
Abstract
Ribozymes that act as polymerases and nucleotide synthases are known experimentally, even though no fully self-replicating system has yet been found. If the RNA World hypothesis is true, ribozymes must have arisen initially from within a random abiotic polymerization system. To investigate the origin of the RNA world, we studied a mathematical model of a chemical reaction system describing RNA polymerization. It is supposed that, in absence of ribozymes, polymerization occurs at a small spontaneous rate, and that in the presence of polymerase ribozymes, polymerization occurs at a faster rate that is proportional to the ribozyme concentration. Chains must be longer than a minimum threshold length in order to have the possibility of acting as ribozymes. The reaction system has two stable states that we term dead and living. The dead state is controlled by the small spontaneous rate and has negligible concentration of ribozymes. The living state has high concentration of ribozymes, and the reaction rates are determined by the ribozymes; thus, the system is autocatalytic. Concentration fluctuations in a finite volume can cause a transition to occur from the dead to the living state, that is, an origin of life occurs within this model. We also consider ribozymes that catalyze nucleotide synthesis. We show that living and dead states arise in the presence of synthase ribozymes in the same way as for polymerases. It has been proposed that recombination reactions are a way of generating long RNA chains in the early stages of life. We show that if the possibility of random reversible recombination reactions is added to our model, this does not lead to an increase in long polymer concentration. Thus, if recombination is fully reversible, there is no autocatalytic state controlled by recombination. Nevertheless, recombination can play an important role in ribozyme synthesis if there is an additional process that keeps the recombination reactions out of equilibrium. We modeled a case studied experimentally in which building block strands of moderate length associate due to RNA secondary structure formation. A recombination reaction then occurs between these strands to form a longer sequence that catalyzes its own formation via the recombination reaction. This system has an autocatalytic state, and it is possible for it to arise within our random polymerization system. If complexes formed by associations of shorter strands can act as catalysts without the requirement that the strands be covalently linked, this would alleviate the need for synthesis of very long strands; hence, it makes the emergence of an autocatalytic system from an abiotic random polymerization system much more likely. Key Words: RNA world—Ribozymes—Origin of life. Astrobiology 11, 895–906.
1. Introduction
The reaction scheme that we discuss in this paper incorporates three kinds of reaction: synthesis of monomers from precursors, polymerization by sequential addition of monomers, and recombination, as shown in Fig. 1. We will refer to ribozymes that catalyze these reactions as synthases, polymerases, and recombinases, respectively. In this paper, we present mathematical models of this reaction system and show that these three types of ribozyme can all be autocatalytic, that is, they can increase the rate of their own formation. Before introducing the mathematical models, we will discuss what is known about these ribozymes experimentally.
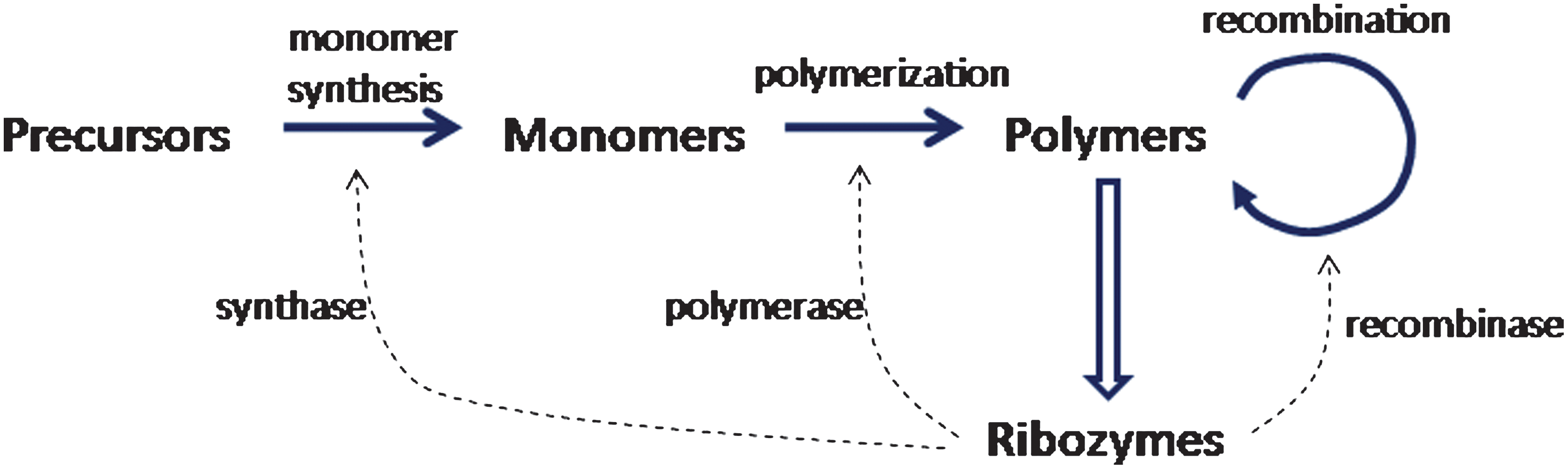
Schematic diagram of an autocatalytic RNA system involving monomer synthesis, polymerization, and recombination. Color images available online at
Polymerases spring immediately to mind when discussing the origin of life because they have the ability to synthesize new RNA strands. In principle, if a polymerase could synthesize another strand of the same length as itself, it could make another copy of itself and, hence, be self-replicating. Polymerases that have been studied in the laboratory use a template strand and synthesize a new complementary strand by sequential addition of complementary monomers. Several polymerases of this kind have been developed (Johnston et al., 2001; Lawrence and Bartel, 2005) by in vitro evolution. A recently developed polymerase is able to synthesize strands of up to 95 nucleotides in length (Wochner et al., 2011), although the polymerase itself has a length of around 170 nucleotides. These studies provide important support for the concept of the RNA world, even though they still fall short of self-replication. An RNA-based organism would also have to synthesize its own nucleotides from simpler precursor molecules found in the environment. Ribozymes capable of catalyzing uridine nucleotide synthesis from uracil and ribose have also been developed in the laboratory (Unrau and Bartel, 1998).
In a living system in the RNA world, ribozyme synthesis would be autocatalytic. However, if ribozymes were involved in the origin of life, then the first ribozymes must have arisen by spontaneous chemical reactions rather than by reactions catalyzed by other ribozymes. Abiotic synthesis of nucleotides is possible but difficult, as reviewed by Joyce and Orgel (2006), although a new method of abiotic pyrimidine synthesis has recently been proposed (Powner et al., 2009). It has also been found that ribose sugars are stabilized by minerals such as borate, which might help to explain how ribose could be selected from a prebiotic mixture of similar sugars (Benner et al., 2010). Progress has also been made in understanding the possible prebiotic mechanisms of RNA polymerization. Mineral-catalyzed synthesis of RNA oligomers from activated nucleotides has been known for some time (Ferris et al., 1996). Recent work has shown that RNA polymerization can be facilitated by cycles of wetting and drying within a matrix of lipid bilayers (Rajamani et al., 2008; Olasagasti et al., 2011). RNA polymerization from nucleoside cyclic monophosphates has also been observed (Costanzo et al., 2009). Although we do not know how well any of these studies match the details of the environmental conditions on early Earth, these results give support to the idea that some form of prebiotic polymerization of relatively long RNAs could have occurred.
We expect that RNA strands must be moderately long in order to have sufficient complexity to function as ribozymes. For example, simple hammerhead ribozymes that catalyze RNA cleavage are of a length of 40–50 nucleotides (Scott et al., 1995), while more complex polymerases are around 170 nucleotides (Wochner et al., 2011). Random abiotic polymerization will typically give rise to length distributions that decrease exponentially, that is, long polymers will be extremely rare with respect to monomers and short oligomers. Any factors that shift the length distribution toward longer chains are therefore potentially helpful in increasing the likelihood of generation of ribozymes. It has been proposed that recombination reactions can act in this way (Lehman, 2008). The type of recombination that is relevant is non-homologous crossing over between two single strands. Recombination can produce a strand that is substantially longer than either of the input strands, for example, two strands of length n can produce a strand of length 2n−1 and a single nucleotide. However, the reverse reaction can also occur, that is, long chains can be destroyed by recombination as well as created. In this paper, we investigate the effect of recombination of the length distribution in more detail.
