
Abstract
The polymerization of amino acids leading to the formation of peptides and proteins is a significant problem for the origin of life. This problem stems from the instability of amino acids and the difficulty of their oligomerization in aqueous environments, such as seafloor hydrothermal systems. We investigated the stability of amino acids and their oligomerization reactions under high-temperature (180–400°C) and high-pressure (1.0–5.5 GPa) conditions, based on the hypothesis that the polymerization of amino acids occurred in marine sediments during diagenesis and metamorphism, at convergent margins on early Earth. Our results show that the amino acids glycine and alanine are stabilized by high pressure. Oligomers up to pentamers were formed, which has never been reported for alanine in the absence of a catalyst. The yields of peptides at a given temperature and reaction time were higher under higher-pressure conditions. Elemental, infrared, and isotopic analyses of the reaction products indicated that deamination is a key degradation process for amino acids and peptides under high-pressure conditions. A possible NH3-rich environment in marine sediments on early Earth may have further stabilized amino acids and peptides by inhibiting their deamination. Key Words: Origin of life—Peptide formation—Pressure—Deamination. Astrobiology 11, 799–813.
1. Introduction
Although peptide formation from amino acid monomers is an endothermic reaction at room temperature and atmospheric pressure, it is thermodynamically favored to proceed at high temperatures (Shock, 1992). This fact has motivated many researchers to study the oligomerization of amino acids experimentally in high-temperature aqueous solutions, simulating hydrothermal systems near divergent seafloor margins (Huber and Wächtershäuser, 1998; Imai et al., 1999; Ogata et al., 2000; Kawamura et al., 2004; Cox and Seward, 2007; Lemke et al., 2009; Sakata et al., 2010). However, as mentioned above, because amino acid decomposition occurs rapidly at high temperatures (e.g., > 200°C), a hydrothermal origin of life that depends on peptides may be problematic (Miller and Bada, 1988; Cleaves et al., 2009). Therefore, a new approach is required to resolve this problem.
Nakazawa et al. (1993) and Nakazawa (2008) hypothesized that sub-seafloor sedimentary environments, where sediments are dehydrated at elevated temperatures and pressures during diagenesis and metamorphism, may have provided more favorable conditions for the polymerization of amino acids. The clay minerals in these sediments may also have played an important role in concentrating amino acids via surface adsorption (e.g., Bernal, 1949; Lambert, 2008). During diagenesis, marine sediments can be subjected to temperatures of ∼150°C and pressures of ∼200 MPa at 5 km below the seafloor, assuming a typical modern geothermal gradient. When these sediments are subjected to moderate-temperature and high-pressure metamorphism during their subduction with oceanic plates (e.g., blueschist), the temperatures and pressures reach 150–400°C and 0.5–1.5 GPa, respectively. Accompanying the reduction in porosity during these processes (e.g.,<20%; Bray and Karig, 1985), the water content of the sediments decreases significantly. In contrast to the hydrothermal model, a sub-seafloor sedimentary environment on early Earth may have allowed the concentration and oligomerization of simple organic compounds in compressional tectonic settings at convergent margins, where pressure could have promoted dehydration reactions. Based on this hypothesis, Ohara et al. (2007) conducted a series of experiments in which glycine was oligomerized at high temperature (150°C) and pressures (5–100 MPa). Peptides up to decamers were produced from solid glycine in the absence of a catalyst, and the starting amino acid was still detectable up to 32 days.
The importance of organic processes in compressional tectonic settings on early Earth has also been addressed by Holm and Neubeck (2009), who suggested that hydration/dehydration processes during the subduction of oceanic plates support the synthesis of various organic compounds (including amino acids, purine, and ribose) and their precursors (including HCN and formaldehyde). During plate subduction, these organic compounds may have experienced very high temperatures and pressures (e.g., 300–400°C and 2–3 GPa, respectively). The important role of pressure in the origin of life was also addressed by Hazen et al. (2002), who suggested that pressure increases the stability of organic molecules at high temperature. However, the stability and decomposition processes of amino acids are poorly understood under these conditions. Therefore, we investigated (i) the stability of amino acids (glycine and alanine) under high-temperature (from 180°C to 400°C) and high-pressure conditions (from 1.0 to 5.5 GPa); (ii) the effects of temperature and pressure on the oligomerization of different amino acids in the solid state; and (iii) the factors that control the stability of amino acids and peptides under the high-temperature and high-pressure conditions. We used somewhat excessive experimental conditions compared with the temperature and pressure conditions expected in diagenetic/metamorphic environments to accelerate the reactions and investigate the processes that occur during the oligomerization and decomposition of amino acids on a laboratory timescale.
2. Materials and Methods
2.1. Starting materials and experimental procedures
We used glycine or
All experiments were conducted with 1500 and 2000 t belt-type presses at the National Institute for Materials Science, which have been used in numerous high-temperature and high-pressure studies (e.g., Taniguchi and Yamaoka, 2001; Taniguchi et al., 2004). The experimental cell structure is illustrated in Fig. 1. The gold capsule containing the amino acid powder was put into a graphite heater, together with a pressure medium: NaCl+10 wt % or 20 wt % ZrO2. The assembled cell was placed between the anvils in the press and then compressed. The pressure was calibrated by using the phase transitions of Bi, Tl, and Ba with increasing pressure in a previous test run (Hong et al., 1999). Heat was generated by passing a current through the graphite heater. The reaction temperature was estimated from the relationship between the applied voltage and the measured temperature, acquired in a previous test run. After the samples were heated at a rate of ∼50°C/min, a reaction was assumed to have started when the temperature reached a set value. The estimates of temperature and pressure made with these methods had standard errors of ±20°C and ±0.2 GPa, respectively.
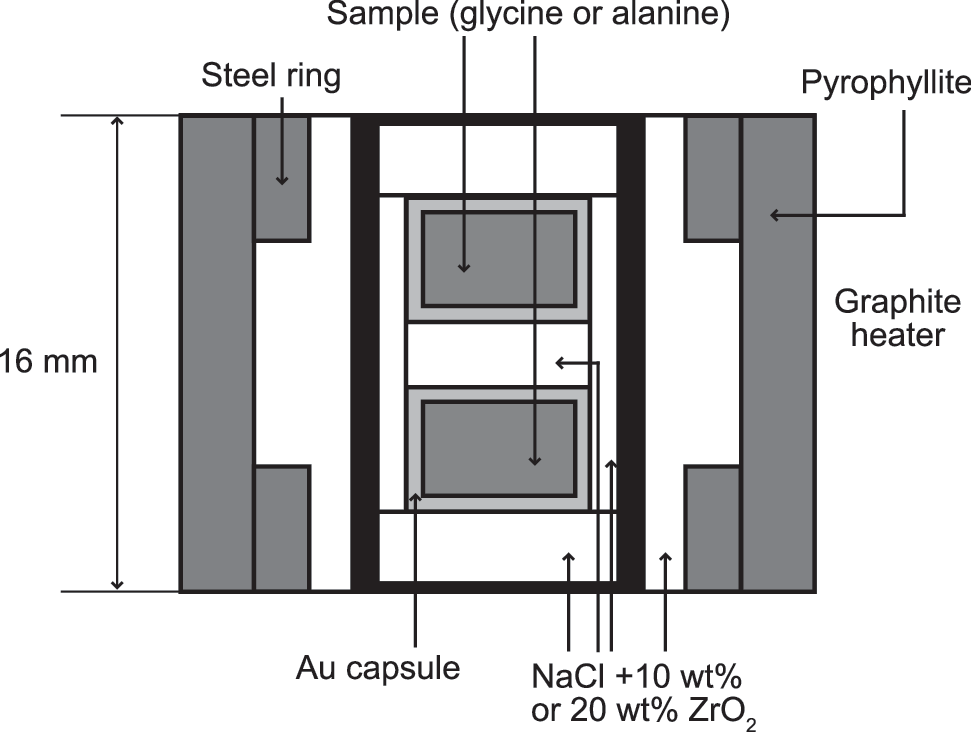
Schematic representation of the cell structure used in the experiments.
