
Abstract
Although not yet identified in the interstellar medium (ISM), N-heterocycles including nucleobases—the information subunits of DNA and RNA—are present in carbonaceous chondrites, which indicates that molecules of biological interest can be formed in non-terrestrial environments via abiotic pathways. Recent laboratory experiments and ab initio calculations have already shown that the irradiation of pyrimidine in pure H2O ices leads to the formation of a suite of oxidized pyrimidine derivatives, including the nucleobase uracil. In the present work, NH3:pyrimidine and H2O:NH3:pyrimidine ice mixtures with different relative proportions were irradiated with UV photons under astrophysically relevant conditions. Liquid- and gas-chromatography analysis of the resulting organic residues has led to the detection of the nucleobases uracil and cytosine, as well as other species of prebiotic interest such as urea and small amino acids. The presence of these molecules in organic residues formed under abiotic conditions supports scenarios in which extraterrestrial organics that formed in space and were subsequently delivered to telluric planets via comets and meteorites could have contributed to the inventory of molecules that triggered the first biological reactions on their surfaces. Key Words: Pyrimidine—Nucleobases—Interstellar ices—Cometary ices—Molecular processes—Prebiotic chemistry. Astrobiology 12, 295–314.
1. Introduction
Small N-heterocycles, including pyrimidine, purine, and nucleobases, have been extensively sought in the interstellar medium (ISM) in the gas phase, but to date none of them have been detected (Simon and Simon, 1973; Kuan et al., 2003, 2004; Charnley et al., 2005; Brünken et al., 2006). Only an upper limit of a few 1014 cm−2 could be derived for the column density of pyrimidine from these observations (Kuan et al., 2003). However, because of the ubiquity of polycyclic aromatic hydrocarbons (PAHs) and polycyclic aromatic nitrogen heterocycles (PANHs) in galactic and extra-galactic interstellar/circumstellar environments (Allamandola et al., 1989; Puget and Léger, 1989; Roelfsema et al., 1996; Galliano et al., 2008), N-heterocycles including pyrimidine- and purine-based species are expected to be present in space where they can condense on the surfaces of cold, icy grains in dense molecular clouds (Sandford et al., 2004; Bernstein et al., 2005). These species are probably formed from the polymerization of small molecules such as acetylene (C2H2), nitrogen atoms being incorporated via the substitution of acetylene by cyanic acid (HCN) (Ricca et al., 2001). The detection of purine- and pyrimidine-based compounds in carbonaceous chondrites (Hayatsu, 1964; Folsome et al., 1971, 1973; Hayatsu et al., 1975; van der Velden and Schwartz, 1977; Stoks and Schwartz, 1979, 1981; Callahan et al., 2011), whose extraterrestrial origin was recently confirmed by isotopic analysis (Martins et al., 2008), strongly supports the existence of non-terrestrial abiotic chemical pathways for their formation under astrophysical conditions.
Recent laboratory simulations have shown that UV photo-irradiation of pyrimidine in pure H2O ices leads to the formation of a large suite of pyrimidine derivatives, including 4(3H)-pyrimidone, a precursor of the nucleobase uracil, and uracil itself (Nuevo et al., 2009). Both of these compounds have been reported in the Murchison, Murray, and Orgueil carbonaceous chondrites (Folsome et al., 1971, 1973; Lawless et al., 1972; Stoks and Schwartz, 1979). Ab initio quantum calculations have shown that 4(3H)-pyrimidone (and/or its tautomer 4-hydroxypyrimidine) and uracil are expected to be the most stable singly and doubly oxidized pyrimidine derivatives formed from the UV photo-induced oxidation of pyrimidine in pure H2O ice (Bera et al., 2010). These calculations also indicated that the presence of H2O as a matrix is essential for the formation of oxidized compounds because it participates in the proton abstraction from the intermediate compounds to the final, stable products.
Ammonia (NH3), another interstellar ice component (Lacy et al., 1998), may also react with pyrimidine upon irradiation and yield new pyrimidine derivatives, including the nucleobase cytosine in the presence of H2O. To examine this chemistry, we performed laboratory experiments of UV photo-irradiation of several NH3:pyrimidine and H2O:NH3:pyrimidine ice mixtures with different relative proportions under astrophysical conditions. The organic residues formed in those experiments were analyzed with chromatography techniques to search for the presence of pyrimidine derivatives, including the nucleobases uracil and cytosine, as well as other non-cyclic compounds of prebiotic interest such as urea and small amino acids. The detection of nucleobases and other interesting species in our residues is compared with previous experimental studies and analyses of meteorites, and their presence and photo-stability in astrophysical environments are discussed from an astrobiological point of view.
2. Experimental Methods
2.1. Ultraviolet photo-irradiation of ices at low temperature
Sample preparation was carried out with the same experimental setup as described in detail in Nuevo et al. (2009) for the study of H2O:pyrimidine mixtures. Gas mixtures were deposited onto a pre-baked (500°C) aluminum foil attached to a cold finger mounted inside a vacuum chamber (background pressure at low temperature: 2.2–5.3×10−8 mbar) and cooled to 15–40 K by a closed-cycle helium cryocooler. H2O vapor (taken from a liquid purified to 18.2 MΩ cm by a Millipore Direct-Q UV 3 device), NH3 gas (Matheson, anhydrous, 99.99% purity), and pyrimidine vapor (taken from a liquid, Aldrich, 99% purity) were mixed in a glass line (background pressure ∼10−6 mbar). NH3:pyrimidine mixtures with relative proportions 10:1, 20:1, 40:1, and 100:1, and H2O:NH3:pyrimidine mixtures with relative proportions of 20:2:1 and 20:1:1, were prepared and transferred into 1.9 L glass bulbs. Ratios between components were determined by their partial pressure with an accuracy of 0.05 mbar.
In each experiment, a total of 32.0–38.9 mbar (∼2.7–3.2 mmol) and 33.7–39.4 mbar (∼2.8–3.3 mmol) for NH3:pyrimidine and H2O:NH3:pyrimidine mixtures, respectively, were deposited onto the cold (15–40 K) substrate and simultaneously photo-irradiated with a microwave-powered H2 discharge UV lamp for durations ranging from 21.5 to 53 hours. This lamp emits Lyman-α photons (121.6 nm) and a continuum centered at∼160 nm with an estimated total flux of about 2×1015 photons cm−2 s−1 (Bernstein et al., 1999; Elsila et al., 2007), which is considered to simulate the UV radiation field from surrounding stars and protostars in astrophysical environments. In terms of photon dose, such experiments correspond to an ice photo-irradiation of about 104 years and 107–109 years in the diffuse and dense ISM, respectively (Mathis et al., 1983; Prasad and Tarafdar, 1983; Shen et al., 2004). The ratios of the number of photons per deposited molecule ranged from 0.25 to 0.6 for both the NH3:pyrimidine and H2O:NH3:pyrimidine mixtures.
After simultaneous deposition/irradiation, each sample was slowly warmed to 220 K under static vacuum, at which time the sample finger was pulled out of the vacuum chamber, and the Al foil was removed and put in a pre-baked (500°C) glass vial. Samples for high-performance liquid chromatography (HPLC) analysis were dissolved in 500 μL of H2O (Millipore, 18.2 MΩ cm resistivity), whereas samples for gas chromatography coupled with mass spectrometry (GC-MS) analysis were either kept dry in pre-baked vials or recovered from the residues dissolved in H2O. All these vials were kept in a freezer (−20°C), and the samples were thawed before analysis at room temperature.
Two additional sets of experiments were performed for the NH3:pyrimidine=40:1 and H2O:NH3:pyrimidine=20:2:1 mixtures. The first set was performed under similar conditions as described above except that the cold finger temperature was set to 120 K to simulate conditions of icy Solar System bodies. The second set of experiments was performed with the use of a CaF2 window to filter out Lyman-α photons, in order to assess the effect of the UV photon wavelength on the formation of photo-products. Those four samples were analyzed with both HPLC and GC-MS.
