
Abstract
Understanding the abiotic fixation of nitrogen and how such fixation can be a supply of prebiotic nitrogen is critical for understanding both the planetary evolution of, and the potential origin of life on, terrestrial planets. As nitrogen is a biochemically essential element, sources of biochemically accessible nitrogen, especially reduced nitrogen, are critical to prebiotic chemistry and the origin of life. Loss of atmospheric nitrogen can result in loss of the ability to sustain liquid water on a planetary surface, which would impact planetary habitability and hydrological processes that shape the surface. It is known that NO can be photochemically converted through a chain of reactions to form nitrate and nitrite, which can be subsequently reduced to ammonia. Here, we show that NO can also be directly reduced, by FeS, to ammonia. In addition to removing nitrogen from the atmosphere, this reaction is particularly important as a source of reduced nitrogen on an early terrestrial planet. By converting NO directly to ammonia in a single step, ammonia is formed with a higher product yield (∼50%) than would be possible through the formation of nitrate/nitrite and subsequent conversion to ammonia. In conjunction with the reduction of NO, there is also a catalytic disproportionation at the mineral surface that converts NO to NO2 and N2O. The NO2 is then converted to ammonia, while the N2O is released back in the gas phase, which provides an abiotic source of nitrous oxide. Key Words: Nitrogen fixation—Mars—Earth—Terrestrial—Abiotic—Prebiotic—Nitrogen—Planetary habitability—Biosignatures. Astrobiology 12, 107–114.
1. Introduction
N
For example, the current martian atmosphere contains only 0.2 mbar of nitrogen, while early Mars is thought to have had 10–1000 times the current pressure (McKay and Stoker, 1989), which would have been enough to support the presence of liquid water on the surface (Pieri, 1980; Carr, 1981; Squyres, 1984; Christiansen, 1989; Mancinelli and Banin, 2003; Bell et al., 2004; Christensen et al., 2004; Herkenhoff et al., 2004; Klingelhöfer et al., 2004; Squyres et al., 2004a, 2004b). Two possible outcomes for the fate of nitrogen are sequestration in the regolith and ejection into space by impactors (which is known as impact erosion; see Chyba, 1990; Mancinelli, 1996; Mancinelli and Banin, 2003; Manning et al., 2009). For example, pathways that allow nitrogen to be converted to ammonia could have caused nitrogen to become sequestered in clay minerals (Bada and Miller, 1968; Mancinelli et al., 1998; Bishop et al., 2002). Such formation of reduced ammonium would also have provided a source of nitrogen for prebiotic chemistry and for the formation of life. To understand early Mars, the potential for prebiotic chemistry and life on Mars, and when it was that Mars may have had the capacity to sustain life, a critical question must be considered: What happened to Mars' nitrogen?
Nitrogen fixation by atmospheric shock heating and subsequent chemistry is one important route for nitrogen fixation. On terrestrial planets with a nonreducing atmosphere (Mattioli and Wood, 1986; Wood and Vigo, 1989; Delano, 2001), that is, a CO2/N2 atmosphere with little or no CO and H2, shock heating leads to NO formation (Chameides, 1979; Yung and McElroy, 1979; Chameides and Walker, 1981; Miller and Schlesinger, 1983; Borucki and Chameides, 1984; Fegley et al., 1986; Kasting, 1990; Zahnle, 1990; Navarro-González et al., 1998, 2001; Nna Mvondo et al., 2001; Miyakawa et al., 2002). Thus, chemical routes that fix NO into forms that are chemically accessible for the origin of life or into forms that become sequestered into the crust become important. It is known that NO can become photochemically converted to nitrate and nitrite, which can subsequently be reduced to ammonia (Summers and Chang, 1993; Summers and Lerner, 1998; Summers and Khare, 2007). Since both nitrate and nitrite can be reduced by iron(II), in the form of FeS, we became interested in whether FeS could lead to the direct reduction of NO to ammonia. The direct reduction of NO could be the most direct route to convert NO to reduced nitrogen on Mars, early Earth, and other terrestrial planets.
2. Materials and Methods
2.1. Apparatus and experimental method
The flask used in the experiment is shown in Fig. 1. FeS (50–400 mg/mL, Sigma-Aldrich -100 mesh, 99.9%) was added to the flask under nitrogen. Then nitrogen-purged high-purity water (Millipore, MilliQ Gradient reverse osmosis system), with added sodium bicarbonate to establish the CO2/bicarbonate buffer needed to establish the desired pH (0–5 g), was added. The aqueous phase was freeze-pump-thaw degassed four times. The vessel was then filled from a tank containing the gas mixture (CO2/N2, 20%/80%, with 1% NO and CO) to a pressure of ∼760 torr, and the system was allowed to react. Additional experiments, in which FeS was added to a flask where the aqueous phase had already been deoxygenated and the nitrogen purge gas replaced with the reaction gas mixture, showed that the order of addition did not affect results. Experiments were conducted in either a 350 mL vessel with 10–20 mL of water or a 40 mL vessel with 1 mL of water (particularly with experiments with isotopically labeled water). For experiments in which pH was studied, samples from the same batch of FeS were used.