We conducted two series of experiments. In the first series (Series 1), the pressure and reaction time were fixed at 2.5 GPa and 2 h, respectively, and the temperature was varied from 180°C to 400°C to investigate the effects of temperature on the stability and oligomerization of amino acids. In the second series (Series 2), the temperature was fixed at either 180°C (Series 2-1) or 250°C (Series 2-2), while the reaction time was varied from 2 to 24 h at two different pressures (1.0 or 2.5 GPa for Series 2-1 and 2.5 or 5.5 GPa for Series 2-2), to investigate the effects of pressure on the stability of amino acids and the kinetics and degree of oligomerization of the amino acids. After the experiment, the samples were recovered and then weighed before the seal was broken to ascertain that the system was closed during the experiments. The weights of the samples changed no more than 2 mg (i.e.,<1.5% of the total weight) after each run. Some of the loss may be attributable to a weighing error or the leakage of volatile compounds (e.g., water, CO2, NH3) produced during the experiments. However, since the losses were less than 1.5% of the total weight, it was assumed that they did not affect the experimental results. After the reaction products were recovered, they were stored at −20°C.
2.2. Analysis of reaction products
The recoveries of the starting amino acids and the yields of peptides produced were analyzed by liquid chromatography/mass spectrometry (2695 separation module and Quattro micro API; Waters Corp.). For the peptide analyses, a reverse-phase column (Atlantis T3, 2.1 mm i.d. ×150 mm, 3 μm; Waters Corp.) was used at 30°C with an eluent mixture of A, an aqueous solution containing an ion-pairing reagent (5 mM undecafluorohexanoic acid), and B, acetonitrile. The A:B ratios were 98:2 and 85:15 for the glycine and alanine peptide analyses, respectively. The flow rate was 2 mL/min. Samples for the analyses were prepared by dissolving ∼5 mg of the reaction product in 1 mL of eluent and then centrifuging the sample at 15,000 rpm for 30 min to remove any possible insoluble residue. Positive-mode electron spray ionization was used to ionize the products. The peptides in the reaction products were identified and quantified from their peaks on selected-ion chromatograms by comparing the reaction products with solution samples containing known concentrations of peptide standards (Fig. 2). The starting amino acids were also analyzed to confirm that the amino acids were not contaminated with the peptides that were the targets of this study. For the amino acid analyses, a hydrophilic interaction liquid chromatography column (Zorbax HILIC Plus, 2.1 mm i.d.×100 mm, 3.5 μm; Agilent Technologies, Inc.) was used with an eluent mixture of A:B=10:90, where A was 10 mM ammonium formate solution and B was acetonitrile. The other conditions were the same as those for the peptide analyses. The recoveries of the starting amino acids in some reaction products were also analyzed with another high-performance liquid chromatography system (L-7100, Hitachi, Ltd.) with post-column derivatization using o-phthalaldehyde (Ohara et al., 2007). The recoveries (%) of the starting amino acids were calculated as 100 × (moles of amino acid in the reaction products)/(moles of starting amino acid). The yields of peptides (%) were calculated as 100×(moles of peptide detected in the reaction products)/(maximum moles of peptide that could be produced from the initial amount of amino acids). For example, in the case of Run# 13-1, 1.35 mmol of starting alanine was used, from which 0.5×1.35 mmol of the dimer could be produced. Because 1.01 μmol of the dimer was actually detected in the reaction product, the yield of the dimer was calculated to be 100×(1.01×10−6)/(0.5×1.35×10−3)=0.15%.
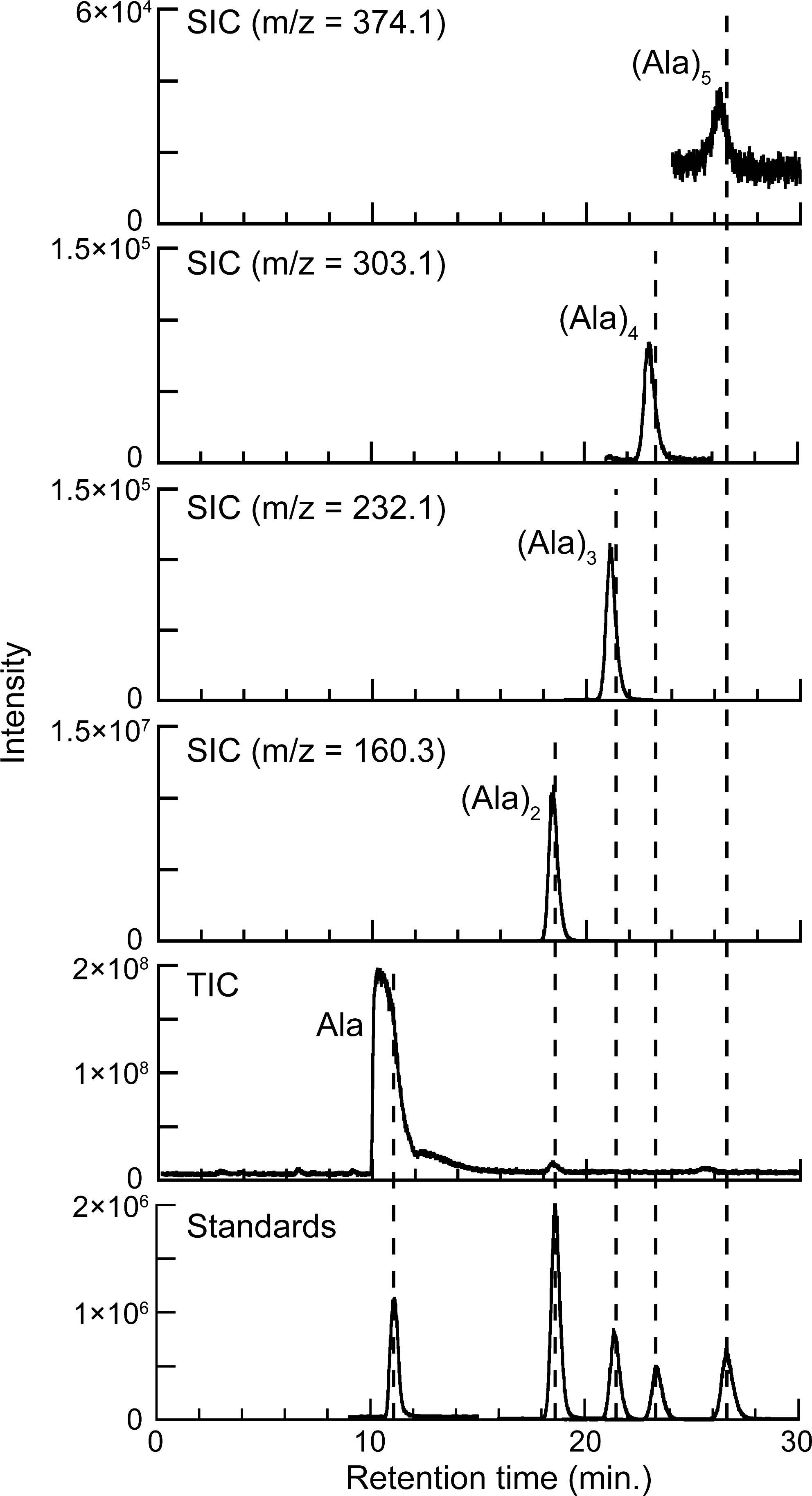
Liquid chromatography/mass spectrometry chromatograms of the reaction products of an alanine experiment (250°C, 5.5 GPa, 2 h) and a standard solution containing alanine and alanine peptides (∼5 μM, from monomers to pentamers). Alanine peptides were identified and quantified on selected-ion chromatograms (SIC) for mass-to-charge ratios (m/z) corresponding to each peptide. The presence of the starting alanine is shown on a total-ion chromatogram (TIC), which was integrated in the range of m/z 80–400. The yield of the starting material was quantified separately with a different method (see Materials and Methods section in the text). The slight differences in the retention times of the peptides between the reaction products and the standard solutions result from the high concentration of alanine in the reaction product.