Finally, for each group of experiments—NH3:pyrimidine ices and H2O:NH3:pyrimidine ices—two types of control experiments were performed: (1) controls in which ices were deposited but not UV irradiated and (2) controls in which the H2 lamp was turned on but no ice was deposited.
2.2. Infrared analysis of the samples
NH3:pyrimidine=25:1 and H2O:NH3:pyrimidine=20:2:1 ice mixtures were deposited, as separate samples, onto an infrared-transparent zinc selenide (ZnSe) window at 14 and 15 K, respectively. An initial infrared (IR) spectrum was measured from each ice sample before any irradiation. The ices were then photo-irradiated with a H2 UV lamp, and IR spectra were recorded at intervals until total exposures of 188 and 200 min were obtained for the NH3:pyrimidine and H2O:NH3:pyrimidine ices, respectively. Photo-destruction of pyrimidine was monitored via one of its strongest IR bands near 1400 cm−1 (βCH, νCN) (Destexhe et al., 1994), since this feature was the least subject to blending with other IR features. After completion of the UV exposures, the ice samples were then slowly warmed (at 2 K min−1), and their IR spectra were obtained at intervals until the samples reached room temperature.
Infrared spectra were taken at a resolution of 1 cm−1 with a Bio-Rad Excalibur Series Fourier-transform IR spectrometer equipped with a mercury-cadmium-telluride (MCT) detector cooled to 77 K with liquid nitrogen. Spectra were recorded in the mid-IR range between 4000 and 650 cm−1 (2.5–15.4 μm) and ratioed to appropriate background spectra taken of the blank sample window before ice deposition.
2.3. HPLC and GC-MS analysis of residues at room temperature
H2O-dissolved samples for HPLC analysis were injected into a Hewlett Packard/Agilent 1100 Series device and separated in a Phenomenex Luna 5u Phenyl-Hexyl column (size: 250 mm×4.60 mm, inner diameter: 5 μm), with a volume of 5 μL for each independent run. Separated compounds were detected by a diode-array UV detector that recorded signals at 220, 245, 256, 280, and 300 nm. The method used (solvent gradients) for these runs and the preparation of the pH=5 ammonium formate buffer are described elsewhere (Nuevo et al., 2009). Peaks in sample chromatograms were identified by comparison of both their retention times and UV spectra with purchased standards dissolved to 10−3 M in H2O (Millipore, 18.2 MΩ cm resistivity) and injected by using the same method as the samples.
Two types of extracts were prepared for GC-MS analysis: dry foils were shaken with 100 μL of ethyl acetate (CH3COOCH2CH3, Fisher Scientific, Optima grade), whereas 100 μL of the residues dissolved in H2O were extracted for analysis. Both ethyl acetate and H2O extracts were transferred to pre-baked (500°C) vials and dried under vacuum in a desiccator for 2 h. We then added 50 μL of a 3:1:1 mixture of N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide (MTBSTFA) with 1% of tert-butyldimethylchlorosilane (tBDMCS) (Restek), dimethylformamide (Pierce, silylation grade solvent), and pyrene (Sigma-Aldrich, analytical standard, 100 ng μL−1 dissolved in cyclohexane) to each dried residue. The vials were then heated to 100°C for 1 h to convert α-hydrogen moieties, such as OH and NH2, into their tert-butyldimethylsilyl (tBDMS) derivatives (MacKenzie et al., 1987; Casal et al., 2004; Schummer et al., 2009). Each tBDMS group added to a compound increases its mass by 114 atomic mass units (amu).
Separation was carried out with a Thermo Trace gas chromatograph coupled to a DSQ II mass spectrometer with a splitless injection, a Restek Rxi-5ms column (length: 30 m; inner diameter: 0.25 mm; film thickness: 0.50 μm), an injector temperature of 250°C, and a helium (carrier gas, ultra pure; Air Liquide) flow of 1.3 mL min−1. The method (temperature gradient) used is described in detail elsewhere (Nuevo et al., 2009). Masses were recorded in the 50–550 amu range, and data analysis was performed with Xcalibur software (Thermo Finnigan). Peaks in sample chromatograms were identified by comparison of both their retention times and mass spectra with the same standards as used for the HPLC analysis, derivatized the same way as the samples.
The standards of pyrimidine, its oxidized derivatives, as well as several other pyrimidine derivatives, are the same as those used for the study of UV-irradiated H2O:pyrimidine ices (Nuevo et al., 2009). Oxidized derivatives include 2-hydroxypyrimidine, 4(3H)-pyrimidone, uracil, 4,6-dihydroxypyrimidine, barbituric acid, and isobarbituric acid. Pyrimidine N-oxide (Aldrich, 97% purity), a pyrimidine molecule to which an oxygen atom is attached to one of the nitrogen atoms (Fig. 1a), was also searched for. Other standards measured include: (1) Amino-bearing pyrimidines: 2-aminopyrimidine (Aldrich, 97% purity), 4-aminopyrimidine (Aldrich, 98% purity), 2,4-diaminopyrimidine (Aldrich, 99% purity), 4,5-diaminopyrimidine (Aldrich, 95% purity), and 2,4,6-triaminopyrimidine (Aldrich, 97% purity). (2) Amino- and hydroxy-bearing pyrimidines: cytosine (Aldrich, 97% purity), isocytosine (Sigma, ≥99% purity), 5-aminouracil (Aldrich, 98% purity), 6-aminouracil (Aldrich, 97% purity), 2-amino-4,6-dihydroxypyrimidine (Aldrich, 98% purity), and 2,4-diamino-6-hydroxypyrimidine (Aldrich, 96% purity). (3) Other pyrimidine derivatives: 2,2′-bipyrimidine, 2-pyrimidinecarbonitrile, orotic acid (see Nuevo et al., 2009), 1,4,5,6-tetrahydropyrimidine (Aldrich, 97% purity), 2-amino-5-nitropyrimidine (Aldrich, 98% purity), and 5-nitrouracil (Aldrich, 98% purity). (4) Other N-heterocycles: hydantoin (Aldrich, 98% purity), pyridine (Sigma, Biotech grade, ≥99.9% purity), and purine (Aldrich, 98% purity). (5) Non-cyclic molecules: urea (Sigma-Aldrich, ACS reagent, ≥99.0% purity); the proteinic amino acids glycine, L-alanine, L-serine (Pierce, lot No. 20065); and the non-proteinic amino acid N-formylglycine (Fluka, ≥98% purity).