Recombination in RNA has been studied experimentally. Lutay et al. (2007) showed that recombination between RNA strands can occur via a cleavage and a subsequent ligation reaction. The cleavage leaves a 2′,3′-cyclic phosphate at the end of one of the fragments, which undergoes a ligation reaction with the 5′ end of another strand. Hayden and Lehman (2006), Draper et al. (2008), and Hayden et al. (2008) studied a recombination system derived from the group I intron of Azoarcus sp. BH72, which is one of the smallest known self-splicing introns (Kuo et al., 1999). The Azoarcus ribozyme can function as a recombinase by two mechanisms (Draper et al., 2008). In the simplest of these, known as tF2, two strands come together due to RNA secondary structure formation, and the ribozyme catalyzes a transesterification reaction between the ends of these strands. A requirement of this reaction is that one of the strands ends with the sequence CAU, which is complementary to the internal guide sequence GUG in the ribozyme. The reaction creates a long sequence with a hairpin loop at the point of connection plus a single nucleotide. It was found that the ribozyme can be broken into four fragments that can self assemble into a trans complex that has almost the same structure as the fully connected ribozyme. If the fragments contain the CAU sequence in the required places, then they can be linked together by the tF2 reaction. One copy of the ribozyme can therefore create another copy by linking the four fragments in the trans complex; that is, synthesis of the ribozyme is autocatalytic.
The Azoarcus recombinase system is somewhat similar to a ligase system studied by Lincoln and Joyce (2009), although the latter case works in two steps. One strand catalyzes the ligation of two fragments to form a complementary sequence. The complementary sequence then catalyzes the ligation of two other fragments that produce the original strand. In both these experiments, the formation of the full-length strand from the fragments is autocatalytic, but in both cases it is necessary to supply the correct fragments to keep the reaction going. The fragments themselves are fairly long and would therefore be expected to be rare in an abiotic random polymerization system. Nevertheless, these experiments give proof that autocatalytic systems of RNA can exist. Our aim in this study was to develop a theory of how autocatalytic RNA systems might have emerged initially from within a random polymerization system.
This paper builds on our previous paper (Wu and Higgs, 2009), in which we introduced a model to study the role of polymerase ribozymes in particular. It was assumed that polymerization could occur spontaneously at a low rate and could also be catalyzed by ribozymes at a higher rate, if polymerases arose. The reaction system has two stable states, which we call dead and living. The dead state has negligible concentration of ribozymes, and polymerization occurs only at the small spontaneous rate. The living state has high concentration of ribozymes, and the reaction rates are determined by the ribozymes; thus, the system is autocatalytic. The concentration of polymers decreases exponentially with length in both states, but in the living state the mean length is much longer and the concentration of long polymers is much higher than in the dead state. For an origin of life to occur, it is necessary for the system to switch from the dead to the living state. In an infinite volume, the dead state is stable forever. In a finite volume, however, concentration fluctuations occur, and these can cause a transition from the dead to the living state. We showed that the transition occurs most easily in a system of moderate volume. If the system is too large, the dead state remains stable indefinitely. If the system is too small, it is very unlikely that the initial catalysts will be generated by the spontaneous reaction.
In the current paper, we consider synthase and recombinase ribozymes in addition to polymerases. We show that, in the presence of synthases, there are once again two stable states. The behavior of the synthase model is very similar to the polymerase case. The recombinase case is somewhat different, because the recombination reaction is inherently reversible. In a general model that includes fully reversible recombination, we find that there can be no autocatalytic state that is controlled by the recombination reaction. However, we then consider a more specific model of the Azoarcus recombinase discussed above and show that, due to the association of the fragments by RNA secondary structure formation prior to the recombination reaction, the recombination reaction is no longer fully reversible. In this case, an autocatalytic state controlled by the recombinase is also possible.
2. Basic Model of Polymerization
We start with a basic system of chemical reaction equations that describes the synthesis of biopolymers such as RNAs. We suppose that the reactions for monomer synthesis and polymerization occur in a local region that is separated from the external environment, but which can exchange molecules with the environment. For example, the local region could be the interior of a lipid vesicle or a cavity in a porous rock or the surface of a catalytic mineral. The basic reactions are that precursor molecules, or “food,” F, are synthesized into monomers, A, and that monomers can polymerize by repeated addition to the end of growing chains.
A denotes any kind of nucleotide monomer, and An
denotes any chain of length n monomers. The model does not keep track of specific sequences of different kinds of monomers. The rate constants for synthesis and polymerization are denoted s and r, respectively. Initially, we treat these as fixed parameters, but later we suppose that these rates are functions of the concentration of biopolymer catalysts. Food molecules enter and exit the system at rate u, and there is a fixed concentration F
0 outside the system. Monomers and polymers exit the system at rate u but do not enter the system because the concentration is taken to be negligible outside. The following set of differential equations describes the change of concentrations of F, A, and An
:
In Eq. 4, P is the total polymer concentration of all lengths n≥2.
Solving Eqs. 3
–5 in the steady state, we find that
where
Hence
From Eq. 3, we have
Substituting into Eq. 4, we have
This is a cubic equation for A, for which there is only one real and positive root. Hence, A and all other quantities can be calculated for any given parameters r, s, u, F 0.
3. Autocatalytic Feedback in Polymerization
According to Eq. 7, the polymer length distribution decays exponentially. Unless z is very close to 1, the concentration of long polymers will be very small. Nevertheless, long polymers are relevant because we suppose that only polymers of length greater than, or equal to, a minimum length L can act as catalysts. The long polymer concentration is
A and z are obtained by numerical solution of Eq. 11. Figure 2 shows PL as a function of polymerization rate r with fixed values of all the other parameters. This follows an S-shaped curve. We now suppose that r can be written as the sum of two components, r=r0 +krPL, where r 0 is a small spontaneous rate (i.e., not driven by biopolymer catalysts) and kr is the rate constant for the polymerization reaction catalyzed by the long polymers. The catalyzed rate is krPL, which is proportional to the long polymer concentration. If a small fraction f of long polymers is a catalyst and the effect per catalyst is k 0, then kr=k 0 f. The relationship between r and PL can be written as
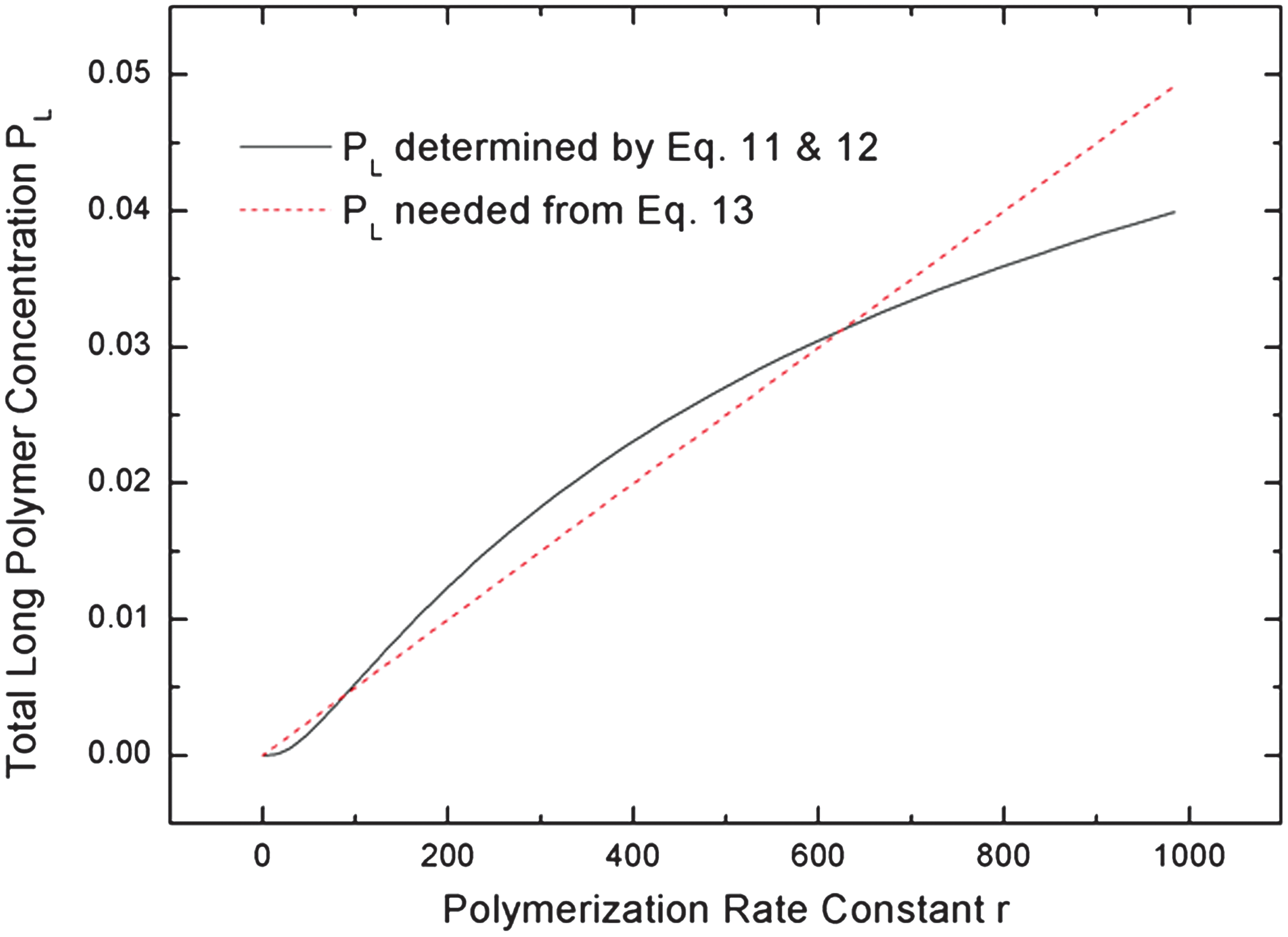
which is a straight line on Fig. 2. Valid solutions for PL are where the line intersects the S-shaped curve. Depending on the values of r 0 and kr, the line may intersect the curve either once or three times. The most interesting region of the parameter space is where the line intersects three times, as in Fig. 2. In this case, it may be shown that the upper and lower intersection points correspond to stable solutions of the differential equations and the middle intersection point is unstable. Once we know the value of r and PL at the intersection point, we can determine the values of A and z that correspond to this point from Eqs. 8 and 12. Hence, we know the polymer length distribution in the presence of autocatalytic feedback.