Some reaction products were also analyzed to investigate the decomposition processes of the amino acids under the experimental conditions used, based on the (a) elemental composition, (b) IR absorption spectra, and (c) stable isotope compositions of the reaction products. A portion of each reaction product was freeze-dried for 24 h to remove any volatile components. The change in the elemental composition of a sample during freeze-drying was estimated by analyzing the elemental contents (C, N, and H) before and after the process with an elemental analyzer (EA; Carlo Erba 1108). The remaining fraction of the analyzed samples was assumed to be oxygen because no other element was present in the starting materials. The IR absorption spectra of the starting amino acids and reaction products were obtained with a Spectrum 400 FT-IR/FIR (Perkin Elmer). The samples were mixed with powdered KBr (for IR; Wako Pure Chemical Industries, Ltd.) in a sample/KBr ratio of ∼0.2 wt %, and then ground to make a pellet. The IR spectra were obtained at a resolution of 2 cm−1 between 500 and 3500 cm−1 wavenumber regions. The N isotope compositions of the starting materials and the freeze-dried reaction products were determined with an EA (Fisons NA-1500)–isotope ratio mass spectrometer (Finnigan delta S) at Kyushu University, converting N in the reaction products to N2 gas. The C isotope compositions of the starting materials and freeze-dried reaction products were determined with an EA (Calro Erba 1108)–isotope ratio mass spectrometer (Finnigan MAT 252) at Tohoku University, converting C in the reaction products to CO2 gas. The N and C isotope compositions are reported with the conventional δ notation as the per mill (‰) deviation of the isotope ratios (15N/14N and 13C/12C) of the sample relative to those of atmospheric nitrogen and Pee Dee belemnite (PDB) carbonate standards, respectively, using the following equations:
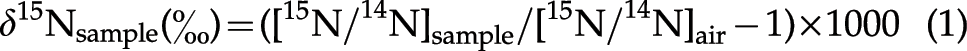
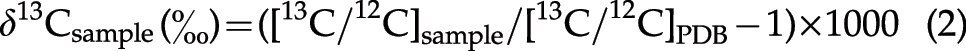
The differences between the isotope compositions of the reaction products at various reaction times (
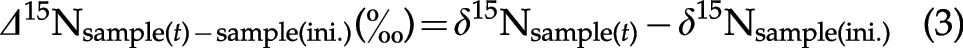
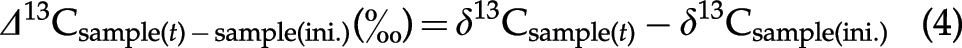
The reproducibilities of these analyses were within±0.3‰ (2σ) for δ15N and±0.4‰ (2σ) for δ13C.
3. Results
3.1. Recovery of the starting amino acids and yields of the peptides produced
The analysis of the reaction products by liquid chromatography/mass spectrometry revealed that peptides up to pentamers were formed from both glycine (Gly)5 and alanine (Ala)5 (Fig. 2). A series of experiments showed that (1) the amino acids were more stable at higher pressures and lower temperatures; (2) longer peptides tended to form at lower temperatures, shorter reaction times, and higher pressures; and (3) the yields of peptides at a fixed temperature and reaction time were higher at higher pressures.
In the Series 1 experiments, reaction temperature was varied from 180°C to 400°C, with a reaction time of 2 h and a pressure of 2.5 GPa. A large difference in the recovery of the starting amino acids was observed between 250°C and 300°C (Fig. 3). The recovery of the starting amino acids (both glycine and alanine) decreased significantly, and no peptides were detected at temperatures above 250°C. When we compared the runs at 180°C and 250°C, the experiments at lower temperatures produced longer peptides [e.g., up to pentamers (Gly)5 in the glycine experiment and tetramers (Ala)4 in the alanine experiment].
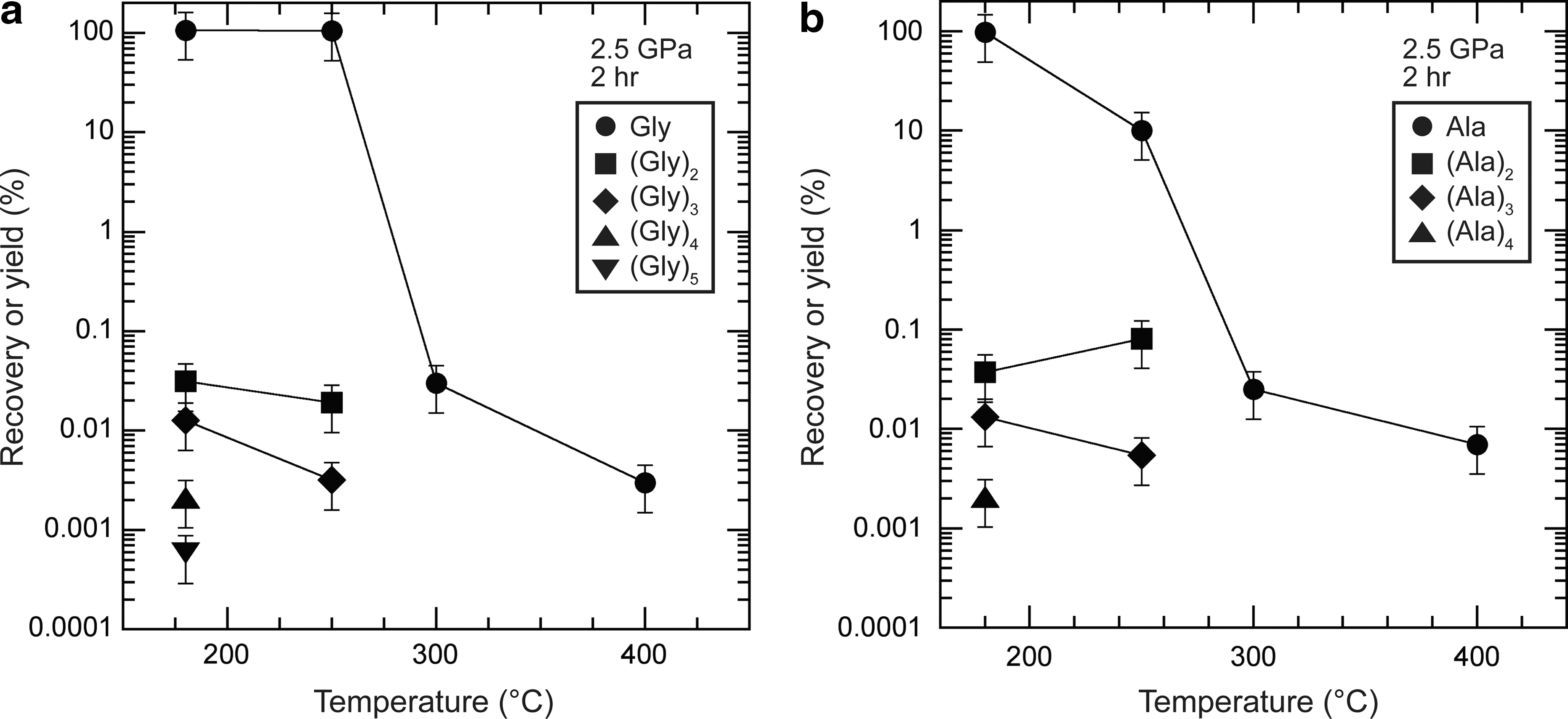
Recoveries of the starting amino acids and yields of the peptides produced as a function of temperature at 2.5 GPa after 2 h, when using either (
In the Series 2-1 experiments, no significant reduction in the recovery of the starting amino acids was observed up to 12 h at 1.0 GPa in either the glycine or alanine experiment (Fig. 4a and 4b). However, there was a measurable reduction in the recovery at 24 h (∼1%). At lower pressures, the peptides detected were up to trimers in both the glycine (Gly)3 and alanine (Ala)3 experiments after heating for 2 h. Whereas the yields of alanine peptides remained relatively constant over 24 h, the yields of glycine peptides increased from 12 to 24 h. At higher pressure (2.5 GPa), the starting amino acids were stable over 24 h (Fig. 4c and 4d). The peptides detected were up to pentamers in the glycine experiment (Gly)5 and tetramers in the alanine experiment (Ala)4 after heating for 2 h. The yields of all the peptides were higher in the 2.5 GPa experiments than in the 1.0 GPa experiments, except for those treated for 24 h.