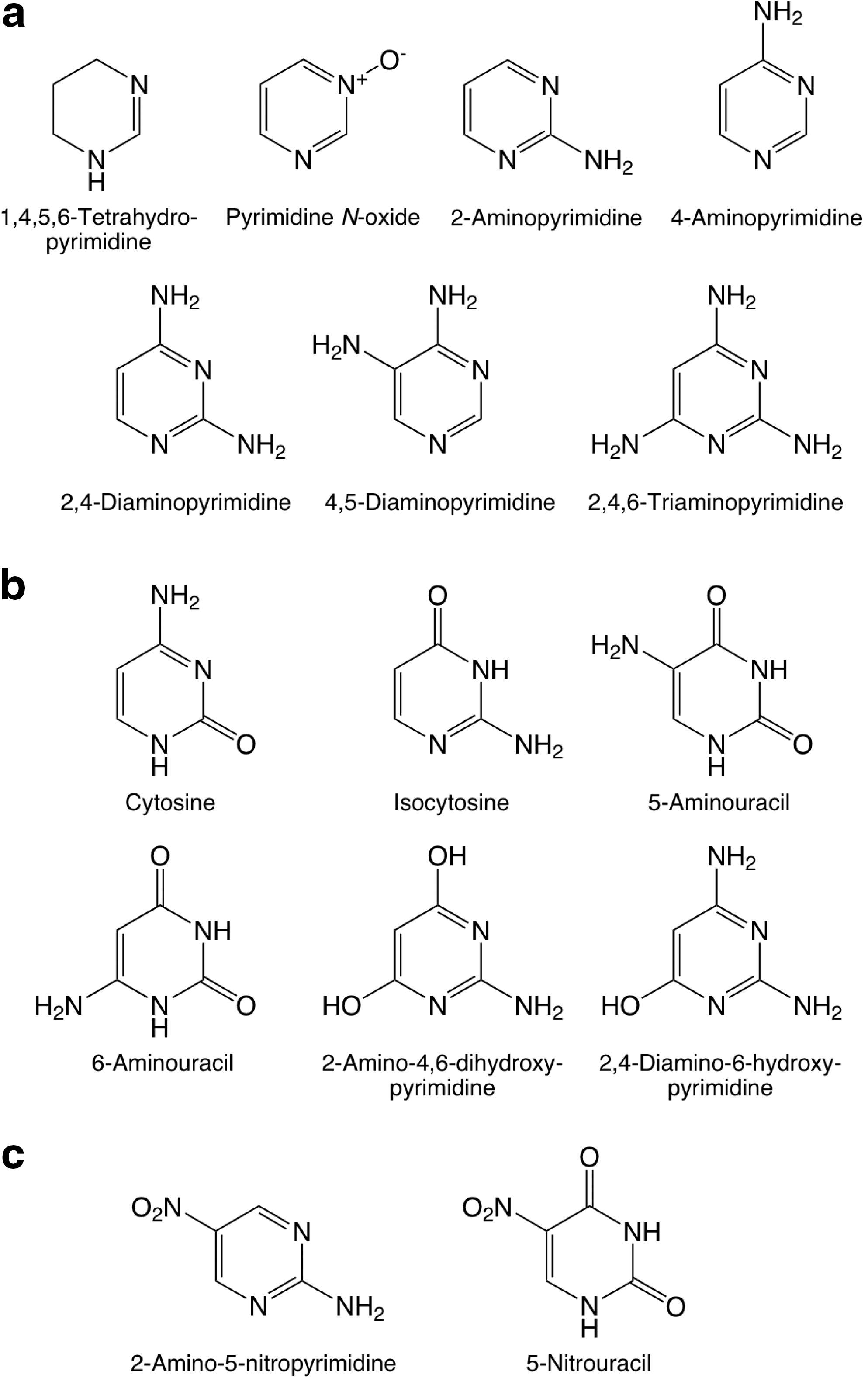
Molecular structures of the standards of pyrimidine derivatives searched for in this study:
The molecular structures of the pyrimidine derivatives listed above are given in Fig. 1. The structures of hydantoin, pyridine, purine, urea, glycine, L-alanine, L-serine, and N-formylglycine are given in Fig. 2. The structures of all other compounds can be found elsewhere (Nuevo et al., 2009).
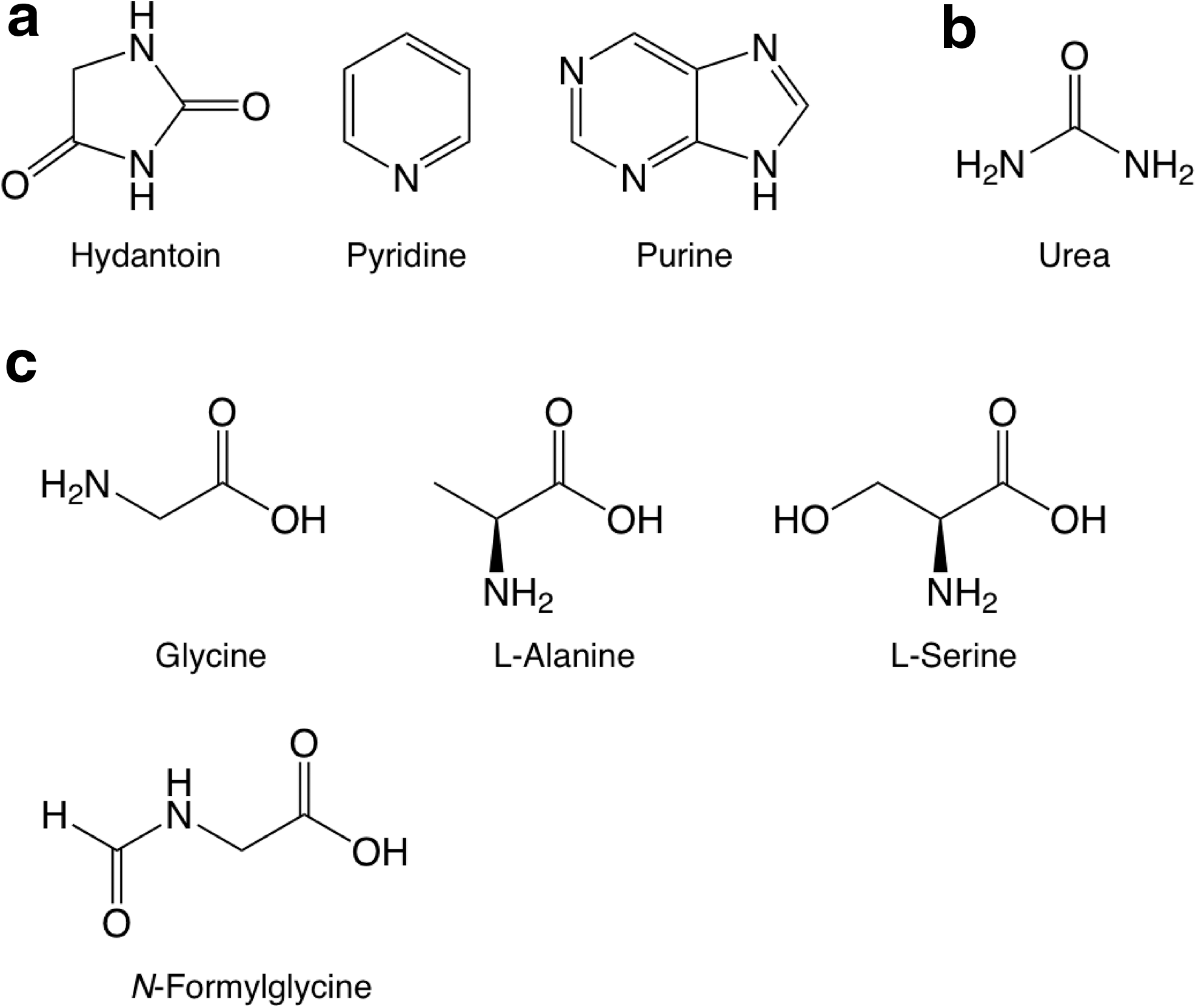
Molecular structures of
3. Results
3.1. Infrared spectroscopy
Spectra of the pre-irradiated ice samples for the NH3:pyrimidine=25:1 and H2O:NH3:pyrimidine=20:2:1 ices are shown on the top and bottom traces of Fig. 3, respectively.