In Fig. 3, we show the length distribution at the two stable solutions corresponding to the intersection points in Fig. 2. Both these solutions are exponential distributions (straight lines on the log plot), but the decay rates are different because there are two different values of z. We refer to these solutions as living and dead states. In the dead state, z is much less than 1, and PL is very small. Hence, polymerization is almost entirely controlled by the spontaneous rate, r≈r 0. In the living state, z is only slightly less than 1, and PL is large. In this case, krPL>> r 0, and the total polymerization rate is almost entirely controlled by the catalytic term. The presence of long polymer catalysts leads to a high rate of polymerization of more long polymers. Hence, this state is autocatalytic, and that is why we refer to it as the living state.
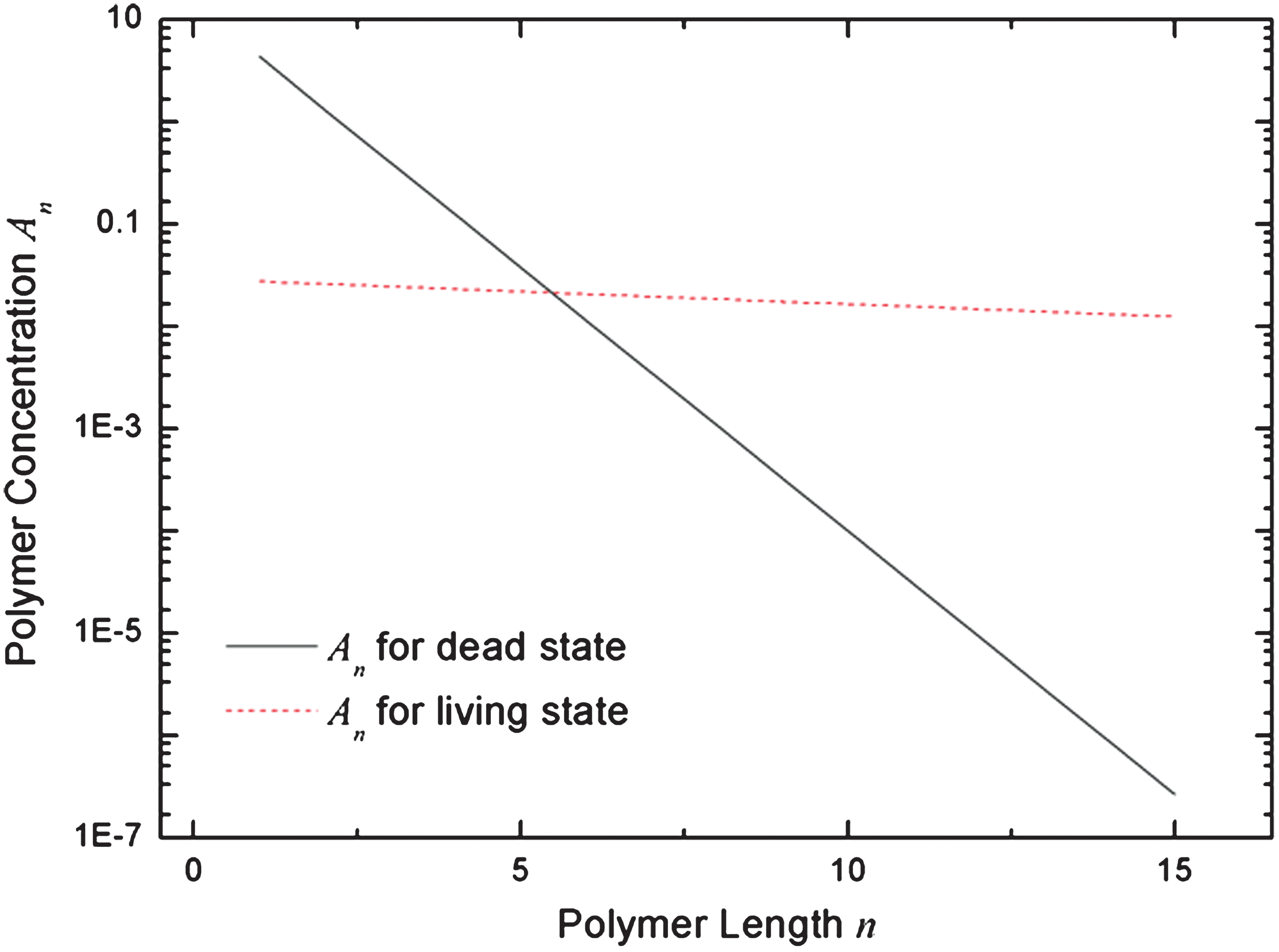
Distribution of polymer lengths at the two stable stationary state solutions of the model with feedback in polymerization as in Fig. 1. For the dead state, A=4.3901, z=0.3051. For the living state, A=0.02746, z=0.9450. Color images available online at
Figure 4 shows the phase diagram of this model in the space of r 0 and kr , illustrating the region (enclosed by the solid lines) where both living and dead states are stable. Points on the solid boundary lines correspond to points in Fig. 2 where the line is tangential to the S-shaped curve, that is, where the central unstable solution merges with either the upper or the lower stable solution. Outside the solid boundary lines, there is only one solution—either living or dead, but not both. If we move around the outside of Fig. 4, we move smoothly from a state with very low PL, which is clearly dead, to a state with very high PL, which is clearly living. As this change is continuous, it is somewhat arbitrary where to draw the boundary between the regions with only dead and only living solutions. One choice is to make use of the point of inflection in the S-shaped curve in Fig. 2. According to this definition, the solution is taken to be living if the intersection point lies above the point of inflection and dead if it lies below. The dotted line in Fig. 4 shows the boundary between the only-living and only-dead regions of the phase diagram obtained by using the point of inflection rule. An alternative choice is to define the solution as living if polymerization occurs predominantly by catalysis (krPL/r 0>1) and dead if polymerization occurs predominantly by the spontaneous rate (krPL/r 0<1). The boundary line according to this definition is when krPL/r 0 =1, which is also shown on Fig. 4. Although the position of the dotted boundary is a matter of definition, the position of the solid boundaries is unambiguous, because the behavior changes suddenly from a single stable state to two stable states as we cross these boundaries.