Diagram of experimental apparatus.
2.2. Product analysis
Gas-phase samples were backfilled into an IR gas cell and measured by Fourier transform infrared (FTIR) spectroscopy. All FTIR spectra were corrected for the pressure change. Analysis of ammonia concentrations was performed on the solution after reaction was over (2–3 h). Analyses were usually by colorimetric methods (Verdouw et al., 1978; Summers, 2005), but some analyses were also done by ion chromatography. Analysis by ion chromatography was done at ambient temperature on a Dionex DX-100 instrument with a Dionex CS-12 column (and a Dionex CG-12 guard column) and 20 mM methane sulfonic acid as the eluent. In experiments analyzed by ion chromatography, all sodium salts were replaced with potassium salts to prevent interference with ammonium detection. Samples were filtered through a 0.2 μm filter prior to injection. Since ammonia and ammonium are in rapid equilibrium when in aqueous solution, the distinction between the two species becomes complicated. We use “ammonia” to refer to the total concentration of ammonia as both NH3 and
2.3. Isotopic labeling experiments
To verify product peak identification in the IR spectra and elucidate the reactions and mechanisms taking place, isotopic labeling experiments were conducted. These were carried out as described above (using the 40 mL flask), substituting a reactant or solvent enriched in a particular isotope (relative to natural abundance). For example, isotopically labeling the water might involve use of water with oxygen atoms that are 100% 18O, instead of the 0.2% natural abundance. If a reaction is run in such water and a product is observed to be isotopically enriched, then the enriched atoms on the solvent molecules would have been the source of those atoms on the product. Conversely, lack of enrichment of the product would show that those atoms did not come from the enriched atoms in the water.
Isotopic substitution can also verify that vibrational peaks are due to the assigned molecules (and vibrations therein). A bond using atoms of a heavier isotope will vibrate at a different frequency and show a different peak in the IR spectrum, compared to the same bond using atoms of a lighter isotope. Changing the relative isotopic abundances present in the molecules in that sample will change the relative intensity of the different peaks for the different isotopes engaged in that bond. Thus, we can correlate the observed peaks with the atoms and bonds being affected.
Reactions were conducted where the water was enriched in 18O and 2D, where bicarbonate/carbon dioxide was enriched in 13C, and where NO was enriched in 18O. No difference was observed with 13C or 2D labeling. NO did not exchange with H2O over the timescale, ∼1.5 h, of these experiments (which can be verified by the lack of increased intensity for N18O peaks in the IR spectra), so transfer of label between NO and the water did not occur during these experiments. Conversely, in experiments where N18O was used, the isotopically labeled gas mixture was produced by exposing NO to
3. Results
3.1. Formation of ammonia and yield
The products of shock heating, NO and CO, were exposed to an aqueous suspension of FeS under a CO2 and N2 atmosphere. The formation of ammonium in the aqueous phase was observed (see experimental section). For example, in one experiment with 95 mg/mL of FeS suspended in water at pH 7.3, 64% of the NO that reacted was converted to ammonia. NO was consumed at a rate of 5×10−8 mol g−1 s−1, and ammonium was correspondingly formed at a rate of 3×10−8 mol g−1 s−1. The formation of ammonia from NO reduction was also measured by ion chromatography. After reaction of NO with 57 mg/mL of FeS at pH∼6.6, ammonia was detected with a yield of 40%. If no FeS is present, formation of ammonium is not detected.
In Table 1, the yield of ammonia over a series of experiments (with 185 mg/mL FeS in each experiment) is shown. The highest yields occur near neutral pH, and the yield declines as conditions become more acidic or alkaline (particularly in the alkaline direction).
Reaction time, 2–3 h.
pH is approximate because no bicarbonate was added; the pH was buffered only by FeS.
These experiments were all performed using FeS from the same batch.
3.2. Gas phase products
Figure 2A shows the changes that occurred in the gas phase during this reaction. (Peaks due to water have been mathematically subtracted, except where noted.) One interesting observation in many experiments is the formation and subsequent disappearance of peaks due to NO2 (at 1602 and 1629 cm−1).