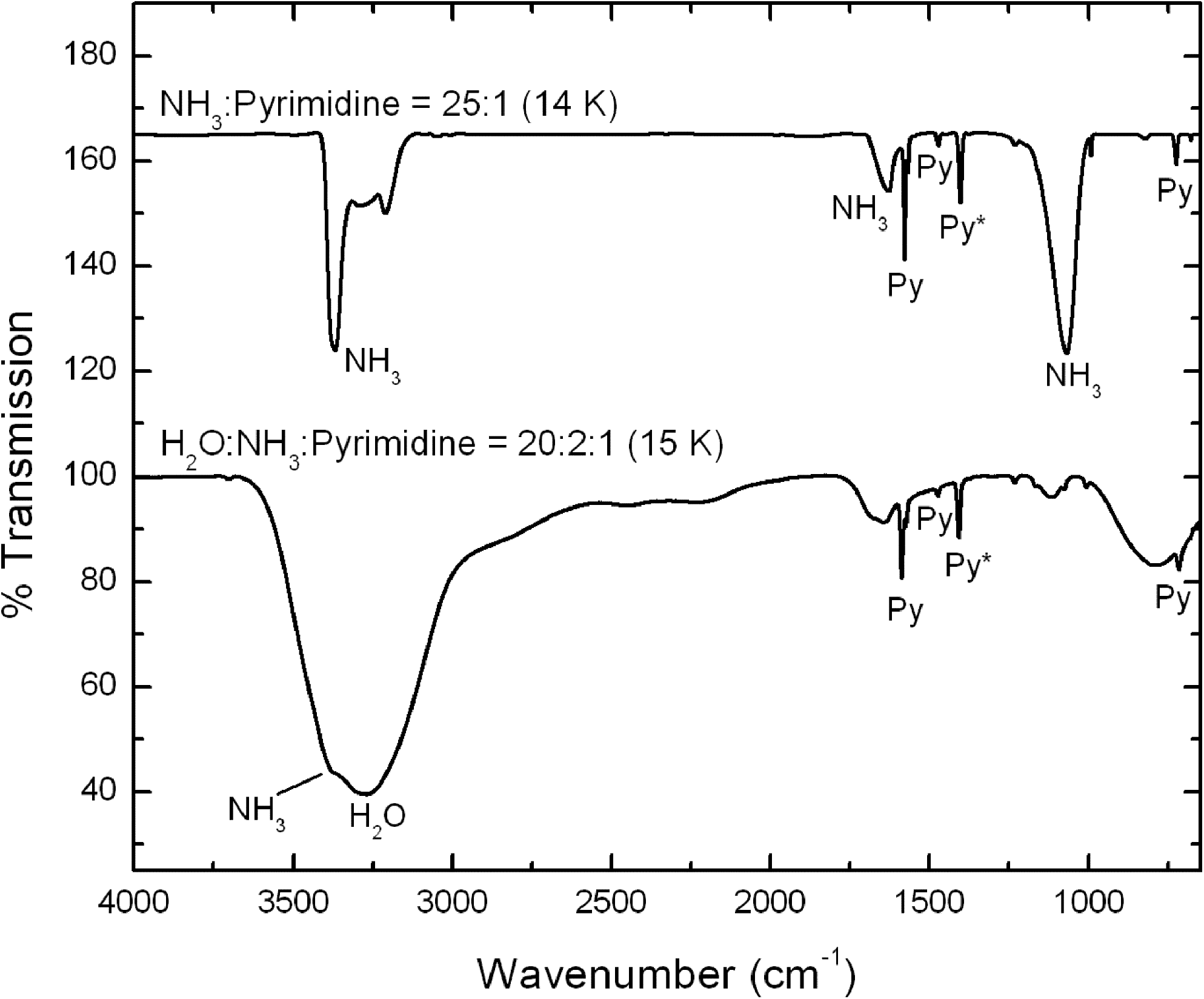
The 4000–650 cm−1 IR spectra of an NH3:pyrimidine=25:1 mixture deposited at 14 K before UV photo-irradiation (top trace, offset for clarity) and an H2O:NH3:pyrimidine=20:2:1 mixture deposited at 15 K before UV photo-irradiation (bottom trace). Infrared bands labeled with “Py” are due to pyrimidine in the ices, the band labeled “Py*” being that used to estimate the photo-destruction efficiency of pyrimidine.
The column density of NH3 in the NH3:pyrimidine=25:1 ice mixture was determined to be 2.2×1018 molecules cm−2 from the integrated area of the ammonia N–H stretching absorbance feature at ∼3372 cm−1, with an integrated absorbance of A=1.1×10−17 cm molecule−1 (d'Hendecourt and Allamandola, 1986). Assuming an NH3/pyrimidine abundance ratio of 25, this corresponds to a column density of pyrimidine prior to UV exposure of ∼8.8×1016 molecules cm−2. The column density of H2O in H2O:NH3:pyrimidine=20:2:1 ice sample was determined to be ∼2.7×1017 molecules cm−2 from the integrated area of the O–H stretching feature at ∼3273 cm−1, with an integrated absorbance of A=1.7×10−16 cm molecule−1 (Hudgins et al., 1993), although it should be noted that this band is blended with the N–H stretching band of NH3 and therefore represents an upper limit. Assuming an H2O/pyrimidine ratio of 20, this corresponds to a pyrimidine column density of ∼1.3×1016 molecules cm−2.
The spectra of both the NH3:pyrimidine and H2O:NH3:pyrimidine ices show two main changes that occurred as the samples were irradiated at low temperature: (1) a decrease in the absolute strengths of the pyrimidine bands relative to those in the spectra of the original unirradiated sample, which indicates that photolysis caused the destruction and/or conversion of some of the pyrimidine, and (2) the appearance of a few new, weak bands associated with photo-products. Subsequent warming of both ices resulted in few spectral changes beyond the normal small shifts of band positions and profiles as the warming ices annealed until the sublimation temperatures of NH3 and H2O were reached. At this point, the majority of the spectral features of the original ice samples disappeared. The spectra show that, by the time the samples reached room temperature, only weak features due to a remaining residue were present. Spectra of the residues that remained after the warming of the irradiated NH3:pyrimidine and H2O:NH3:pyrimidine ices are shown in the top and bottom traces of Fig. 4, respectively.
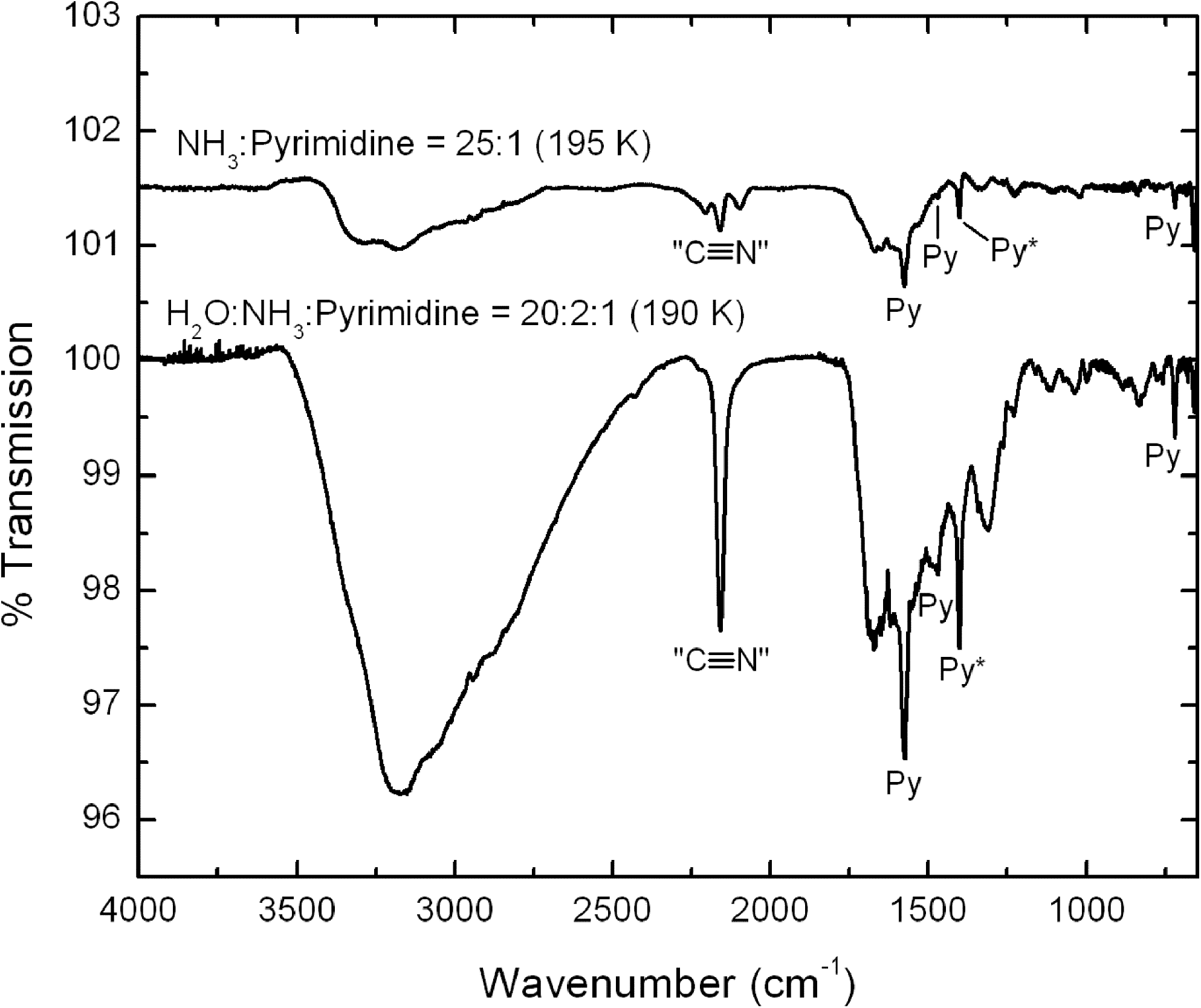
The 4000–650 cm−1 IR spectra of residues that remain after the sublimation of the main ice components of an irradiated NH3:pyrimidine=25:1 ice mixture (top trace, offset for clarity) and an irradiated H2O:NH3:pyrimidine=20:2:1 ice mixture (bottom trace). Infrared bands labeled with “Py” are due to unreacted pyrimidine in the residues, the band labeled “Py*” being that used to estimate the photo-destruction efficiency of pyrimidine.