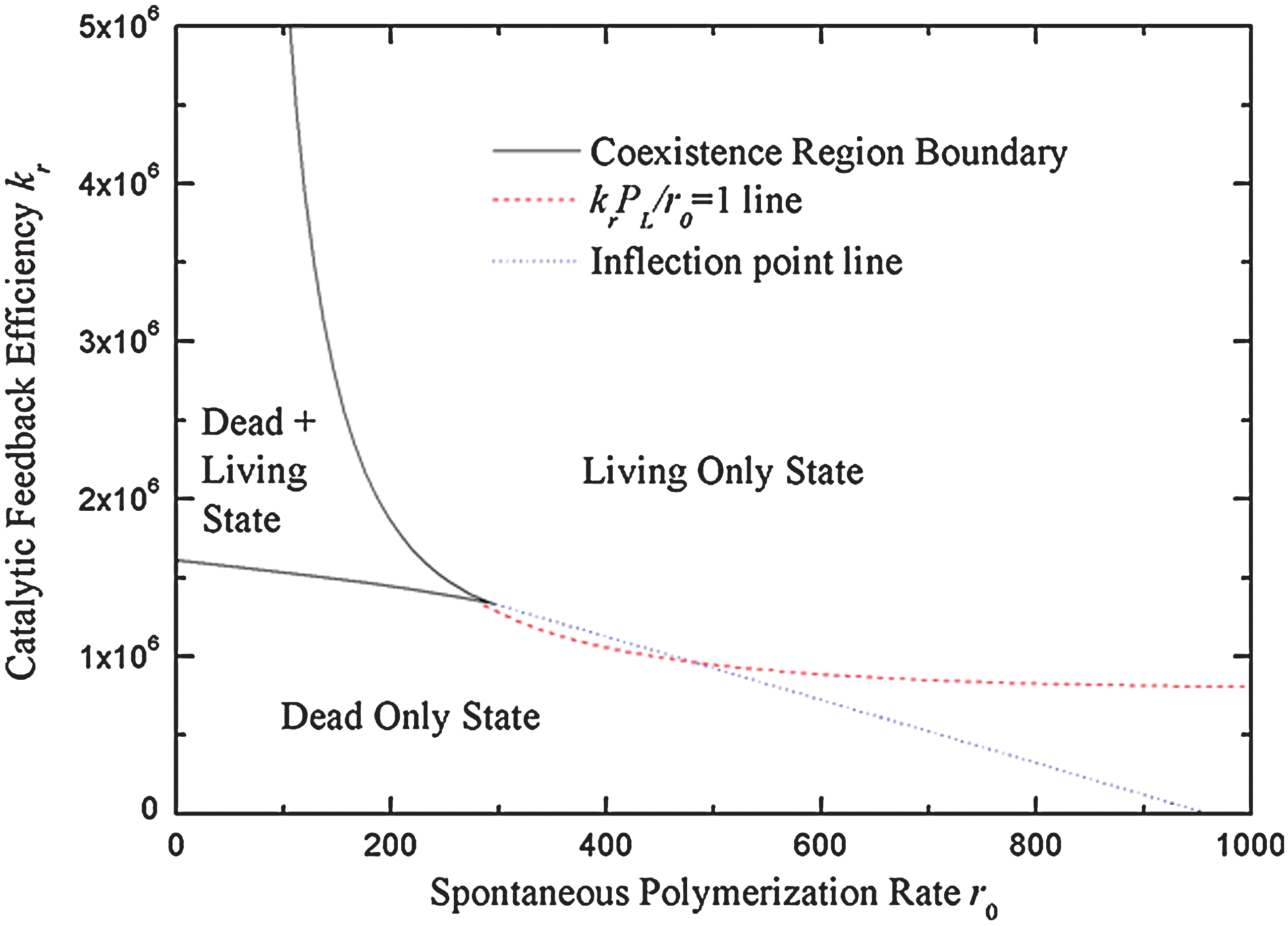
Phase diagram of the reaction system as function of feedback efficiency kr
and spontaneous polymerization rate constant r
0, when u=1, s=10, F
0=10, L=50. Color images available online at
The model described here is very similar to that given in our previous paper (Wu and Higgs, 2009), but we wish to point out two slight differences. In the previous paper, we included a step of formation of activated monomers, representing nucleotide triphosphates or imidazoles, as in the experiments of Ferris et al. (1996). For simplicity, we have eliminated this step in the present paper, and we treat monomer synthesis as a single step. The results are very similar to before, and the conclusions do not depend on whether we include the activation step. The second difference to our previous paper is that we have treated the food molecule concentration F as a variable with its own dynamics given by Eq. 3, whereas previously we kept F fixed at a constant. This change is necessary in order to consider synthase ribozymes, as we do in the next section. Wu and Higgs (2009) went on to discuss the stochastic dynamics of the model with polymerase ribozymes. The main result was that, in a finite volume, concentration fluctuations can drive the system from the dead state to the living state, that is, an origin of life can occur in this model. The stochastic dynamics of the model considered here should be very similar to the case in our previous paper; therefore, we will not consider the dynamics further here. In the present paper, we will proceed to compare the model above, in which feedback is in the polymerization reaction, with models in which feedback occurs by either nucleotide synthesis or recombination.
4. Autocatalytic Feedback in Nucleotide Synthesis
We now return to the basic model of Eqs. 3 –5, and we suppose that the polymerization rate, r, is simply a constant. However, we consider the possibility that long polymers can act as nucleotide synthases instead of polymerases. Figure 5 shows the concentration of long polymers as a function of s, when r and the other parameters are constant. This is another S-shaped curve that is similar to Fig. 2. We introduce feedback into nucleotide synthesis by setting s=s 0+ksPL, where s 0 is a small spontaneous synthesis rate and ks is the rate constant for synthesis catalyzed by the long polymers. The analysis now proceeds as before. The solutions of the model in the presence of feedback must be where the S-shaped curve intersects the line:
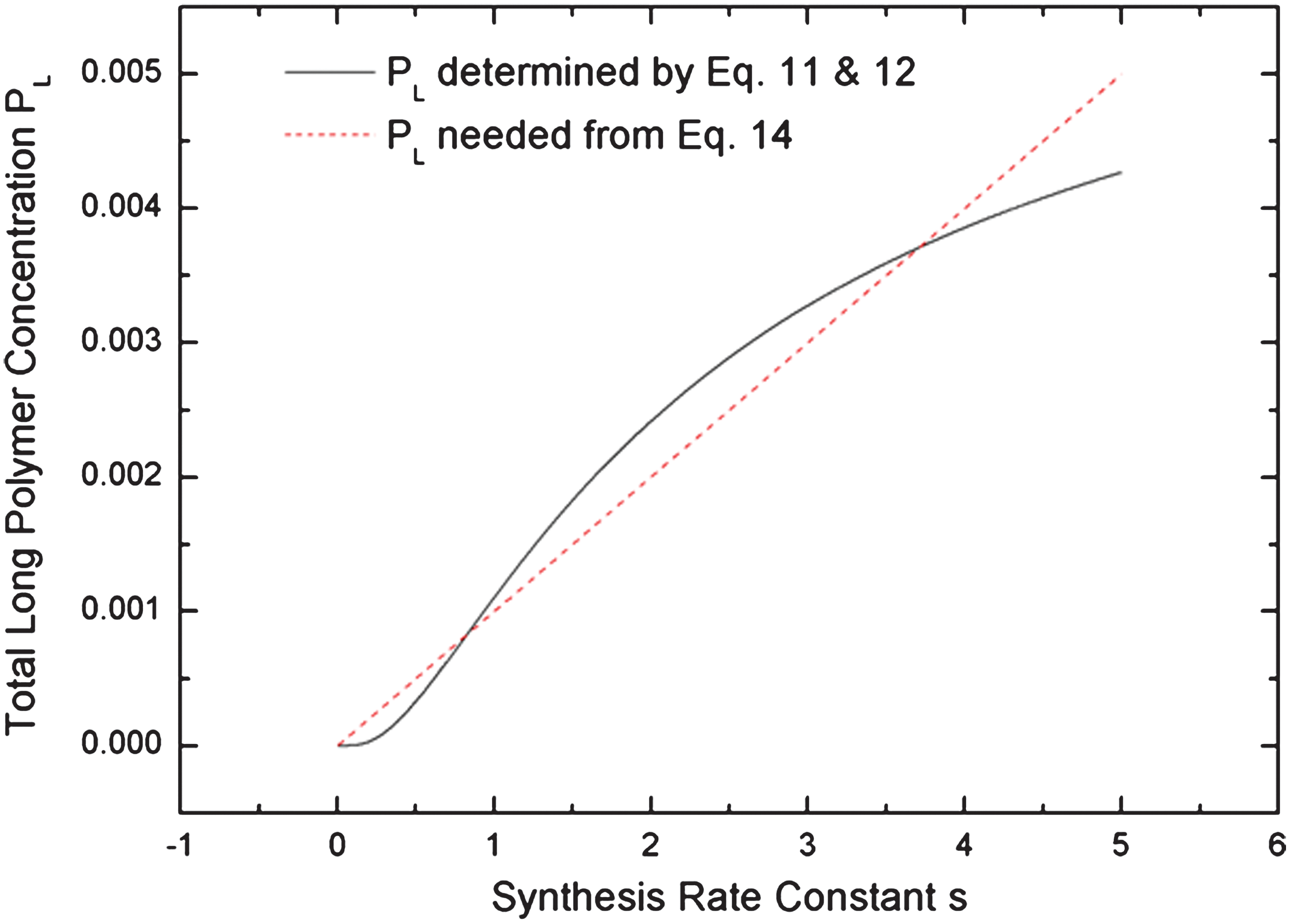
The phase diagram in the s 0 and ks parameter space (Fig. 6) has a similar shape to the previous case in the r 0 and kr parameter space (Fig. 4). There is a living state, in which monomer synthesis is controlled by the catalytic rate and ribozyme concentration is high, and a dead state, in which monomer synthesis is controlled by the spontaneous rate and ribozyme concentration is very low. In a finite volume, concentration fluctuations can cause the system to jump from the dead to the living state, as before.