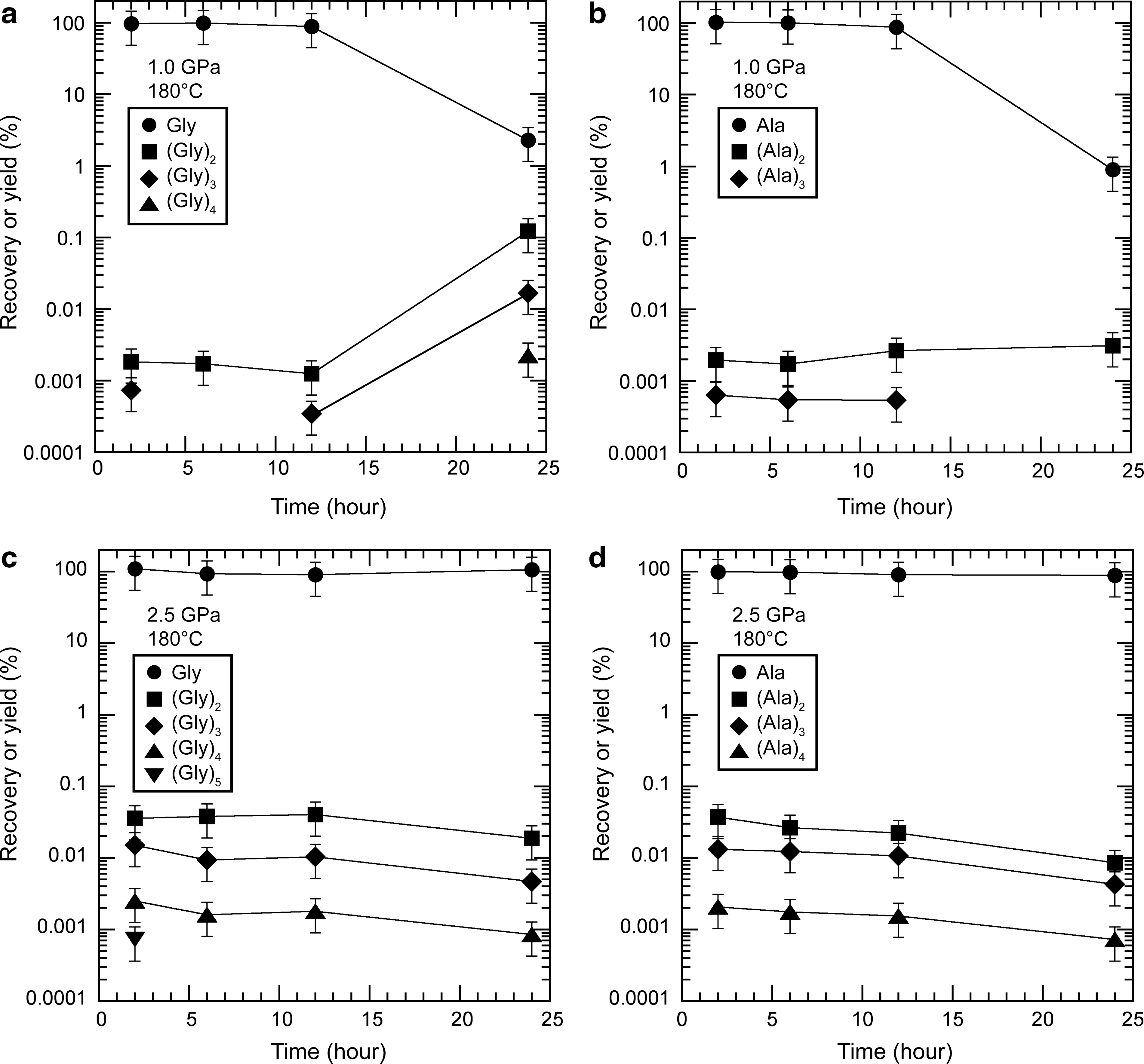
Recoveries of the starting amino acids and yields of the peptides produced as a function of the reaction time at 180°C in (
In the Series 2-2 experiments, when the reaction time was varied from 2 to 24 h at 250°C and 2.5 GPa, peptides were formed in the first 2 h in both the glycine and alanine experiments (Fig. 5a and 5b). However, the yields of peptides decreased significantly after 6 h in both the glycine and alanine experiments, which corresponds to the reduced recovery of the starting amino acids. When the experiments were conducted at 5.5 GPa, the starting amino acids were still present after 24 h in both the glycine and alanine experiments (Fig. 5c and 5d). The peptides were formed within 2 h and reached steady-state values (or more precisely, quasi–steady-state values). Hereafter, we use the term “steady state” to mean quasi–steady state (Lasaga, 1998). The yields increased significantly in both the glycine and alanine experiments compared with those at 2.5 GPa. Furthermore, no clear reduction in the yield of peptides was observed over 24 h.
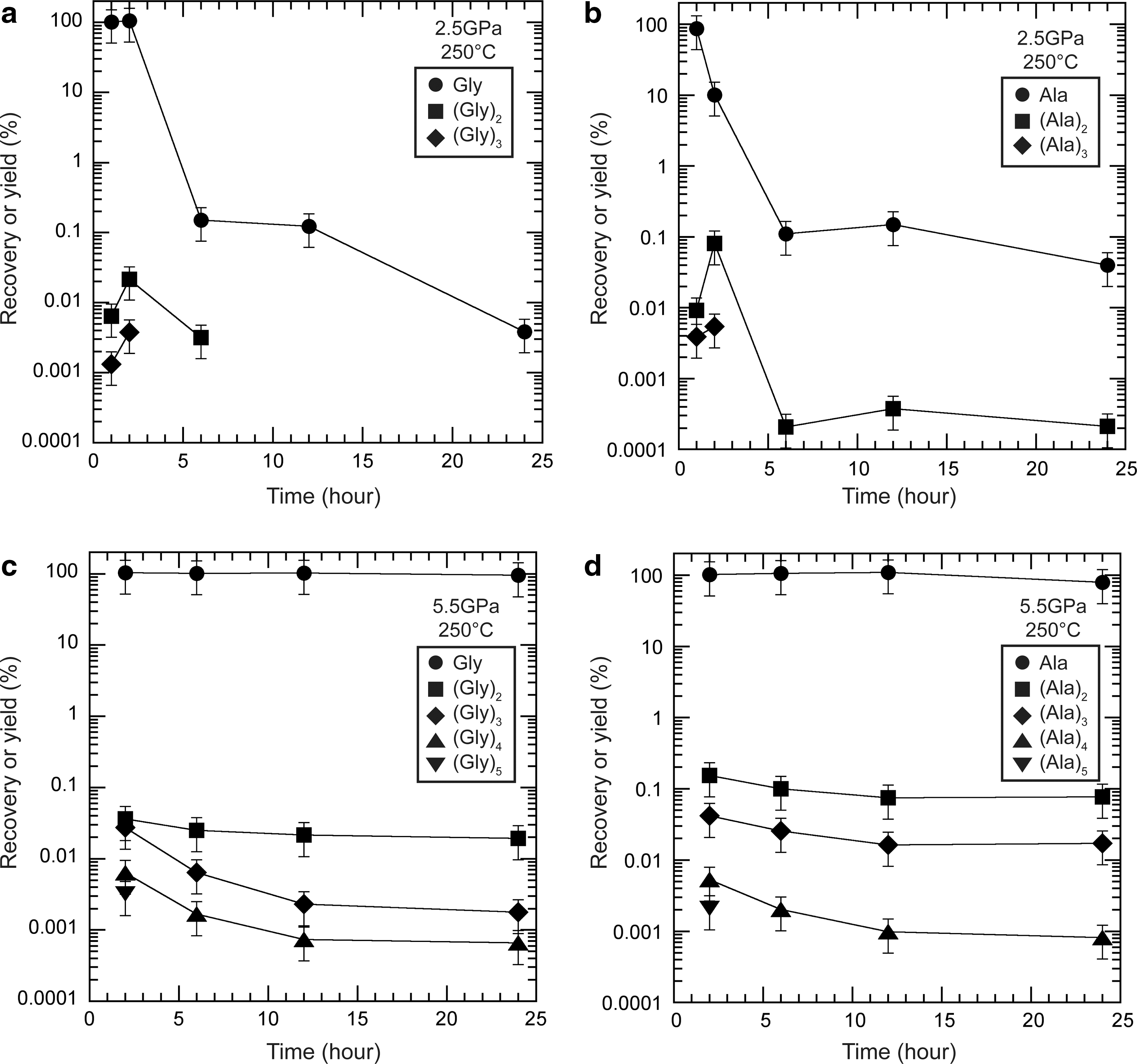
Recoveries of the starting amino acids and yields of peptides produced as a function of the reaction time at 250°C in (
3.2. Elemental compositions of the reaction products
The elemental compositions of the reaction products in Series 2-2 were determined with an EA both before and after freeze-drying. Although the weight of the reaction products at 5.5 GPa did not change significantly, some 2.5 GPa reaction products (e.g., produced at 400°C for 2 h or at 250°C for 24 h) lost as much as 50% of their initial weight. Freeze-drying the reaction products tended to increase the fraction of C, which indicates that one or more of the other elements (N, H, and O) were contained in volatile components and preferentially lost during freeze-drying. The freeze-dried samples of the Series 2-2 experiments at the lower pressure (variable reaction times at 250°C and 2.5 GPa) showed a reduction with time in the N/C ratio (from 0.5 to ∼0.25 for glycine and from 0.33 to ∼0.2 for alanine) and in the O/C ratio (from 1.0 to ∼0.4 for glycine and from 0.67 to ∼0.3 for alanine), but the samples after 6 h showed almost no change in these ratios (Fig. 6). Whereas no change in the N/C or O/C ratio was observed in the freeze-dried samples in the glycine experiments at the higher pressure (5.5 GPa), these ratios decreased slightly under the high-pressure conditions in the alanine experiment (Fig. 6b). The H/C ratios of the reaction products also decreased in accord with the reductions in the N/C and O/C ratios.