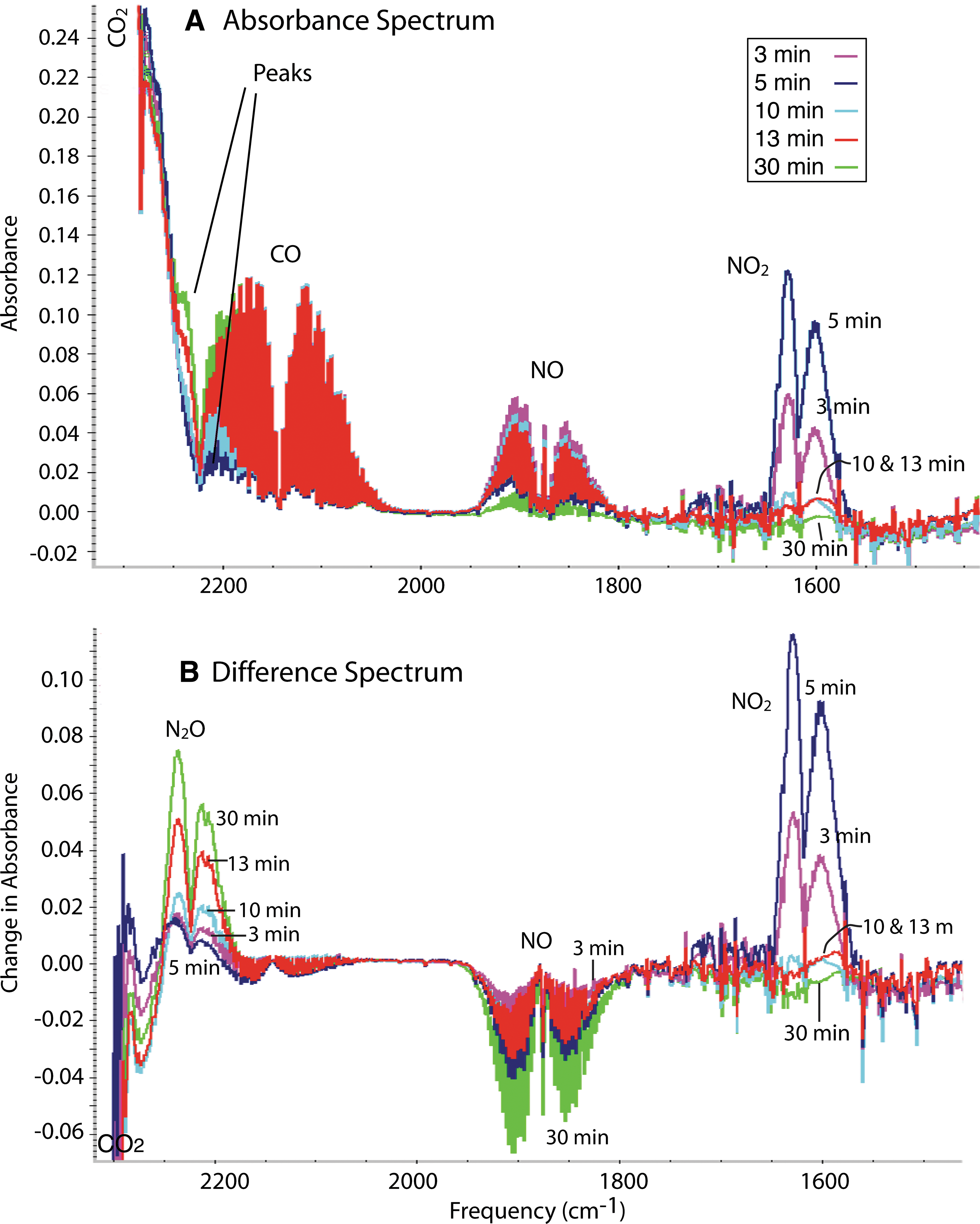
FTIR absorption spectra of gas phases (1% NO, 1% CO, 19% CO2, 79% N2, 0.04 L) that had been allowed to react with a suspension (1 mL) of FeS in water (370 mg/mL). (
There are peaks at 2235, 2210, 1270, and 1300 cm−1. The peaks at 2235 and 2210 cm−1 are hard to see because they are right next to the large CO2 peak, but they become clearer in the difference spectra (Fig. 2B). In a difference spectrum, the initial gas spectrum is subtracted from the spectrum obtained after reaction, showing only the changes in absorbance upon reaction. Species that were produced are seen as positive peaks, and those that were consumed as negative peaks. These peaks are due to the formation of N2O. There is no evidence that CO was consumed or produced in the reaction. The small positive and negative changes in absorbance that can occasionally be seen can be attributed to small errors in subtraction.