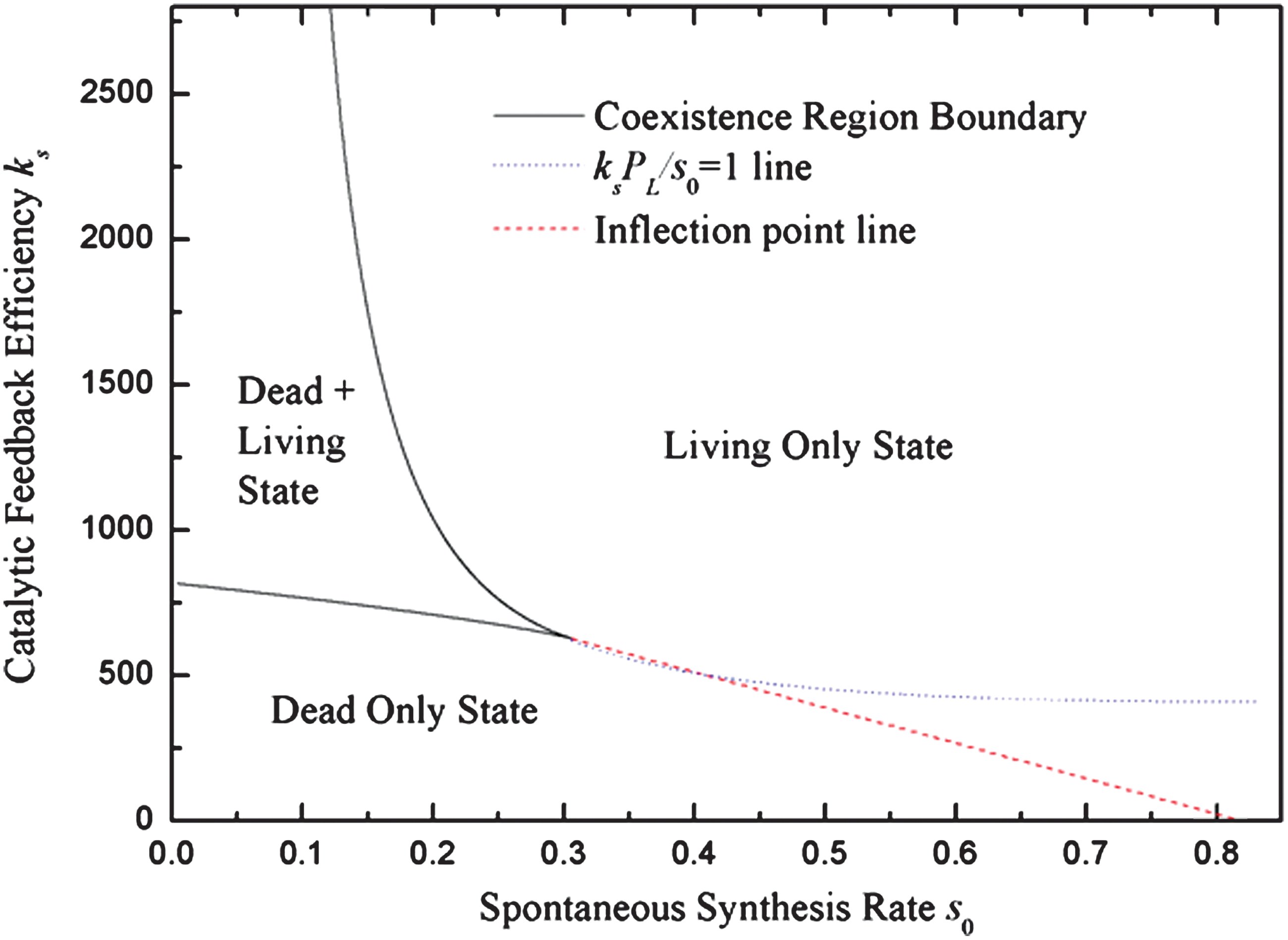
Phase diagram of the reaction system as function of feedback efficiency k and spontaneous synthesis rate constant s
0, when u=1, r=100, F
0=10, L=50. Color images available online at
It should be noted that it is important to treat the food concentration F as a variable in the case where feedback is in nucleotide synthesis. When s is high, F is depleted and becomes much less than F 0, as seen from Eq. 10. In this case, the rate of food entering the system limits the overall concentration of monomers and polymers and insures that the top part of the S-shaped curve in Fig. 4 flattens out in the limit of large s. If the food concentration was treated as a constant, there would be no maximum limit to the rate of nucleotide synthesis, and the concentration of monomers and polymers would grow indefinitely once ribozymes appeared, which would be unrealistic.
5. Effect of Recombination on the Polymer Length Distribution
According to our argument, an origin of life can only occur if spontaneous reactions are able to generate the first few ribozymes that initiate the autocatalytic feedback. If r 0 and s 0 are both small, then presumably we would need to wait very long times for rare fluctuations to occur, as discussed in the dynamical sections of Wu and Higgs (2009). Any factors that increase the likelihood of forming polymers long enough to be ribozymes will increase the probability of an origin of life. It has been proposed that recombination reactions can act in this way (Lehman, 2008). Therefore, we wish to add the possibility of recombination between the polymer chains in our model.
Initially, we will consider a closed reaction system where the only reactions possible are recombinations of the type shown in Fig. 7, where a chain of length n reacts with a chain of length m to produce chains of length n−k+h and m−h+k. We suppose that the rate constant for both forward and backward reactions is ρ. These rates should be equal because the same kind of chemical bond is being made and broken in each case. We suppose that 1≤k≤n, so there are n places where a chain of length n can recombine.