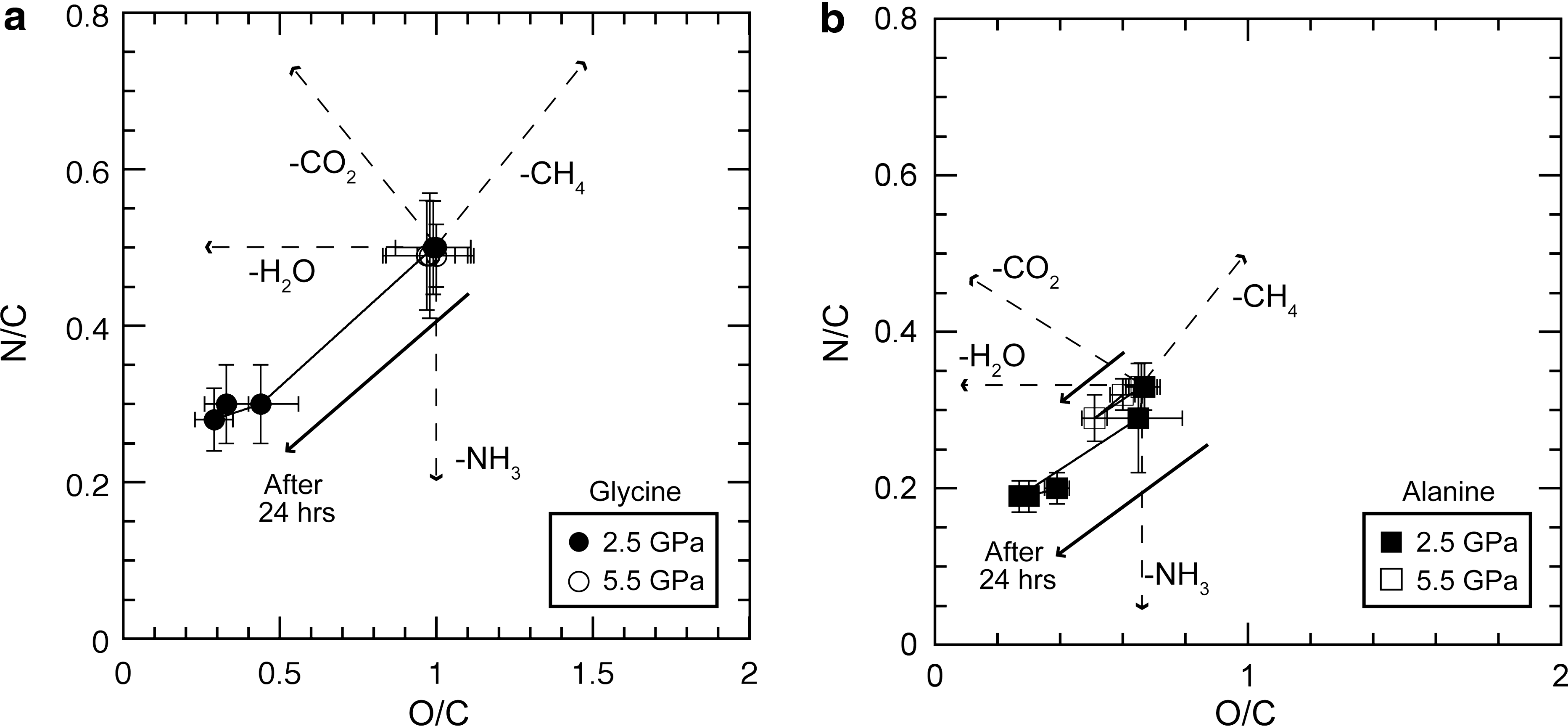
N/C–O/C diagrams for the products after various reaction times (2–24 h) in (
3.3. IR spectra
The IR spectra of the starting amino acids and reaction products were used to identify the functional groups that remained after the experiments. The presence of functional groups in our samples is indicated by peaks at the following wavenumbers: ∼1500, 2610, and ∼3100 cm−1 for the amino group and ∼1380 and ∼1630 cm−1 for the carboxyl group (Fig. 7a and 7c). Note that there is a ∼10 cm−1 shift in those peaks between glycine and alanine and that the two strongest peaks at 1595 and 1630 cm−1 were probably contributed by both the amino group and the carboxyl group. The assignments were based on previous experimental (Tsuboi et al., 1958; Rubinsztain et al., 1984; Garcia et al., 2008; Suriya Kumar et al., 2008) and theoretical studies (Fischer et al., 2005; Kumar et al., 2006; Chowdhry et al., 2008) of the IR responses of solid amino acids. Other functional groups containing N–C=O bonds (e.g., amide groups or peptide bonds) were not distinguished from amino groups or carboxyl groups because a solid amino acid tends to have complex inter- and intramolecular hydrogen bonding between the two functional groups (Garcia et al., 2008).
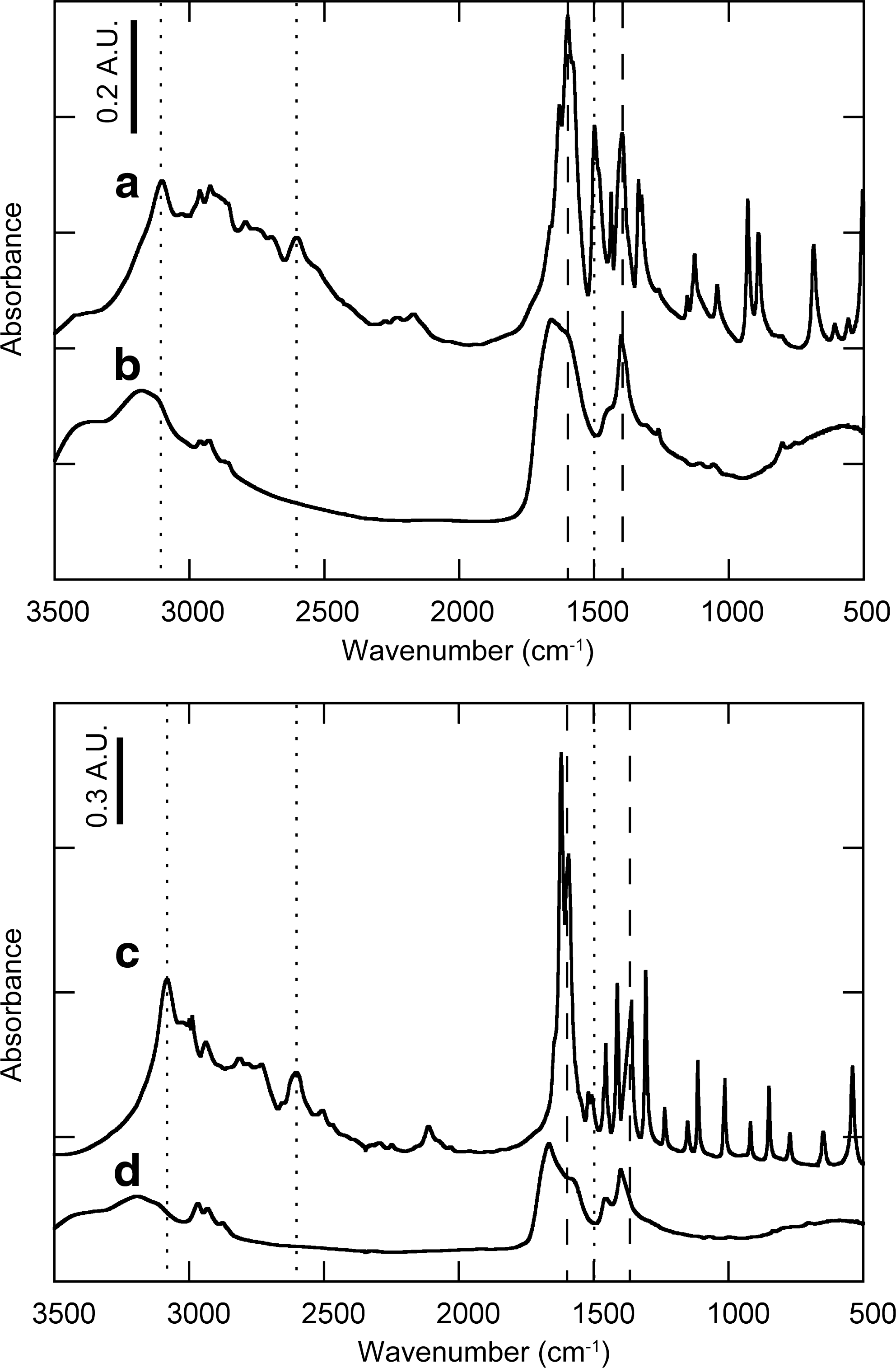
IR spectra of (
The IR spectra of the reaction products at 2.5 GPa in the Series 2-2 experiments (variable reaction times at 250°C) show that all the peaks observed in the starting amino acids either disappeared or were reduced in intensity (Fig. 7b and 7d). The prominent peaks observed in the IR spectra of the reaction products are at ∼1400, ∼1460, ∼1600, and ∼1670 cm−1. Among these, the peaks at ∼1400 and ∼1670 cm−1 were attributed to the carboxyl group (Rubinsztain et al., 1984), which indicates that there were significant amounts of carboxyl group left in the reaction products. However, these peaks are shifted to larger wavenumbers by ∼30 cm−1, which may have resulted from the dehydration of the carboxyl group (transforming it to a carbonyl group). Conversely, no amino groups were identified in the reaction products. A few other peaks were also observed between 2850 and 3000 cm−1, which were attributed to CH n vibrations (Kumar et al., 2006; Garcia et al., 2008). The reaction products after 6, 12, and 24 h at the lower pressure in the Series 2-2 experiments (both glycine and alanine) produced almost the same IR spectra.