3.1.1. Pyrimidine photo-destruction
The photo-destruction efficiency for pyrimidine in these ices was determined by monitoring the decreasing strength of its band near 1400 cm−1 (labeled “Py*” in Figs. 3 and 4) as a function of UV exposure. The half-lives for the photo-destruction of pyrimidine with our lamp on the NH3:pyrimidine=25:1 and H2O:NH3:pyrimidine=20:2:1 ices were measured to be 510 and 270 min, respectively (Table 1). These are 2–3 orders of magnitude longer than pyrimidine in a pure argon matrix (0.93 min; Peeters et al., 2005) and significantly longer than the values measured for H2O:pyrimidine ices (38 min; Nuevo et al., 2009), which implies that the photo-destruction of pyrimidine in NH3-containing ices is much less efficient.
Estimated according to the following UV photon fluxes: laboratory, 2×1015 photons cm−2 s−1 (Elsila et al., 2007); diffuse interstellar medium (DISM), 8×107 photons cm−2 s−1 (Mathis et al., 1983); dense clouds (DC), 1×103 photons cm−2 s−1 (Prasad and Tarafdar, 1983); Solar System, 3×1013 photons cm−2 s−1 (Peeters et al., 2005).
Data from Nuevo et al. (2009).
Data from Peeters et al. (2005).
Assuming an optically thin ice and a first-order decay (Cottin et al., 2003), these photo-destruction efficiencies can be used, with the UV flux rates given in Table 1, to predict the half-life of pyrimidine in these ices if present in various astrophysical environments. We derived half-lives of pyrimidine in our NH3:pyrimidine=25:1 ice of about 19,000 yr, 1,900 Myr, and 570 h in the diffuse ISM, dense clouds, and Solar System (at 1 AU), respectively. Corresponding photo-destruction half-lives of pyrimidine in an H2O:NH3:pyrimidine=20:2:1 ice are about 10,300 yr, 1,030 Myr, and 300 h, respectively (Table 1).
While the photo-destruction rates of these two ices differ, both lead to the same qualitative conclusions. Pyrimidine in NH3-containing ices will likely be thoroughly reprocessed if these ices are present on the exposed surfaces of Solar System bodies. However, individual dense interstellar clouds typically last for timescales of tens of millions of years, which is far shorter than the estimated pyrimidine photo-destruction half-life under these conditions and suggests that pyrimidine in NH3-rich ices will not be efficiently reprocessed in these objects. Finally, such ices would not be stable in the diffuse ISM.
3.1.2. Pyrimidine photo-products
A list of the principal IR bands seen in the spectra of the irradiated samples of our NH3:pyrimidine=25:1 and H2O:NH3:pyrimidine=20:2:1 ices, together with some possible identifications, are summarized in Table 2. The residues at ∼190 K show many similarities and a few significant differences.
Sill et al. (1980); bHudgins et al. (1993); cSandford and Allamandola (1990); dSchutte and Greenberg (1997); eDemyk et al. (1998); fPalumbo et al. (2000); gCotton and Zingales (1961); hBernstein et al. (1997); iSandford et al. (1988); jGerakines et al. (2004); kBurgdorf et al. (2010); lGaigeot and Sprik (2003); mGreen (1962); nMuñoz Caro and Schutte (2003); oEvans et al. (1991); pThompson and Jacox (2001); qDelwiche et al. (1993); rMaier and Endres (1999); sStolkin et al. (1977); tHashiguchi et al. (1984); uMcCluskey and Frei (1993); vShimanouchi (1972); wMaier and Endres (2000); xSteiner et al. (1979); yPettersson et al. (1999).
The band marked with an asterisk (*) was used to determine the photo-destruction half-life of pyrimidine. The assignments in the lower part of the table are only tentative.
During irradiation of the H2O:NH3:pyrimidine ice, new, weak features were produced near 2341, 2167, and 2136 cm−1 (Table 2). All these features are common in irradiated ices that contain mixtures of C-, O-, and N-containing molecules. The feature at 2341 cm−1 is due to CO2 (Sandford and Allamandola, 1990), while that at 2136 cm−1 is due to CO (Sandford et al., 1988). The feature near 2167 cm−1 is probably due to OCN− or isonitriles, or both (Cotton and Zingales, 1961; Bernstein et al., 1997, 2000; Schutte and Greenberg, 1997; Demyk et al., 1998; Palumbo et al., 2000), and labeled “C≡N” in the IR spectra of the residues (Fig. 4).
The principal new feature that appears during irradiation of the NH3:pyrimidine ice absorbs near 2086 cm−1 (Table 2). This feature is probably due to HCN or larger nitriles (Bernstein et al., 1997; Gerakines et al., 2004; Burgdorf et al., 2010). It should be noted that the spectrum of this irradiated ice also shows the presence of very weak CO2 and OCN− bands, which indicates that a very small amount of oxygen was present in this ice sample. This oxygen probably comes from trace amounts of H2O in our vacuum system (H2O is our primary contaminant when the system operates in the 10−8 mbar pressure range).
As the irradiated NH3:pyrimidine and H2O:NH3:pyrimidine ices were warmed and their original NH3 and H2O:NH3 matrices sublimed away, ions, radicals, and neutrals in the ices became mobile and may have reacted to form more complex species. Spectra of the resulting residues at 190–195 K (Fig. 4) contain a number of weak absorption features, some due to unreacted pyrimidine and others due to new photo-products. The spectra of both residues show many similarities and suggest the presence of new functional groups involving C–N bonds including nitriles (–C≡N), isonitriles (–N≡C), OCN−, as well as one or both of HCN and CN− (Table 2).
There are, however, several distinct differences between the spectra of the two residues. For example, the NH3:pyrimidine residue contains a band at 2098 cm−1, which is thought to be due to HCN or other nitrile-containing compounds, and a weak band at 1515 cm−1 that may be due to
3.2. Liquid chromatography
3.2.1. NH3:pyrimidine mixtures
Although several NH3:pyrimidine mixtures with ratios ranging from 10:1 to 100:1 were irradiated in this study, the discussion will mainly focus on the results obtained for the 40:1 mixtures. The total HPLC chromatogram (λ=256 nm) of a residue produced from the UV photo-irradiation at 18–29 K of an NH3:pyrimidine=40:1 mixture irradiated for ∼24 h is shown in the top trace of Fig. 5a. The bottom trace corresponds to a blank sample in which a similar mixture was deposited on the Al foil but not UV irradiated. The only peak visible in the chromatogram of the blank sample, at a retention time (Rt ) of 18.11 min, is due to unreacted pyrimidine. All other peaks in the irradiated sample chromatogram are due to products formed from photo-processes at low temperature and/or during warm-up. As was the case for the UV irradiation of H2O:pyrimidine ices (Nuevo et al., 2009), the chromatograms of NH3:pyrimidine samples showed no significant differences regardless of whether they were injected the day they were produced or up to 160 days later.