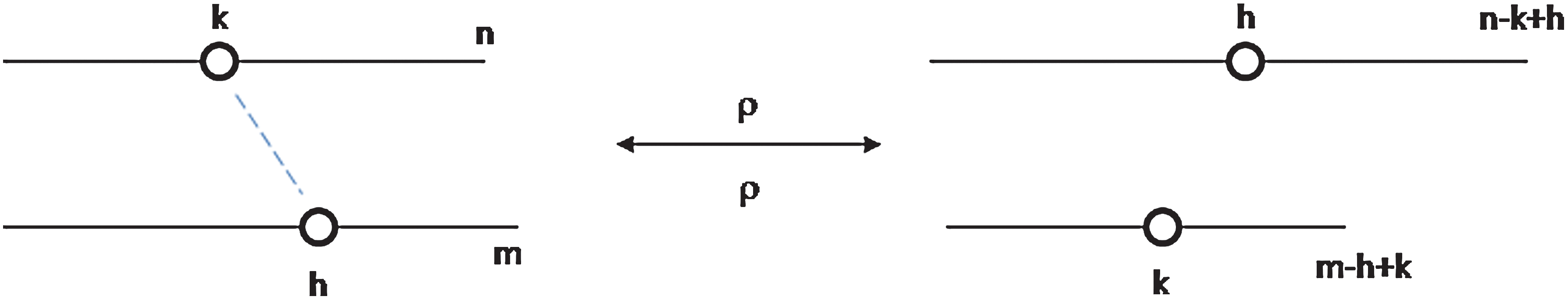
A possible random recombination reaction. Color images available online at
In a closed system with only recombination, the total number of monomers is fixed, but the length distribution can change. The rate of change of polymer concentration for any given n is
where λ
n
and μ
n
are the total rates of destruction and creation of chains of length n. From Fig. 7, we have
and
Figure 8 shows a simulation of the distribution following Eq. 15, beginning from the case where all chains are length n=10. Recombination causes this distribution to broaden and eventually tend to an exponential distribution of the form An=Azn− 1. To see that the exponential distribution is stationary, consider the pair of reactions in Fig. 7. The forward and backward rates are equal if the distribution is exponential:
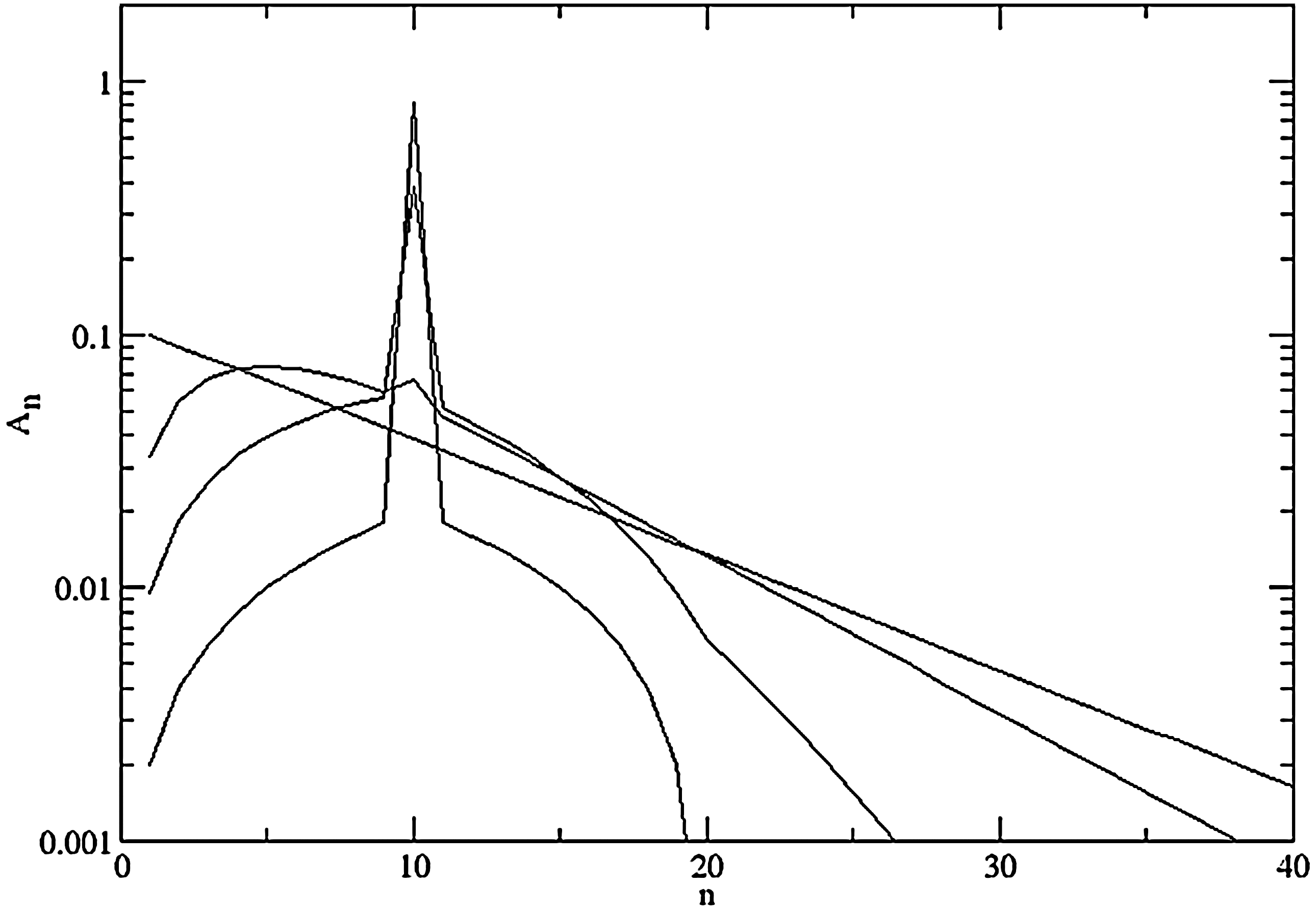
The effect of recombination on the polymer length distribution in a closed system. Beginning with only chains of length 10, the distribution is shown at three finite times and also in the long time limit, where it converges to an exponential with mean length 10.
It also follows that the total rates of creation and destruction of chains are equal if the distribution is exponential:
The mean length from the exponential distribution is 1/(1−z). As the mean length was 10 initially, and as recombination does not change the mean length, it must be the case that z=0.9 in this example. However, from Eqs. 18 and 19, we see that any exponential distribution, irrespective of z, will be stable under recombination. The value of z is determined by the initial conditions and not by the recombination rate ρ.
We now consider the effect of adding recombination to our polymerization reaction system. Equation 3 stays the same, and Eqs. 4 and 5 are modified to
In absence of recombination, the stationary distribution is exponential, with a value of A and z determined from Eqs. 8 and 11. As λ n =μ n for any exponential distribution, it follows that the stationary distribution of Eqs. 20 and 21 is unchanged by the presence of the recombination terms, that is, the polymer length distribution depends on s and r as before, but it does not depend on ρ. If ribozymes arise in this system that catalyze recombination, we might try to introduce feedback in the recombination rate by writing ρ=ρ 0 +kρ PL, as we did before for s and r. However, this will make no difference to the length distribution. The presence of recombinases does not increase the rate of forming more recombinases. Therefore, if recombination is completely reversible, there cannot be an autocatalytic state that is initiated by recombinases.
6. A Non-Reversible Recombinase
In the introduction, we discussed the experiments on the Azoarcus ribozyme that have demonstrated that autocatalysis can arise in a recombinase system (Hayden and Lehman, 2006; Draper et al., 2008; Hayden et al., 2008). This might seem to contradict the result of the previous section; however, there is really no contradiction because in the experimental system recombination is not fully reversible. Figure 9 shows a schematic diagram of a non-reversible recombination reaction that is intended to model the case of the Azoarcus ribozyme. Two distinct RNA strands, X 1 and X 2, which we will call building blocks, associate together by formation of RNA secondary structure to form a complex, B. This brings the ends of the strands into close proximity. Recombination then occurs between the end of one strand and the penultimate nucleotide of the other strand, resulting in a longer strand and a single nucleotide. The longer strand is denoted C for “catalyst.” It is a recombinase that specifically catalyzes the recombination of X 1 and X 2. In other words, the formation of C is autocatalytic. In the experiment, there are four building block strands and three recombination reactions needed to create the catalyst, but in the following model we will consider only two building blocks, as this is sufficient to illustrate the point.
Schematic diagram of association between two RNA strands followed by a recombination reaction giving rise to a longer strand and a single nucleotide.