3.4. C and N isotope analyses
The C and N isotope compositions of the starting amino acids and reaction products were analyzed to investigate how C and N are released from amino acids under high-temperature and high-pressure conditions. In the Series 2-2 experiments, no change in the isotope compositions was observed at the higher pressure (5.5 GPa). At the lower pressure (2.5 GPa), the δ15N values of the reaction products were up to ∼7‰ heavier relative to those of the starting amino acids (Fig. 8a). In the glycine experiments, the N isotope compositions of the reaction products appeared to reach a constant value after 6 h that was ∼7‰ heavier than that of the starting glycine. In contrast, in the alanine experiments, the N isotope compositions decreased with time, from ∼6‰ heavier than that of the starting alanine at 2 h to 3‰ heavier at 24 h. However, the N lost as volatile compounds, estimated from the elemental compositions before and after freeze-drying and the total weight loss during freeze-drying, remained roughly constant after 6 h in both the glycine and alanine experiments (Fig. 8b).

(
The C isotope compositions of the reaction products also showed different trends in the glycine and alanine experiments (Fig. 8c). Although up to 20% of C was estimated to have been lost as volatile components (Fig. 8d) in the alanine experiments, the isotope composition did not change, except for a sample at 2 h that was slightly (∼1‰) enriched in 13C. Conversely, at 2.5 GPa, the reaction products in the glycine experiments were depleted in 13C. The δ13C values of the reaction products were ∼4‰ lighter than that of the starting glycine.
4. Discussion
4.1. Oligomerization of amino acids under high-pressure conditions
In the Series 2 experiments, the recovery of the staring glycine and alanine significantly increased with increasing pressure from 1.0 to 2.5 GPa at 180°C (Series 2-1; Fig. 4) and from 2.5 to 5.5 GPa at 250°C (Series 2-2; Fig. 5), which indicates that amino acids are more stable at higher pressures, at least under anhydrous conditions. However, even under high-pressure conditions (e.g., 2.5 GPa), the amino acids did not survive at temperatures above 250°C (Fig. 3) after 2 h, which suggests that the stability of amino acids is very sensitive to temperature and that pressure alone may be unable to stabilize amino acids over 250°C. In further experiments carried out at 5.5 GPa and 300°C for 2 h, the recovery of both glycine and alanine was also low (<1%).
When the pressure was increased in the Series 2 experiments, the yields of all the peptides produced from glycine and alanine increased (Figs. 4 and 5). At higher pressures, these amino acids were oligomerized up to pentamers, (Gly)5 and (Ala)5. The formation of alanine pentamers was particularly noteworthy because no previous study has reported the formation of alanine peptides longer than tetramers in the absence of a catalyst. For example, the dimerization of an amino acid can be written as
where R1 indicates the amino acid side chain (H for glycine, CH3 for alanine). In aqueous solution, the dimerization of amino acids is not thermodynamically favored under increased pressure because of the increase in the total molar volume (Shock, 1992). However, in our study, pressure did not suppress the formation of peptides. We attribute the formation of peptides to (1) the increased stability of the amino acids; (2) the elevated temperature; and (3) the low water activity. Because oligomerization is sensitive to temperature, the presence of amino acids for a long period of time at high temperature is important for the formation of peptides, as was suggested by Lemke et al. (2009) in an aqueous system. Whereas factors (2) and (3) have also been suggested in previous studies (e.g., Meggy, 1953, 1956), we propose that pressure plays a key role in stabilizing amino acids and peptides at temperatures greater than 100°C under anhydrous conditions. We will discuss the factors that control the stability of amino acids at high pressure in the next section. These results are also consistent with a previous study by Ohara et al. (2007), in which they oligomerized glycine powder at 150°C and 100 MPa. Compared with our study, they produced longer oligomers (up to decamers) at a lower temperature and with a longer reaction time (e.g.,∼32 days). Therefore, we suggest that moderate temperatures (e.g., 100–150°C) favor the production of longer peptides when the reaction time is prolonged.
The exact reaction mechanisms underlying the oligomerization of amino acids have not been clarified by these experimental studies. However, the results of all our experiments show that the yields of (Gly) n+1 and (Ala) n+1 were 2–10 times smaller than those of (Gly) n and (Ala) n , except for (Gly)2 and (Ala)2, which were 2–5 orders of magnitude smaller than the recoveries of their monomers (Figs. 3, 4, and 5). This may be due to the formation of cyclic dimer peptides, diketopiperazine (DKP). Although the formation of DKP was confirmed in both the glycine and alanine experiments, they were not to be quantified in this study. The large difference between the yields of dimers and the recoveries of their monomers may also indicate that not all the starting amino acid contributed to the oligomerization reaction because oligomerization occurred at local sites (e.g., at the grain boundary or in the aqueous phase). Although our experiments commenced with no free water molecules, water was probably involved in the reactions during the experiments, produced due to the dehydration of the amino acids (Fig. 6). The presence of small amounts of water may play a role in facilitating the transport of the reactants and products. For example, Meggy (1953) showed that DKP powder was polymerized at 180°C when small amounts of water (molar ratio of water/DKP≈0.5) was added to the system. Therefore, if the oligomerization actually occurred in the aqueous phase in our experimental systems, the yields of peptides may be improved by the addition of a small amount of water.
In the Series 2-2 experiments, the yields of glycine and alanine peptides were the highest at 2 h at both 2.5 and 5.5 GPa (Fig. 5). These experimental results indicate that the effect of pressure on the kinetics of peptide formation (e.g., Reaction 5) is small. In contrast, temperature significantly affected the rate of peptide formation. In the Series 2-1 experiments, the yield of glycine peptides increased after 24 h at 1 GPa (Fig. 4a). Although the same trend was not observed in the alanine experiment, this raises the possibility that the yields of the peptides would have increased at 180°C in the 2.5 GPa experiments (Fig. 4c and 4d) if the reaction time had been extended to more than 24 h. The effect of temperature on the kinetics of peptide formation has also been discussed by Sakata et al. (2010), who showed that the rate constant for the dimerization of glycine (Reaction 5) at 250°C is more than one order of magnitude higher than that at 180°C (activation energy of 88 kJ/mol for Reaction 5), which also supports the possibility that our peptide yields would have increased given more time. It is also consistent with the study of Ohara et al. (2007), in which they produced the highest yields of glycine peptides after heating for 8 days at 150°C. However, the reverse reaction of amino acid dimerization, peptide hydrolysis, did not appear to be as fast as reported by Sakata et al. (2010), which is attributable to the low water activity in our experiments, as follows:
At 180°C and 1.0 GPa (Fig. 4a), the recovery of the starting glycine decreased significantly, and the yields of the peptides increased, which suggests that the hydrolysis of the peptides was slow enough to be negligible. Conversely, at 250°C and 2.5 GPa (Fig. 5a and 5b), the yields of peptide decreased, with a reduction in the recovery of the starting amino acids. This may be attributable to the hydrolysis of the peptides to amino acids that was accelerated by the temperature increase and/or the thermal decomposition of the peptides occurring coincidently with the thermal decomposition of the starting amino acids.
