of a given set of amino acids. This schematic diagram illustrates three hypothetical alphabets (
Here, we replace variance with a measure that gives the highest adaptive value to an alphabet that divides a large range into even intervals (Fig. 2C). Our rationale is that an alphabet that combines both range and even distribution of objects within this range is one that minimizes the discrepancy between any ideal properties favored for a particular site within a protein sequence and the nearest available amino acid.
To quantify this revised scoring system for each amino acid property, we developed a parameter we refer to hereafter as “coverage.” Coverage is calculated by first sorting a given amino acid set into order of increasing magnitude and then calculating (i) its “breadth” as the difference between maximum and minimum values (i.e., the statistical property of “range”) and (ii) its “evenness of spread” (the sample variance, σ2, of the intervals between consecutive pairs of members, i.e., greater evenness = smaller variance).
Although it might seem tempting to combine these two features (range and evenness) into a single calculation, to do so would introduce unwarranted assumptions—that we know the relative adaptive value for trading off evenness against range. By retaining the two values as separate entities, our measurement of coverage is conservative; it accepts one amino acid alphabet as better (more adaptive) than another only if it exhibits greater range and greater evenness.
2.4. Calculating the expected characteristics for a random alphabet of amino acids
Building from these assumptions, we performed three specific tests: we compared (in terms of coverage) (i) the full set of 20 genetically encoded amino acids for size, charge, and hydrophobicity with equivalent values calculated for a sample of 1 million alternative sets (each also comprising 20 members) drawn randomly from the pool of 50 plausible prebiotic candidates (Fig. 1); (ii) the subset of 8 prebiotic amino acids that became part of the standard alphabet with random sets of 8 drawn from this same pool of 50 prebiotic candidates; and (iii) the full set of 20 standard amino acids with sets of 20 drawn from the larger pool of 76 candidates.
For each test, we measured coverage for a random sample of 10,000 amino acid sets, recording the fraction of random sets that exhibited better coverage than the standard amino acids. We repeated each test 100 times to obtain the 95% confidence interval for the mean percentage (μ) of random alphabets with a coverage greater than the coded set: (μ-1.96σ/√n, μ-1.96σ/√n), where n is 100, and σ is the standard deviation of the 100 percentage values.
3. Results
When we compared the coverage of the standard alphabet of 20 amino acids for size, charge, and hydrophobicity with equivalent values calculated for a sample of 1 million alternative sets (each also comprising 20 members) drawn randomly from the pool of 50 plausible prebiotic candidates, results showed that the standard alphabet exhibits better coverage (i.e., greater breadth and greater evenness) than any random set for each of size, charge, and hydrophobicity, and for all combinations thereof (Fig. 3, top value). In other words, within the boundaries of our assumptions, the full set of 20 genetically encoded amino acids matches our hypothesized adaptive criterion relative to anything that chance could have assembled from what was available prebiotically.

The mean (μ) and 95% confidence interval for the percentage of random alphabets with a coverage greater than the coded set for (i) 20 (Coded) amino acids from a pool of 50 (Abiotic\non-α class) candidates (top value); (ii) 8 (Abiotic ∩ Coded) amino acids from 50 (Abiotic\non-α class) possibilities (middle); and (iii) 20 (Coded) from 76 [(Abiotic\non-α class) ∪ Biosynthetic] amino acids (bottom) in each of the three properties of charge (pI), size (van der Waals volume), and hydrophobicity (logP). (μ-1.96σ/√n, μ-1.96σ/√n), where n is 100 and σ is the standard deviation of the 100 percentage values. Color images available online at
By itself, this result is not compelling in that many members of the standard alphabet do not occur within the Murchison pool of prebiotic candidates. Indeed, a widespread view is that life began genetically encoding proteins with a restricted subset of prebiotically plausible amino acids, subsequently incorporating additional amino acids through a series of biosynthetic “inventions” (see, e.g., Wong, 2007; Freeland, 2009; Higgs and Pudritz, 2009; Cleaves, 2010). We therefore analyzed a putative starting point for genetic coding by calculating the coverage of the 8 prebiotically plausible amino acids that are found within the standard 20 and comparing with alternative sets of size 8 drawn randomly from the pool of 50 prebiotic candidates. Once again, for each molecular property, evolution's “choice” appeared significantly nonrandom. Indeed, for any combination of two chemical properties (e.g., size and hydrophobicity, but not charge) <1% random sets showed better coverage, and <0.1% of the random sets appeared more adaptive across all three properties simultaneously (i.e., in charge and size and hydrophobicity) (Fig. 3, middle value).
Taken together, results of these two analyses indicate that life genetically encodes a highly unusual subset of amino acids relative to any random sample of what was prebiotically plausible. However, in the process of evolving new amino acids that extended biochemistry beyond these prebiotic boundaries, the evidence once again suggests that life was able to select from a larger set of possibilities than those we find genetically encoded today (see Methods).