In the pH experiments that produced the data for Table 1, the yield of N2O was independent of pH (within experimental error). In these experiments, the yield was ∼35% (based on N). Without FeS, no reaction was observed, so all gaseous species must have been formed by reactions with, or at, the FeS in the aqueous phase.
3.3. Isotopic labeling
The reaction was also studied with 18O isotopically labeled water and NO. Figure 3 shows the difference spectrum obtained when the reaction was run using
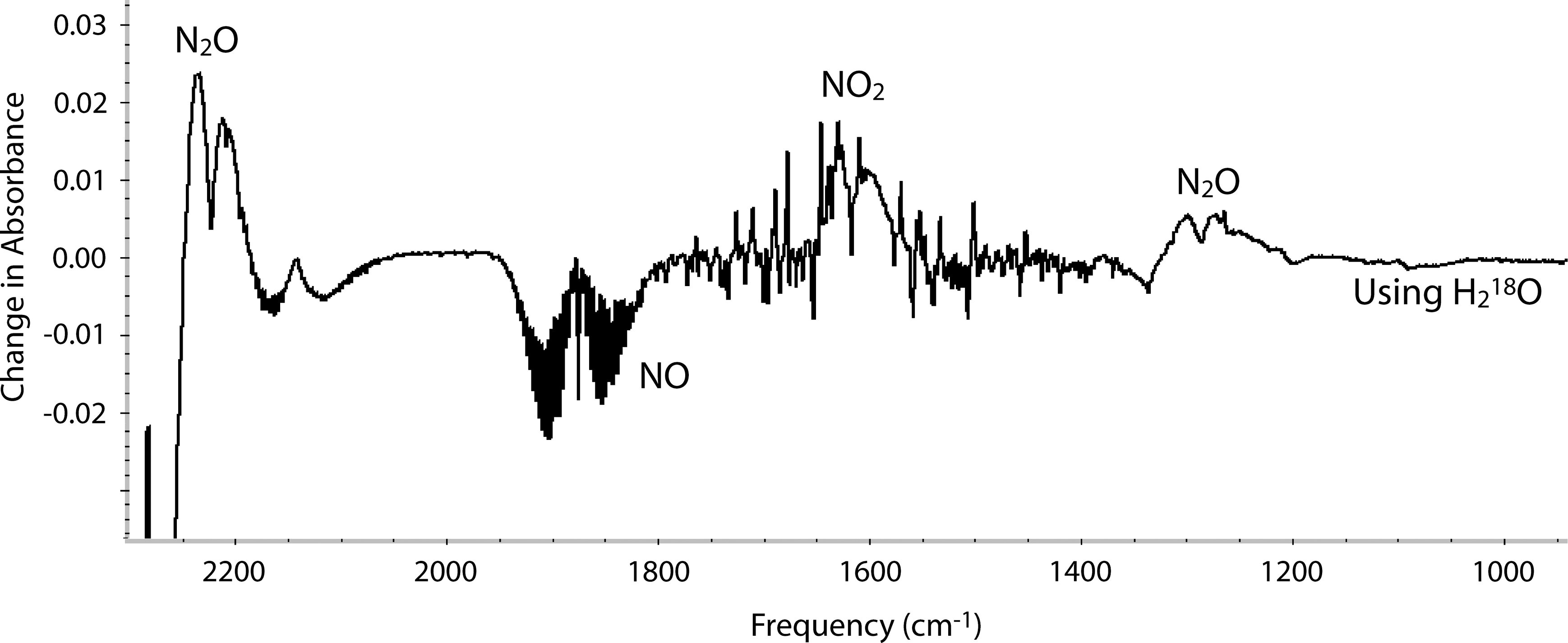
FTIR difference spectra of gas phases (1% NO, 1% CO, 19% CO2, 79% N2, 0.04 L) that had been allowed to react with a suspension of FeS in 18O water (0.5 mL, 100%

FTIR difference spectra of a gas phase (1% NO [50% 18O], 1% CO, 19% CO2, 79% N2, 0.04 L) that had been allowed to react with a suspension of FeS in water (1 mL), 370 mg/mL. All spectra are corrected for the pressure change upon transfer to the IR cell. The spectrum of water vapor has been digitally subtracted. Color images available online at

FTIR difference spectra of gas phases (1% NO, 1% CO, 19% CO2, 79% N2, 0.04 L) that had been allowed to react with a suspension of FeS in water (0.5–1 mL), 370 mg/mL. Experiment used
4. Discussion
A consideration of the gaseous products and the isotope labeling experiments informs about the chemistry that is occurring in conjunction with the formation of ammonia. The transient nature of NO2 formation is not surprising. Nitrogen dioxide is reactive with water (to form nitrate/nitrite; see the work of Gmelin, 1936; Lee and Schwartz, 1981), and it would be expected to be destroyed as it is formed (if it is not reduced to ammonia by FeS). Thus, it ends up being another intermediate species in the reduction of NO to ammonia. Since reaction of NO and, hence, formation of any products from it (such as NO2) is not observed in the absence of FeS (Summers and Khare, 2007), NO2 must be produced by FeS in an aqueous phase. Clearly, however, under the right conditions, some NO2 can escape up into the gas phase. It is quite possible, given similarities to other nitrogen oxides, that NO2 is reduced by FeS to ammonia. However, it is known that, if it is not directly reduced by FeS, it will be converted to nitrite/nitrate (Gmelin, 1936; Lee and Schwartz, 1981), which will, under these conditions, be reduced by FeS (Summers, 2005). Whether it is directly reduced to ammonia by FeS or converted to nitrate/nitrite and then reduced is not clear (and may be a somewhat academic distinction).