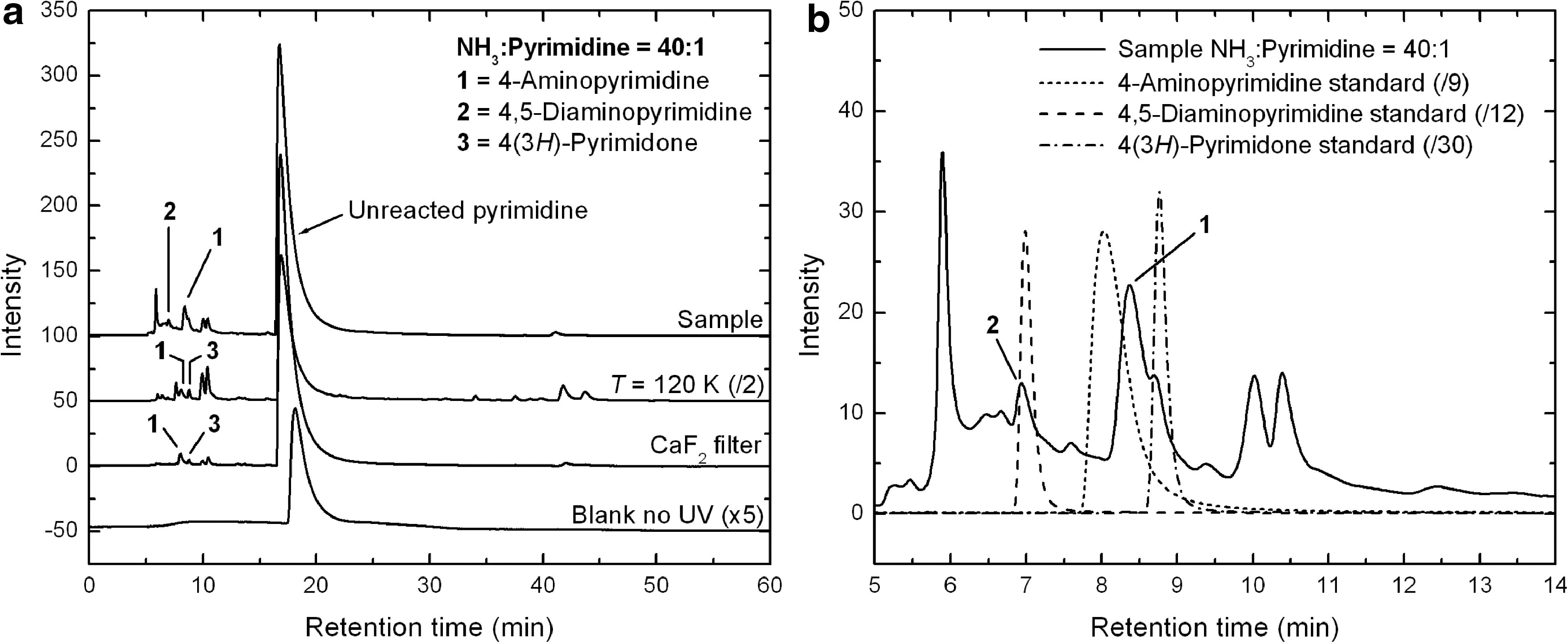
The chromatogram of the irradiated sample shows the same very intense, broad peak at 16.74 min (Fig. 5a, top trace) as in the blank sample, due to unreacted pyrimidine. Its presence indicates that pyrimidine is not efficiently converted into photo-products when mixed with NH3 ice and UV irradiated. This is significantly different from what happens when pyrimidine is mixed with an H2O ice (Nuevo et al., 2009). Peaks assigned to photo-products in the irradiated NH3:pyrimidine sample are concentrated in the 5–15 min retention time region (Fig. 5b) and are weak in intensity. The two strongest peaks assigned to photo-products are ∼6 and ∼10 times weaker than the peak of unreacted pyrimidine.
The few photo-products that could be identified in this residue are listed in Table 4 and labeled
Masses reported here (in atomic mass units) correspond to the mass of the most intense peak for each standard in the GC-MS mass spectra, i.e., the total mass of the derivatized compound (M*) minus the mass of one tert-butyl group ([M* – 57]+ fragment), except for compounds that are not derivatized (see note b). Numbers between parentheses are the number of tBDMS groups attached to the parent molecules.
Compounds not derivatized by the MTBSTFA+1% tBDMCS agent (no tBDMS groups attached). Masses reported here are thus the same as the pure compounds.
The HPLC chromatogram of 2,4,6-triaminopyrimidine displays several peaks among which the peak eluting at 8.21 min is the most intense.
The mass of derivatized purine, and thus the mass of the most intense peak in GC-MS chromatograms, is 1 amu higher than what is expected by adding a tBDMS group to purine.
The HPLC peak of urea is too weak to obtain a clear UV spectrum.
n.d.=Not detected.
Detected in all samples.
Data from Nuevo et al. (2009), unless otherwise stated.
Present work.
Detected at trace levels.
Molecules detected in other samples than the irradiated NH3:pyrimidine=40:1 and H2O:NH3:pyrimidine=20:2:1 samples whose HPLC and GC-MS chromatograms are shown in Figs. 5 –9.
Molecules detected by GC-MS in the same H2O:pyrimidine mixtures as reported in Nuevo et al. (2009) after publication of the paper.
Compounds always eluting at the same retention time as one or more other unidentified species (coeluents).
Oxygen-bearing species detected in NH3:pyrimidine samples (see Sections 3.2 and 4.1).
Finally, a weak peak at 8.89 min, assigned to 4(3H)-pyrimidone and labeled as peak
None of the other compounds searched for (Tables 3 and 4) could be identified in the HPLC chromatogram of the sample, though several peaks show clear profiles and UV spectra. In particular, the intense peaks at 5.90, 10.02, and 10.39 min (Fig. 5b) remain unidentified. They may be due to any or all of the following: isomers whose standards are not available; non-cyclic compounds, as suggested by IR spectra (Figs. 3 and 4); cyclic compounds other than pyrimidine-based species, although pyridine and purine were not detected (Table 4). Additional unidentified, weaker peaks eluting at 41.07 and 43.11 min (Fig. 5a), already observed in H2O:pyrimidine residues (Nuevo et al., 2009), have unique UV spectra compared with the rest of the photo-products. They may be due to molecules formed from rearrangements of pyrimidine into different cyclic structures, with no addition of nucleophilic groups such as OH or NH2, which usually elute at shorter (<15 min) retention times.
The chromatograms of the residues produced from the UV irradiation of an NH3:pyrimidine=40:1 mixture at 120 K and of a similar mixture for which a CaF2 filter was used to cut off Lyman-α (121.6 nm) photons at 18–24 K are shown in the top middle and bottom middle traces of Fig. 5a, respectively. They display qualitatively similar peaks compared with those observed for the NH3:pyrimidine=40:1 sample irradiated at lower temperature with no filter (Fig. 5a, top trace), with different relative intensities. Peaks present are pyrimidine (∼16.89 and 16.84 min for the residues formed at 120 K and with the use of the CaF2 filter, respectively), 4-aminopyrimidine (8.04 and 7.99 min), as well as both groups of unidentified peaks in the 10–10.5 min and 39–44 min retention time ranges. The peak assigned to 4(3H)-pyrimidone (at 8.76 and 8.74 min) is also present.
It is clear from comparison of the chromatograms of the residue formed at 120 K (Fig. 5a, top middle trace) with that of the 18–29 K sample (top trace) that the photochemistry is somewhat more efficient at the higher temperature, since the photo-product peak intensities in the sample irradiated at 120 K are about 1.5 times higher than for the 18–29 K sample. Also, while these different samples show a similar set of peaks, the relative abundance of the products seems to vary. For example, the most abundant (unidentified) photo-product of the sample irradiated at 18–29 K (Rt =5.90 min) is one of the least abundant photo-products in the 120 K sample, whereas the unidentified compounds eluting at 10–10.5 and 39–44 min are formed much more efficiently at 120 K than at lower temperature. In contrast, the use of the CaF2 filter resulted in a significant decrease in the photochemistry efficiency (compare the top and bottom middle traces in Fig. 5a), even after normalization of the photo-product abundances to the number of pyrimidine molecules deposited and to the ratio of the number of photons to the number of pyrimidine molecules deposited. This result suggests that (1) Lyman-α photons play an important role in the photochemistry that takes place in such ices, and (2) photo-products are still formed by lower-energy photons even in the absence of Lyman-α photons, although with a significantly lower efficiency.