We therefore performed a third test to assess whether evolution appears to have selected a set of amino acids with nonrandom coverage within these options. We enlarged the pool of 50 prebiotic candidate amino acids with 26 biosynthetic possibilities to define a new pool of 76 α-amino acids that were available for genetic coding by the time life's standard amino acid alphabet became finalized. We then explored whether evolution appears to have favored specific metabolic inventions that increased coverage for important biochemical properties by comparing the standard alphabet of 20 with a random sample of 1 million alternative sets of equivalent size drawn from an expanded pool of 76 candidates (Fig. 1). Once again the results indicate that evolution selected a highly unusual set of 20 amino acids; a maximum of 0.03% random sets out-performed the standard amino acid alphabet in two properties, while no single random set exhibited greater coverage in all three properties simultaneously (Fig. 3, bottom value).
These results combine to present a strong indication that the standard amino acid alphabet, taken as a set, exhibits strongly nonrandom properties. However, this tells us relatively little about whether the specific amino acids found within the standard alphabet (i.e., the “coded” amino acids) are likely to be present within any plausible, optimal set.
To address this question, we isolated the fraction of “better” amino acid alphabets obtained from each of the tests described above. From these, we noted where coded amino acids occurred and thus plotted a distribution to show the frequency with which we found each coded amino acid within each of these better sets. For example, in the first test, we identified a total of 49,527 randomly chosen alphabets that exhibited better coverage in size than the 8 amino acids of the standard alphabet found in the Murchison meteorite. Of these 49,527 “better alphabets,” we found that 13,479 (27.22%) contained one coded amino acid. We then used the hypergeometric distribution to calculate the probability of finding each number of coded amino acids within a better alphabet under the null hypothesis that all amino acids are equally likely to appear here. Figure 4 plots these observed frequencies and null expectations.

Frequency (%) of coded amino acids found within “better” (more adaptive) alphabets for (
The only instance where the observed frequencies matched the expected frequencies was in the charge category [e.g., we observed 40.0% random sets containing 1 of the 8 coded amino acids found on the Murchison meteorite, while the expected value was calculated under our null hypothesis as 40.2% (Fig. 4A)]. For size, hydrophobicity, and the combination of all three properties, it appears the observed values are skewed toward a higher occurrence of coded amino acids than is expected by chance (Fig. 4A, 4B). In other words, Fig. 4 suggests that an adaptive choice of amino acids is predisposed toward the inclusion of at least some of those used by life on our planet.
4. Discussion and Conclusions
Whether we consider a starting point of genetic coding within (i) the pool of prebiotically plausible amino acids, (ii) the end point of the standard alphabet relative to this prebiotic pool of candidates, or (iii) the process by which evolution escaped these prebiotic boundaries, we see a consistent, unambiguous pattern; random chance would be highly unlikely to represent the chemical space of possible amino acids with such breadth and evenness in charge, size, and hydrophobicity (properties that define what protein structures and functions can be built). Further analysis indicated that, even under this simple criterion, any selection of an optimal amino acid alphabet is likely to include some of those found within contemporary genetic coding.
Clearly, much is left to investigate. For example, the results presented here do not include consideration of factors other analyses have suggested could contribute to defining an optimal set of amino acids, such as “cost of biosynthetic manufacture” (e.g., see Dufton 1997; Akashi and Gojobori, 2002; Cleaves, 2010) or “rotational flexibility around the peptide bond” (e.g., Weber and Miller, 1981; Koca et al., 1994; Chipot and Pohorille, 1998). If meaningful values for such properties could be calculated for noncoded amino acids, then their inclusion could further refine the extent to which we may regard evolution's choice as predictable. In another direction, it remains a wide-open question as to whether the small fraction of alternative alphabets with comparable coverage would permit a very different suite of protein structures.
Given these restrictions, it is remarkable that such a simple starting point for analysis yields such clear results. Further understanding why life on Earth evolved one particular set of amino acids as a fundamental framework for protein biochemistry would inform our expectations for an independent origin of life, whether it evolved naturally elsewhere or is created by scientists in the laboratory.
On this latter point, we note that recent advances in synthetic biology have demonstrated protocols by which scientists may introduce a wide range of artificial amino acids into genetic coding (e.g., see Liu and Schultz, 2010). However, these efforts have thus far been dominated by empirical success (showing us what we can introduce into the genetically encoded amino acid alphabet) and currently lack any theoretical, guiding framework to show us why we might pick specific amino acids for incorporation. As the scientific community approaches the frontier of rationally designed life-forms (in which the fundamental building blocks of protein synthesis are a user-defined set of amino acids), it would be helpful to develop clear, testable ideas about how best to leverage the possibilities of biochemical engineering. We have previously addressed this point in greater detail when discussing the methods by which we seek to address our question (see Lu and Freeland, 2006, and references therein). Given the strength of results derived here from such simple assumptions, we hope that the wealth of unexplored refinements to our approach will promote further synergy between those who seek to understand the origin and evolution of life on our planet and those who seek to engineer new, expanded amino acid alphabets.
Footnotes
Acknowledgments
We acknowledge the access provided to the computational facilities of the Bioinformatics research unit at NUI Maynooth. This research was supported by the National Aeronautics and Space Administration through the NASA Astrobiology Institute under Cooperative Agreement No. NNA09DA77A issued through the Office of Space Science.
Disclosure Statement
No competing financial interests exist.