No incorporation of oxygen from
Since there was no exchange with water during the reaction, it seems highly unlikely that the formation of NO2 proceeded by the cleavage of the N-O bond and formation of a surface bound oxygen species (which would be expected to readily exchange with water). This makes the formation of a NO dimer and its reaction with a third NO the most likely mechanism.
Since N2O is not formed in sufficient amounts to account, by Eq. 1, for all the NO that reacts, it seems unlikely that ammonia formation could be going entirely by the formation of NO2 (and its subsequent conversion to ammonia). Similarly, it seems unlikely that FeS, which will bind and reduce nitrate and nitrite, would also bind NO (well enough to catalyze its disproportionation) but would not reduce it. For that reason, we believe that both the direct reduction of NO to ammonia and also the formation of NO2 (and subsequent conversion to ammonia) occurs.
The pH dependence is similar to the reduction of nitrate/nitrite by FeS (Summers, 2005), which also decreases as the pH rises (probably due to the interference from bicarbonate ions). However, in this case reduction of NO shows a decrease in yield in acidic solutions not seen with nitrate/nitrite.
If we add this route to the already known fixation processes leading from NO, we can construct the following schematic of abiotic nitrogen cycling from atmospheric processes on terrestrial planets (Fig. 6).

Simplified schematic of abiotic nitrogen fixation pathways proceeding from the formation of NO. Color images available online at
5. Conclusions
It has been previously shown that NO could be photochemically converted to nitrate and nitrite by two pathways, one through the gas-phase formation of HNO and the other by the gas-phase formation of NO2 (Summers and Khare, 2007). These results show that another pathway for the abiotic fixation of nitrogen on terrestrial planets exists, the reduction of NO to ammonia by FeS. This pathway probably represents the most efficient route from NO to reduced nitrogen, which makes it of particular prebiotic importance. Such fixation can result in sequestration of nitrogen in the crust. Ammonium would be quite stable sequestered in clays (Bada and Miller, 1968; Mancinelli et al., 1998; Bishop et al., 2002).
On young planets, about the age at which life originated on Earth, it is thought that the amount of continental crust is limited, which leads to a picture of a world with high levels of island volcanism and other types of volcanoes in close proximity to marine/lacustrian environments. It is also interesting to note that, in such environments, hydrothermal activity that produces FeS is possible in close proximity to NO generation by volcanically generated lightning (Navarro-González et al., 1998). Since this pathway does not involve a photochemical step, this means that the NO generated would only need to be transported downward into aqueous phases that contain FeS in order to react. The close proximity of sources of NO and FeS makes this an interesting environment for the formation of prebiotic reduced nitrogen (as illustrated in Supplementary Fig. S1, available online at
FeS also catalyzes the disproportionation of NO into N2O and NO2. The biogenic formation of N2O has been proposed as a signature of biological activity in the atmospheres of extrasolar planets (Segura et al., 2005). However, this brings up the issue as to which abiotic sources of N2O might exist (for example, see; Nna-Mvondo et al., 2005; Summers and Khare, 2007). This chemistry would be an abiotic source of N2O and thus has potential implications for the use of nitrous oxide as a biosignature.
Footnotes
Acknowledgments
The authors would like to thank the NASA Astrobiology Institute (NNA04CC05A) and Exobiology Program for support.
Author Disclosure Statement
No competing financial interests exist.
Abbreviation
FTIR, Fourier transform infrared.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.