The HPLC chromatograms obtained for residues formed from NH3:pyrimidine mixtures with relative ratios of 10:1, 20:1, and 100:1 (not shown here) are qualitatively similar to those given in Fig. 5. This suggests that the relative proportion between NH3 and pyrimidine in the starting mixture within this concentration range has no major effect on the types of photo-products formed, and it only affects the absolute quantities and/or relative yields of these photo-products.
As a general remark, HPLC results show that the UV irradiation of NH3:pyrimidine mixtures is not as efficient for making photo-products as what is observed for H2O:pyrimidine mixtures (Nuevo et al., 2009). However, it is interesting to note that the basic chemistry taking place is similar to what occurs when pyrimidine (Nuevo et al., 2009), small PAHs (Bernstein et al., 1999, 2001, 2002b; Ashbourn et al., 2007), and small PANHs (Elsila et al., 2006) are irradiated in ices.
3.2.2. H2O:NH3:pyrimidine mixtures
The HPLC chromatograms (λ=256 nm) of the residues produced from an H2O:NH3:pyrimidine=20:2:1 mixture that was UV photo-irradiated at 20–32 K for ∼23.5 h (top trace), and two similar mixtures—one irradiated at 120 K (top middle trace) and the other irradiated with the use of a CaF2 filter at 19–28 K (bottom middle trace)—are shown in Fig. 6a. The bottom trace corresponds to an H2O:NH3:pyrimidine=20:2:1 mixture that was deposited on the Al foil but not UV irradiated (blank). Similarly to what was observed for H2O:pyrimidine (Nuevo et al., 2009) and NH3:pyrimidine mixtures, the non-irradiated H2O:NH3:pyrimidine sample did not show any significant peak except that of unreacted pyrimidine (Rt =18.61 min).
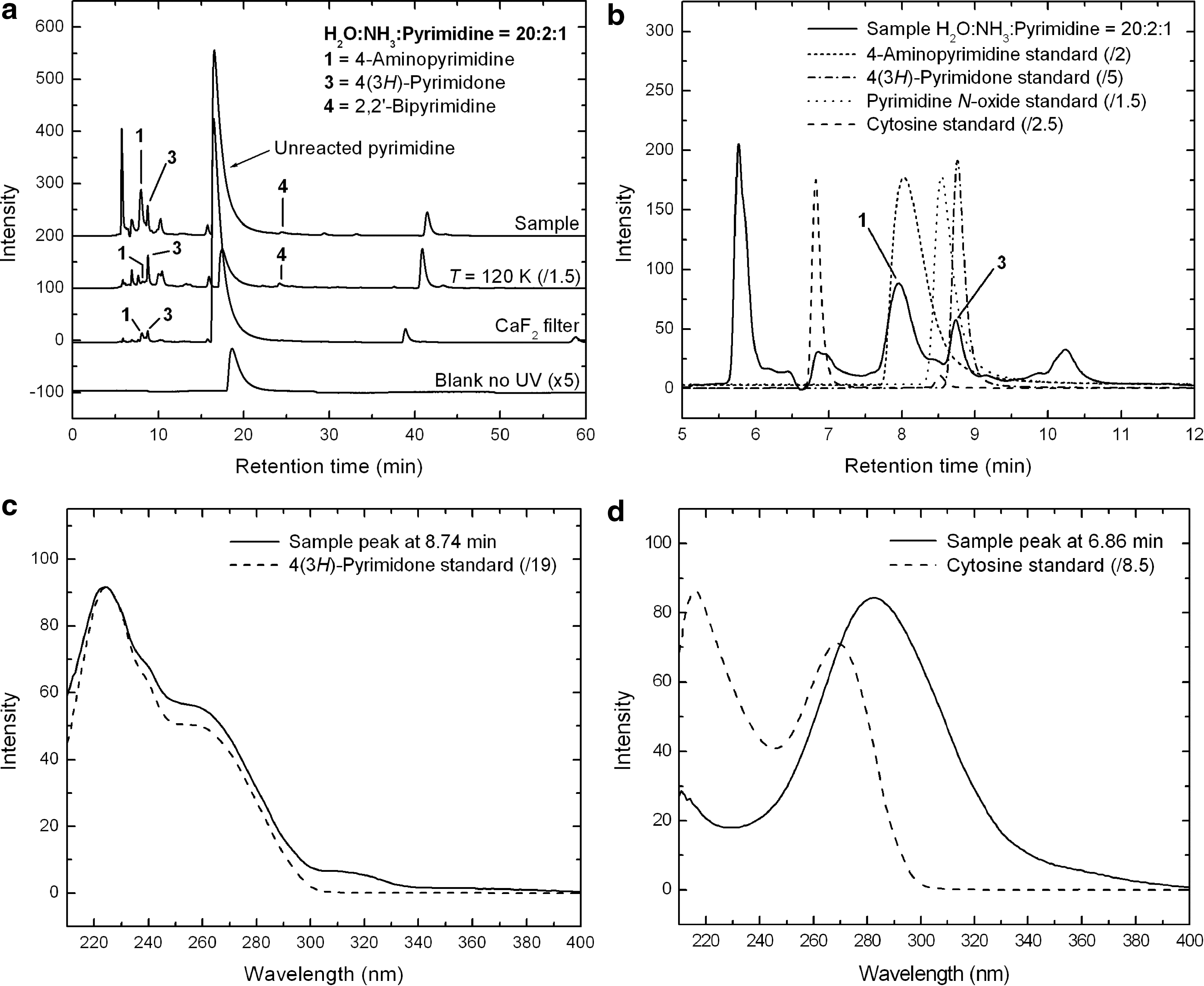
The compounds identified in H2O:NH3:pyrimidine samples are listed in Table 4. The most intense peak observed in the chromatogram of the 20–32 K sample is unreacted pyrimidine (Rt =16.54 min, Fig. 6a). The intensity ratio between the pyrimidine peak and the most intense peaks assigned to photo-products is about 2, which indicates that the presence of H2O in the starting mixture helps to convert pyrimidine more efficiently into photo-products, including aminopyrimidines, by a factor of ∼3.5 compared with NH3:pyrimidine mixtures (Fig. 5). However, since the relative abundance of pyrimidine and the other ice components is about twice as high in the initial H2O:NH3:pyrimidine ice as in the initial NH3:pyrimidine ice, this photo-conversion efficiency ratio might in fact be closer to ∼2. The role of H2O as a catalyst for such reactions is in agreement with theoretical studies of the formation of uracil in H2O:pyrimidine mixtures (Bera et al., 2010) and will be discussed in more detail in Section 4.1.
Among the photo-products identified are 4-aminopyrimidine (peak
A second oxidized pyrimidine derivative was detected in the chromatogram of several H2O:NH3:pyrimidine samples, namely, pyrimidine N-oxide (Rt =8.46 min), though not in the sample presented here. A peak eluting with a retention time close to that of pyrimidine N-oxide is present in its chromatogram (Fig. 6b), but it displays a different UV spectrum. The presence of such a compound is predicted by theoretical calculation in which H2O:pyrimidine mixtures are UV irradiated (Bera et al., 2010). Although its presence in H2O:pyrimidine residues was not initially confirmed (Nuevo et al., 2009), it has since been detected in those residues as well and is reported in Table 4.