We want to know whether this autocatalytic reaction can emerge from within our random polymerization system considered above. Suppose that both building blocks are of length m, and the catalyst is of length 2m−1. Suppose that a fraction x of random sequences of length m is suitable to be each of the building blocks. The rate of formation of each building block by random polymerization is therefore x times the rate of synthesis of chains of length m. It follows that a fraction x
2 of sequences of length 2m−1 are catalysts, and the rate of synthesis of the catalyst by random polymerization is x
2 times the rate of synthesis of chains of length 2m−1. Initially, we ignore the complex formation and treat the formation of C as a single reversible reaction:
Adding this to our random polymerization system gives
Both X
1 and X
2 have the same concentration, denoted X, so we only need one equation for these two. We suppose that the chain length distribution is unaffected by the small fraction x that forms the building blocks, so the monomer and polymer concentrations are still given by Eqs. 7 and 11. From (23) and (24), it can be seen that if the recombination rate is zero, X and C tend to steady state concentrations X
0 and C
0, given by
In this state, X 2 =AC, and the presence of the reversible recombination reaction does not make any difference to the frequency of C.
For the recombination reaction to be a useful way to generate the longer sequence, it is necessary to make it non-reversible, which can be done by adding the complex formation step into the model.
The rate constant for formation of the complex is v, and this is taken to be irreversible because the secondary structure is strong; that is, the backward rate is assumed to be negligible with respect to u, so that the complex is lost from the system before it disassociates. In the complex, the two ends that undergo recombination are close together. The forward recombination reaction is essentially a monomolecular reaction, whereas the reverse reaction is bimolecular. Although the reaction itself is reversible, the rates are no longer equal. Let α be the effective concentration of chain ends close to the bond at which recombination occurs, so the forward recombination rate is ραB. It is possible that α>A, in which case recombination can also be biased toward the creation of longer chains. The dynamical equations become
Here, it is assumed that the complex escapes from the system at the same rate u as the polymers. In the steady state, Eq. 28 can be written as
where ω=v/(rA+u). Hence,
By summing (28
)–(30) in the steady state, we obtain
The simplest way to introduce autocatalytic feedback into the model is to suppose that the recombination rate is proportional to the catalyst concentration: ρ=kC. It would also be possible to add a small spontaneous rate ρ
0
of recombination in absence of C, in the same way as we included s
0 and r
0 in previous sections of this paper. However, C is already formed by random polymerization at a small rate, and this is sufficient to get the autocatalytic system started; therefore, the addition of ρ0 is not essential, and we will leave it out. Substituting ρ=kC into (30) and using (33) to eliminate B, we obtain the following quadratic equation for C:
This has a single positive root for C. Thus X, B, and C are all obtained explicitly. These are shown as a function of k in Fig. 10. The parameters for the random polymerization reaction system are F 0=10, s=10, r=5, and u=1, in which case A= 0.587 and z=0.746. We also chose building block length m=25 and a fraction x=0.01. The stationary concentrations are X 0=5.1×10−6 and C 0=4.5×10−11. The concentrations and reaction rates are in arbitrary units, and their values are unimportant. All that matters is that, as the catalyst is much longer than the building blocks, C 0 is very much less than X 0. The limits C 0 and X 0 are shown as dashed lines in Fig. 10.
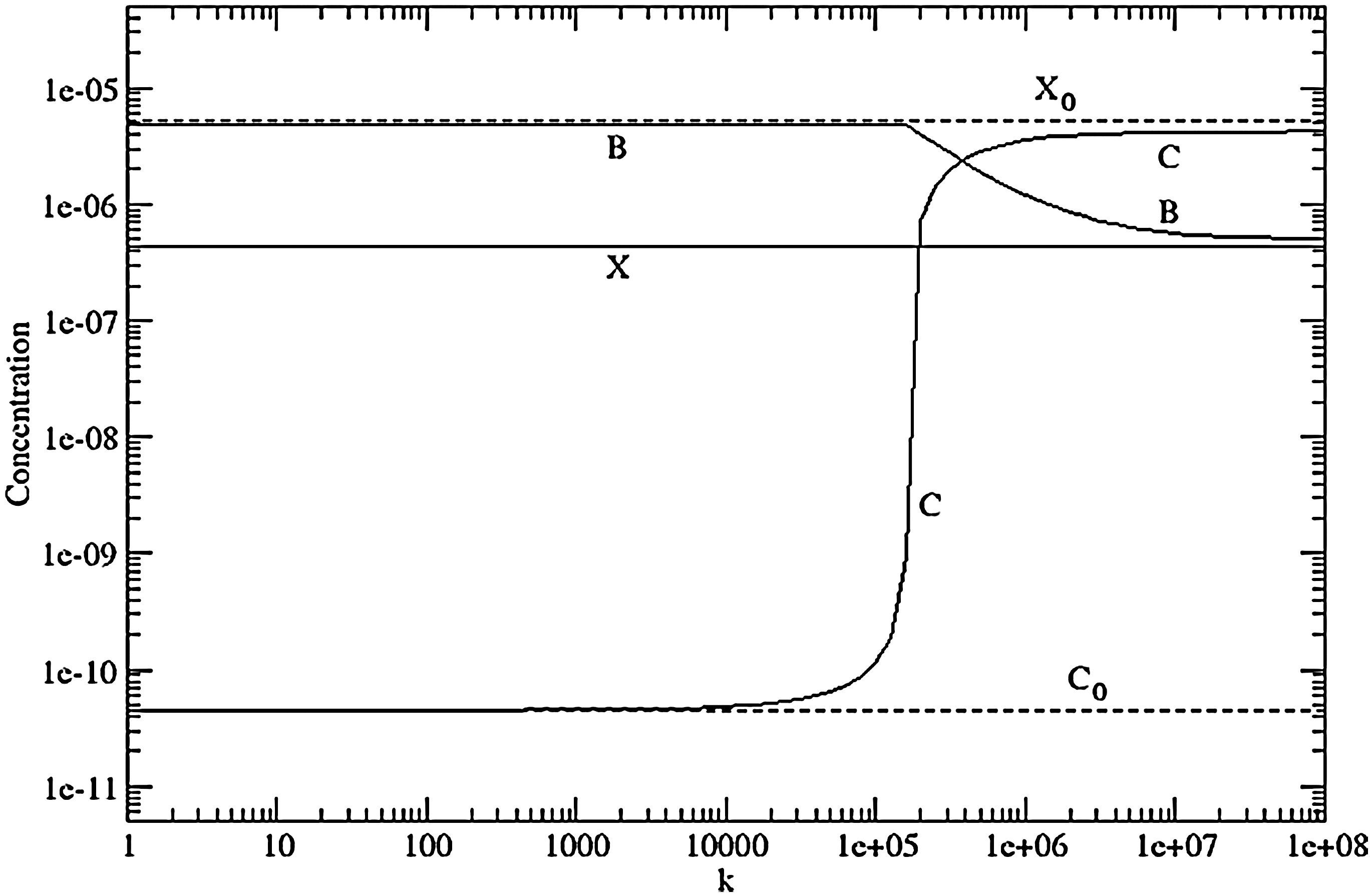
The non-reversible recombinase model in the case where only C is a catalyst. Parameters are F 0=10, s=10, r=5, u=1, v=108, x=0.01, α=5, and m=25.
We now consider formation of the complex B. If X is close to X0 , the ratio of the rates of formation and removal of B is ωX 0. From Eq. 32, if ωX 0 is small, X≈X 0. For complex formation to make a difference, v must be large enough so that ω X 0 >> 1. This means that the complex has time to form before the building blocks escape the system or are destroyed by further random polymerization. In this example, we chose v=108, which gives ωX 0=131. For these parameters, X falls substantially below X 0 because most of the building blocks are converted to the complex. Note that X depends on v, but it does not depend on the recombination reaction; therefore, X is a horizontal line on Fig. 10. If k is small, then the recombination reaction is negligible. Thus B is close to the limiting value of X 0, and C is close to C 0. If k is sufficiently large, the recombination reaction becomes significant. Most of the complex is converted to C, so C approaches X 0, and B decreases. It can be seen that there is a rather rapid switch between the regime where C≈C 0 and the regime where C≈X 0. The value of k required for recombination to switch between regimes is roughly where the formation rate of the catalyst, ραB, is equal to the removal rate, (rA+u)C. This gives ραB≈kCαX 0 ≈(rA+u)C, or k≈(rA+u)/αX 0. In this example α=5, and the predicted value of the switch is k≈ 1.5×105, which is close to what is seen from the exact solution in the graph. Once again, the numbers are arbitrary, but the point is that the required k value is of order 1/X 0. Thus, if the building block concentration is low, then only a very efficient catalyst with very high k will have a noticeable effect.