Our study also shows that the effects of pressure on the stability and oligomerization of amino acids are similar for glycine and alanine. The yields of peptides of both amino acids increased significantly with increasing pressure. In contrast, the effects of temperature on the stability of the peptides differed slightly between glycine and alanine. At 180°C (Series 2-1), whereas alanine peptides were identified up to tetramers (Ala)4, glycine peptides were identified up to pentamers (Gly)5 (at 2.5 GPa for 2 h; Fig. 4 c and 4d). At 24 h and 1.0 GPa (Fig. 4a and 4b), the yields of glycine peptides were greater than those of alanine peptides. These results indicate that, at that temperature, glycine peptides decompose more slowly than alanine peptides. However, at 250°C, the yields of alanine peptides were higher than those of glycine peptides at both 2.5 and 5.5 GPa, which indicates that alanine peptides decompose more slowly than glycine peptides at 250°C. These results suggest that the thermal stability of peptides varies, depending on the constituent amino acids. This may be attributable to their different sublimation temperatures, which stem from their molecular weights, as suggested by Bertrand et al. (2009) to explain the selective survival of amino acids under high-temperature and high-pressure conditions.
4.2. Factors controlling the stability of amino acids
The elemental ratios of the reaction products showed that, at 2.5 GPa and 250°C, both the N/C and O/C ratios decreased with time (Fig. 6). Because this was concurrent with the reduction in the recovery of the starting amino acids (Fig. 5a and b), the reductions in these ratios are attributed to the decomposition of the starting amino acids. The simplified degradation reactions for the amino acids can be written as
or
where R1 indicates the amino acid side chain (H for glycine, CH3 for alanine). Although H was added to balance the reactions in (7) and (9) for simplification, an alternative pathway is the polymerization of the residues by C–C bonding. Based on the trends observed in the elemental compositions of the reaction products (Fig. 6), dehydration (Reaction 7) and deamination (Reaction 9) appear to be the primary processes involved in the decomposition of the amino acids under our experimental conditions. The IR spectra also indicate that deamination occurred and that deamination was probably more prominent than decarboxylation (Fig. 7). The possible formation of carbonyl groups, inferred from the shift in the peaks at ∼1380 and ∼1630 cm−1, is also consistent with a dehydration reaction during the decomposition of the amino acids.
No isotope fractionation occurred in either the glycine or alanine experiments at 5.5 GPa, where the losses of N and C were almost negligible (Fig. 8b and 8d). In contrast, the N isotope compositions in the 2.5 GPa reaction products were heavier than those in the starting materials. This can be explained by a kinetic isotope effect in a simple irreversible process, deamination. Because of the weaker bond strength of the lighter isotope, deamination occurs faster for the lighter isotope, leaving the heavier isotope in the reaction product. This was observed in both the glycine and alanine experiments (Fig. 8a). However, in the alanine experiments, the N isotope composition of the reaction products became ∼2‰ lighter from 2 to 6 h. We attribute this lightening to the reincorporation of degassed ammonium into the reaction products. This can be described as the reverse of the deamination process, toward an equilibrium between the amino acid and ammonia:
Alternatively, N can also be reincorporated into the reaction product by other processes, such as the formation of C=N or nitrogen-containing heterocyclic compounds (e.g., pyridine), which are more stable than amines. We suggest that the latter is the more likely mechanism. Therefore, we can explain the lack of an amino group in the reaction products detected by IR spectroscopy (Fig. 7), even though the N/C ratio in the reaction product was still roughly half that of the starting material (Fig. 6). The formation of nitrogen-containing heterocyclic compounds during the pyrolysis of amino acids has also been suggested by previous researchers (Kato and Tsuchida, 1981; Rubinsztain et al., 1984; Basiuk and Douda, 2000). However, because IR spectroscopy may not be very sensitive to the amino group compared with the carboxyl group, it is still possible that some amino groups persisted in the reaction product throughout Reaction 10.
The C isotope data show a different trend between glycine and alanine at 2.5 GPa. Whereas almost no fractionation of the C isotopes was observed in the alanine experiment, C in the reaction products of the glycine experiment at 2.5 GPa was isotopically lighter than that in the starting material. The depletion of 13C in the reaction product probably reflects the heterogeneity of the C isotope compositions of amino acids. Rustad (2009) used ab initio methods to theoretically calculate the C isotope compositions of various amino acids and showed that the carbon of the carboxyl group is ∼29‰ heavier than the α-carbon in glycine. Therefore, even when a kinetic isotope effect associated with decarboxylation causes several per mill (‰) enrichment of 13C in the remaining carboxyl group, it would have resulted in the depletion of 13C as whole molecules in the reaction product. Based on the estimated C loss, ∼40% of the carboxyl groups may have been lost, assuming that no α-carbon was lost during the experiments. Therefore, decarboxylation must have occurred to some extent.
We suggest that the survival of some carboxyl or carbonyl groups, or both, in the reaction products at high temperatures was attributable to the high pressures applied in the experiments. Under ambient pressure conditions (1 atm), the pyrolysis of amino acids by heat yields CO2 as a major product, which indicates that decarboxylation is the primary process in the decomposition of amino acids (e.g., Rodante, 1992; Li and Brill, 2003). Although the application of pressure seems to effectively prevent decarboxylation during the decomposition of amino acids, it may not slow their deamination compared with their decarboxylation. Deamination has been suggested to proceed through polar transition states (Michels et al., 1996). In such processes, increasing the pressure may facilitate a reaction because of the electrostriction of the solvent (Van Eldik et al., 1989). Sato et al. (2004) also pointed out that deamination markedly occurs in the decomposition of amino acids under moderate-pressure hydrothermal conditions (200–340°C, 20 MPa). Therefore, we propose that deamination is a key process that determines the stability of amino acids and peptides under high-pressure conditions. Consequently, to stabilize amino acids and peptides under geologically relevant conditions and on geologically relevant timescales, deamination must be prevented. In one possible scenario, the enhanced presence of ammonia/ammonium ions in the sediments could have further stabilized the amino acids and peptides during diagenesis/metamorphism, because the N isotope data in this study indicate that ammonium is reincorporated into the reaction products.
4.3. Astrobiological implications of our experimental results
Our experimental study has demonstrated that pressure inhibits the decomposition of anhydrous amino acids but not their oligomerization under high-temperature conditions (e.g.,<250°C). Both glycine and alanine were oligomerized up to pentamers during the experiments. These results indicate that amino acids were possibly oligomerized on early Earth in water-poor, high-temperature, and high-pressure environments. Diagenetic/metamorphic environments containing marine sediments may provide all these conditions. Although the dehydration of marine sediments progresses with decreasing porosity at depth (e.g.,<20% porosity at 3 km below the seafloor; Bray and Karig, 1985), free water molecules are probably still present in diagenetic/metamorphic environments. In these environments, amino acids may have dissolved in the pore fluids or adsorbed onto the surfaces of clay minerals. Therefore, although the results of our experimental study may not be directly applicable to those environments, they give fundamental insight into the stability and oligomerization of amino acids under water-poor, high-temperature, and high-pressure conditions. We should also note that these experiments were conducted in a closed system, although the natural diagenetic/metamorphic environments are expected to have been somewhat open in terms of fluids and volatile compounds. Water escaping from the system may have facilitated the formation of longer oligomers of amino acids with better yields compared with the results of the experiments reported here.
Although much previous research (e.g., Russell and Hall, 1997; Huber and Wächtershäuser, 1998; Shock and Schulte, 1998; Imai et al., 1999; Martin et al., 2008) has proposed that seafloor hydrothermal systems at divergent margins (on and near oceanic ridges) provide suitable conditions for the prebiotic synthesis of organic materials, only a few researchers (Nakazawa et al., 1993; Holm and Neubeck, 2009) have discussed the possible roles of Earth's dynamics at convergent margins in the origin of life. Our experimental study demonstrates the significance of pressure in the stability of organic compounds and their polymerization, and supports this idea. These data may also imply that plate tectonics was a prerequisite for the origin of life on early Earth (Holm and Neubeck, 2009) and possibly elsewhere, although the existence of plate tectonics on early Earth is controversial (e.g., Hamilton, 1998; Harrison et al., 2005; Moyen et al., 2006). Without knowing the detailed geothermal structures of early Earth (e.g., pressure-temperature conditions, subduction rate), it is difficult to estimate the timescale required for amino acids to be converted to more complex organic molecules (e.g., proteins). The cycling of subducted materials at convergent margins may have required more than millions of years (e.g., Muramatsu et al., 2001), which is much longer than our experimental timescale (24 h). Therefore, we applied much higher temperatures and pressures in this study than those expected in diagenetic/metamorphic environments to accelerate the reactions. In natural systems, the reactions (both oligomerization and decomposition) may have proceeded much more slowly under moderate-temperature and moderate-pressure conditions (e.g., < 150°C, <1 GPa).
The concentrations of organic compounds in the ocean on early Earth present a crucial problem for their polymerization because they are considered to have been too low for these reactions to have occurred, as first discussed by Bernal (1949). Instead, these organic compounds may have been concentrated in marine sediments by their adsorption onto clays (e.g., Lahav and Chang, 1976), possibly following their fluid migration in the sediments. Recently, Baaske et al. (2007) showed that single nucleotide molecules can be theoretically concentrated more than 108-fold by thermodiffusion through millimeter-sized pores. Although these researchers proposed pores in hydrothermal vents as the setting in which the concentration of organic materials might occur, we suggest that diagenetic/metamorphic environments may provide such fluid migration on a much larger scale. Therefore, various organic materials, including peptides, could have been differentiated and concentrated in sediments by similar mechanisms (e.g., Wing and Bada, 1991).
Some questions regarding the stability and oligomerization of amino acids in diagenetic/metamorphic environments are not answered by this study. For example, although the effects of various minerals (e.g., clays, oxides) on the stability and oligomerization of amino acids has been investigated by various researchers (see a review by Lambert, 2008), no study has investigated the stability of amino acids under high-pressure conditions in the presence of minerals. Because many organic compounds, including amino acids, are expected to adsorb to the surface of clay minerals or intercalate between layers of clays in marine sediments, the stability and oligomerization of amino acids in these states must be investigated at high temperatures and high pressures.
Other parameters that may significantly affect the stability of amino acids in these environments are the pH and redox conditions, although these conditions in Hadean diagenetic/metamorphic environments are unclear. Increases in pH may increase the stability of amino acids by their speciation from the zwitterionic to anionic forms (Li et al., 2002). The dimerization rate of glycine at high pH is higher than at neutral pH (Sakata et al., 2010). The effects of redox conditions on the stability and oligomerization of amino acids are much less clear than the pH effects. In general, the stability of organic compounds is expected to be higher under more reducing conditions, where the oxidative degradation of organic compounds (oxidation to CO2) does not occur readily. Andersson and Holm (2000) examined the effects of a mineral redox buffer, a pyrite–pyrrhotite–magnetite assemblage, on the decomposition rates of amino acids under hydrothermal conditions (e.g., 200°C and 5 MPa). They showed that the decomposition rates for most amino acids were slowed by the presence of the mineral redox buffer. In contrast, Bada et al. (1995) showed that another mineral redox buffer, a quartz–fayalite–magnetite assemblage, did not affect the decomposition rate of alanine at 240°C. This discrepancy may stem from the lack of equilibrium attained between the mineral assemblages and the surrounding environments at the temperatures examined, and the short durations of the reactions (<100 h). A future experiment with high solid/solution ratios, which better simulate diagenetic/metamorphic environments, may potentially resolve this problem.
The isotope fractionation during the decomposition of amino acids demonstrated in this study may have implications for the use of N isotopes as a biosignature and for determining the N isotope composition of Earth's interior. Our experimental study showed that the devolatilization of amino acids resulted in the enrichment of 15N in the residual organic compounds (Fig. 8a) under high-temperature and high-pressure conditions, which suggests that the N isotope composition of mature organic matter (e.g., kerogen) must be interpreted with consideration of the metamorphic effect (e.g., Papineau et al., 2009). The metamorphic effect on the N isotope composition of
Although our experimental study demonstrated that amino acids are more stable at higher pressures, pressure alone may not stabilize amino acids at high temperatures (>180°C). Our study also suggests that deamination may be a key process determining the stability of amino acids and peptides under high-pressure conditions and that NH3-rich environments could further stabilize amino acids and peptides in sediments. Most researchers now agree that the concentration of ammonia in the atmosphere has been insignificant because of its rapid photochemical decomposition by UV (Kuhn and Atreya, 1979; Kasting, 1982). In contrast, the concentrations of ammonia and the ammonium ion in the ocean could have been elevated on early Earth because of the possible high flux of ammonia into the ocean by active submarine hydrothermal systems (Brandes et al., 1998, 2008; Smirnov et al., 2008) or frequent meteorite impacts, or both (Nakazawa et al., 2005; Furukawa et al., 2009). For example, Smirnov et al. (2008) estimated that the annual flux of ammonium ions from Hadean seafloor hydrothermal systems could have ranged from 5.8×108 to 1.9×1012 mol/year. Based on the results of Nakazawa et al. (2005), meteorite impacts could have supplied as much as ∼1012 mol/year ammonia during the Late Heavy Bombardment (4.0–3.8 Ga). Both of these sources would have provided a much greater ammonium flux than occurs in the modern ocean, although the ammonium flux into the ocean from modern seafloor hydrothermal systems is poorly understood (Tivey, 2007). A possible major sink for these ammonia/ammonium ions, other than atmospheric decomposition, would have been their burial with sediments after their adsorption onto clays. This may also be suggested geologically by the finding of
5. Conclusions
We conducted a series of experiments in which glycine or alanine was placed under high-temperature (180–400°C) and high-pressure (1.0–5.5 GPa) conditions. The experimental results are summarized as follows. (1) The recovery of the starting amino acids increased significantly from 1.0 to 2.5 GPa at 180°C and from 2.5 GPa to 5.5 GPa at 250°C, which indicates that amino acids are more stable under high-pressure conditions. (2) Both glycine and alanine were oligomerized up to pentamers under high-temperature and high-pressure conditions (e.g., 250°C, 5.5 GPa), which indicates that the effect of pressure on the kinetics of peptide formation was small compared with that of temperature. Although the effect of pressure on the oligomerization of the amino acids was similar for glycine and alanine, the effect of temperature differed slightly, which suggests that the thermal stability of peptides varies, depending on their constituent amino acids. (3) Pressure inhibited the decomposition of anhydrous amino acids. Based on the elemental composition and IR spectra of the reaction products, deamination and dehydration were the major processes that occurred during their decomposition under high-temperature and high-pressure conditions. Stable isotope analyses of the reaction products also revealed that both deamination and decarboxylation had occurred. (4) The N isotope compositions of the reaction products indicated that some ammonium released from the amino acids appeared to be reincorporated into the solid, either as an amino group or another nitrogen-containing group. The C isotope compositions of the reaction products indicated that the C in amino acids is isotopically heterogeneous.
These results suggest that compressional tectonic settings at convergent margins may have played an important role in stabilizing amino acids and peptides on early Earth.
Footnotes
Acknowledgments
We are grateful to Y. Kimura and H. Naraoka for allowing us to use their instruments for IR analyses and N isotope analyses, respectively. We thank Y. Ohtomo for her technical assistance and A. Ishida, I. Johnson, A. Lasaga, and M. Oba for their helpful comments, which improved the early manuscript. We also thank three anonymous reviewers and editor S. Cady for their constructive comments on the manuscript. This research was supported by the Global COE program “Global Education and Research Center of Earth and Planetary Dynamics” at Tohoku University and Grants-In-Aid for scientific research from the Japan Society for the Promotion of Science to T.O. (22740342), T.K. (21244080), and H.N. (21340162).
Author Disclosure Statement
The authors have no competing financial interests.
Abbreviations
DKP, diketopiperazine; EA, elemental analyzer.