Here, we summarize the argument of this section so far. If the complex formation step is included in the reaction system, the recombination reaction is no longer exactly reversible. In this case, it is possible for a recombinase C to arise that catalyzes its own formation. The high C regime in Fig. 10 could be classed as a living state because it is autocatalytic. The rate of formation of the catalyst in this regime is almost entirely determined by the catalyzed reaction and not by the spontaneous rate of random polymerization. However, if the building blocks are moderately large, the concentration of the building blocks produced by random polymerization, X 0, is fairly low. For this reason, this system has two important limitations. First, the catalyst does not make more of its own building blocks. The best that it can do is to convert all the existing supply of building blocks to more catalyst. Thus, C can never rise higher than X 0. Second, only catalysts with very high k can reach the autocatalytic regime if X 0 is low. Such catalysts are presumably rare.
We will now introduce one further feature that is also a property of the Azoarcus experimental system. It was found that the complex B was also a catalyst of roughly equal efficiency to the fully formed long strand C (Hayden et al., 2008). This is presumably because the structure of B is almost the same as that of C except for the hairpin loop formed where the building blocks are connected. Therefore, we will consider the case where the recombination rate is proportional to the sum of these concentrations: ρ=k(B+C). Using (33), this means that ρ=k(X
0+C
0−X), and X is already known from (32). Substituting this value of ρ into (30) gives the steady state solution for C:
Figure 11 shows the concentrations of X, B, and C as a function of k in the case where both B and C are catalysts. This differs from Fig. 10 in that C can be several orders of magnitude higher than C0 even when k is low because a substantial amount of C is formed by B acting as a catalyst. When k is large, most of B is converted to C. The situation is then similar to Fig. 10, and C approaches X0 . Thus, if B is also a catalyst, this gets around the limitation that the system only works with very high k, but it does not avoid the limitation that the maximum concentration of catalyst is equal to the building block concentration.

The non-reversible recombinase model in the case where both B and C are catalysts. Parameters are as in Fig. 10.
7. Discussion
A key feature of our model in the case when feedback occurs in either polymerization or nucleotide synthesis is the S-shaped curve of the long polymer concentration as a function of the reaction rate (shown in Figs. 2 and 5). From this curve, it is clear that the phase diagram has regions where there is only one stable solution and regions where there are two stable solutions separated by an unstable solution. A similar S-shaped curve was introduced in a model for the origin of life by Dyson (1999). This was part of the motivation for our model, as we discussed in more detail previously (Wu and Higgs, 2009). However, our model is much less abstract than that of Dyson (1999), because it related to a specific model of RNA polymerization kinetics and is supported by experimental results on ribozymes and abiotic RNA polymerization. In our model, the distinction between the living and the dead states is also very clear: in the dead state, reactions occur principally at their spontaneous rates and the ribozyme concentration is negligible, whereas in the living state, reactions occur principally by catalysis and the ribozyme concentration is high.
The S-shaped curve and the two kinds of stable state seem to be rather general features, as they are found to occur in our original model, in two cases in this paper, and in Dyson's model. This suggests that they may also apply in real-world chemical systems that are evidently much more complicated than any of the models that we can study theoretically. This is important because the idea of the origin of life as a stochastic transition follows directly from the S-shaped curve. The intermediate unstable state acts as a barrier between the dead and the living states. We have shown by stochastic simulations (Wu and Higgs, 2009) that concentration fluctuations can drive the system across the barrier and lead to an origin of life. Although we have not carried out stochastic simulations with the case of the nucleotide synthase ribozymes discussed here, the structure of these models is the same, so we expect that the same phenomena will occur in the stochastic dynamics.
Although the relevance of recombination reactions to ribozyme synthesis has been proposed previously, our paper makes it clear that recombination is only a useful feature if it is not fully reversible and if it is biased toward synthesis of longer chains. In the case we considered, the reaction occurred after the association of the strands to form a complex. This brought the strands into close proximity and increased the forward reaction rate relative to the backward rate. The complex formation step itself was also assumed to be non-reversible. The free energy of RNA structure formation is large compared to the thermal energy kT; therefore, the rate of disassociation of the strands is likely to be very slow. For this reason, we did not include the possibility that the strands might disassociate without undergoing recombination (i.e., B→X 1+X 2). Addition of a small rate of disassociation would not qualitatively change the conclusions. In the case considered, secondary structure formed before recombination, but it is also possible that secondary structure formed after recombination could create a bias toward the formation of longer strands. If a strand folds into a stable secondary structure, a substantial fraction of the nucleotides are in double-stranded conformations that are less likely to interact with other strands than the single-stranded regions. Shorter strands have lower folding free energies and are more likely to be in unfolded configurations than longer strands; therefore, they should potentially be more likely to recombine. If a long strand forms and subsequently folds to a stable structure, this might slow the reverse reaction. Secondary structure formation is also relevant in the ligase system of Lincoln and Joyce (2009). In this case, the building blocks interact with the catalyst strand rather than with each other. This is useful in that it brings the building blocks close together; however, it means that, after the reaction, the newly synthesized strand is strongly associated with the catalyst strand. For further catalytic steps, it is necessary to separate these strands, which might be difficult. In the recombinase case, this problem does not arise because base pairing between the catalyst and the building blocks occurs only at the three-nucleotide guide sequence, rather than along the whole length of the catalyst.
An important feature of the Azoarcus recombinase system (Hayden et al., 2008) is that the trans complex is a catalyst even if the building block strands are not linked covalently. This makes it much more likely that the ribozyme will arise in a random polymerization system, because it is only necessary to synthesize strands of the length of the building blocks rather than the length of the full ribozyme. This property could also be true of any other kind of ribozyme. For example, if there were a trans complex that could act as a polymerase, this would make the origin of an autocatalytic state controlled by the polymerase much easier. In this case, it would not matter very much whether there was an additional recombinase or ligase that linked the building blocks of the polymerase covalently. The system could continue to function with only trans complexes as catalysts.
The polymerase ribozymes envisaged in our model are general, in the sense that they are assumed to increase the rate of polymerization irrespective of the sequence. The Azoarcus recombinase system considered above and the ligase system of Lincoln and Joyce (2009) are specific, in the sense that they work with well-defined building block strands. In one sense, specificity is good, because it insures the product of the reaction is the desired catalyst sequence. On the other hand, specificity is bad because it means that only a tiny fraction of strands in a mixture of random sequences can undergo the reaction. Neither the recombinase nor the ligase in these two examples increase the rate of formation of the building blocks; therefore, they are limited by the supply of their building blocks. The general polymerase would not be limited in this way because it can act on any strand. The corresponding disadvantage is that a completely general polymerase still makes random sequences, so only a tiny fraction of the product is useful. The model investigated here does not keep track of sequences or of the property that RNA polymerization is likely to be template-directed. In the protein world, RNA- and DNA-polymerase enzymes are general, that is, they can act on any sequence; but the sequence specificity arises because they are template directed, and therefore they only act on the specific nucleotide sequences that are present. A template directed RNA-polymerase ribozyme in the RNA world could work in the same way if it could use any available strand as a template.
It is the generality of replication that allows evolution to occur. Once a mechanism has evolved that replicates any sequence, then the sequence can evolve without reinventing the replication mechanism. This is clearly what happened at some point in the history of life on Earth. On the other hand, specific ribozymes functioning with specific substrates might be simpler and more likely to arise initially, although they would be more constrained in terms of their structure and function and would probably be less likely to support evolution in the long term. Understanding the relationship between sequence specificity and generality in autocatalytic systems is an important unresolved issue that will be important for future experimental and theoretical work on ribozymes.
Footnotes
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada. We thank Niles Lehman for helpful comments on recombinase ribozymes.