4,5-Diaminopyrimidine, detected in the HPLC chromatogram of the NH3:pyrimidine=40:1 sample (Fig. 5), was not detected in any of the H2O:NH3:pyrimidine samples with HPLC. However, an additional pyrimidine derivative was found in all H2O:NH3:pyrimidine residues, namely, 2,2′-bipyrimidine (peak
Finally, the biological nucleobase cytosine (4-amino-2-hydroxypyrimidine) was unsuccessfully searched for in the HPLC chromatogram of the H2O:NH3:pyrimidine residue. A peak eluting at a very close retention time to that of cytosine is present (Fig. 6b), but it displays a very different UV spectrum (Fig. 6d). The search for isocytosine (2-amino-4-hydroxypyrimidine), an isomer of cytosine, in HPLC chromatograms was also unsuccessful.
The chromatograms of the residues produced from the UV irradiation of the additional H2O:NH3:pyrimidine=20:2:1 mixtures, one at 120 K (Fig. 6a, top middle trace) and the other at 19–28 K for which a CaF2 filter was used (bottom middle trace), show a similar trend to what is observed for the NH3:pyrimidine residues (Fig. 5a). The 120 K residue shows a significant increase in the efficiency of conversion of pyrimidine into photo-products (Fig. 6a, top middle trace). Similarly to NH3:pyrimidine mixtures, the formation of photo-products for ices irradiated at 120 K is about 1.5 times more efficient than for irradiation at the lower temperature. As expected, the peak intensities of the photo-products relative to that of unreacted pyrimidine are significantly lower when a CaF2 filter is used (compare top middle and bottom middle traces of Fig. 6a), which confirms that photochemical reactions leading to the formation of pyrimidine derivatives are enhanced by the absorption of Lyman-α photons.
With the exception of 4,5-diaminopyrimidine, HPLC chromatograms show that the photo-products formed in H2O:NH3:pyrimidine residues correspond to the sum of the photo-products found in H2O:pyrimidine residues (Nuevo et al., 2009) and those identified in NH3:pyrimidine residues (Table 4). In addition, HPLC chromatograms indicate that the presence of H2O in the starting mixtures seems to increase the efficiency of conversion of pyrimidine into all the photo-products, including aminopyrimidine derivatives (Fig. 6).
3.3. Gas chromatography–mass spectrometry
The use of an independent and complementary analytical technique such as GC-MS allowed us to confirm the presence of compounds identified in the HPLC chromatograms, as well as identify species not detected with HPLC. Mass spectrometry gives us important information about the carriers of GC-MS peaks for which standards are not available by constraining the molecular mass and structure for unidentified species. GC-MS identifications of compounds were performed by comparing single-ion chromatograms (SICs) of samples to standards for the mass of the most intense fragment of the tBDMS derivatives (M* – 57 amu), which corresponds to the derivatized compound (M*) that has lost one tert-butyl (–C(CH3)3) group (57 amu) (Table 3; Casal et al., 2004; Schummer et al., 2009).
3.3.1. NH3:Pyrimidine mixtures
Several samples produced from the UV irradiation of NH3:pyrimidine ices with different relative proportions were analyzed, though we focus here on the results obtained for a residue formed from a 40:1 ice. Figure 7a shows a comparison of the GC-MS total-ion chromatograms (TICs) of a residue formed from the irradiation of an NH3:pyrimidine=40:1 ice mixture at 19–28 K (top trace) with those of residues formed from the irradiation of a similar mixture at 120 K (top middle trace), and from another similar mixture irradiated at 18–24 K with the use of a CaF2 filter (middle trace) (see Section 2.1). The bottom middle and bottom traces correspond to the chromatograms of a non-irradiated NH3:pyrimidine=40:1 ice mixture deposited at 21–33 K (blank no UV) and of the derivatization agent (MTBSTFA with 1% of tBDMCS) (procedural blank), respectively.
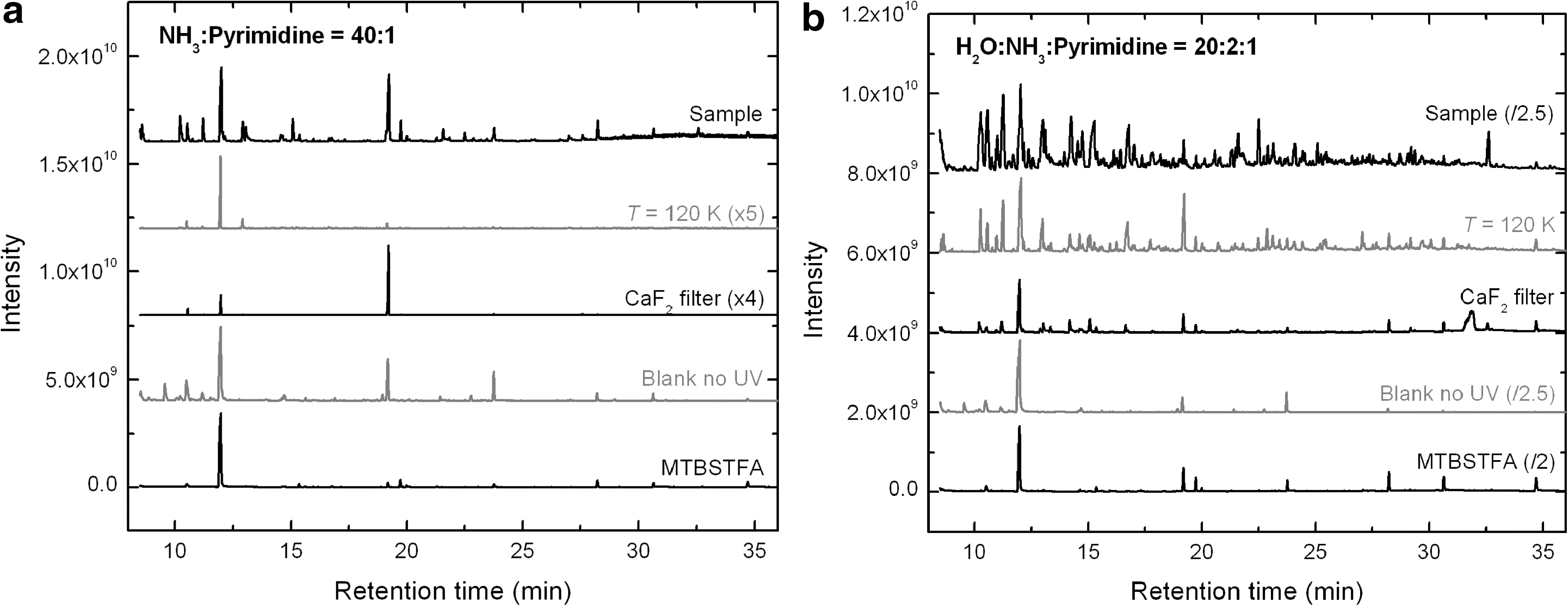
The chromatogram of the 19–28 K residue (top trace) shows a few peaks due to photo-products and derivatization by-products. The main peak at Rt =12.00 min is due to the presence of a compound or fragment released by the derivatization agent (bottom trace), as is also the case for other weaker peaks eluting at 10.55, 19.24, 19.75, 23.78, 28.25, and 30.67 min. These peaks are present in all chromatograms, with relative intensities varying from one sample to another, including in the non-irradiated sample (bottom middle trace). The other peaks observed in the chromatogram of the no-UV blank could not be identified. However, their mass fragmentation patterns indicate that they are not due to pyrimidine derivatives but rather to silicon-bearing compounds which may have originated from the gas chromatograph fused-silica capillary column used for sample separation (column bleeding) or the vials in which samples were dried and derivatized, as well as to rubber-like compounds from the gloves used.
Comparison of the 19–28 K sample (Fig. 7a, top trace) with the sample formed from the irradiation of the ice mixtures at 120 K (top middle trace) indicates that irradiating NH3:pyrimidine ice mixtures at higher temperature inhibits the formation of several compounds, since only very few peaks can be observed in the chromatogram of the 120 K sample. This result is different from what was observed for the equivalent residue analyzed with HPLC (Fig. 5a), for which the amount of photo-products formed was enhanced at 120 K (Section 3.2). It is also different from what was observed for UV-irradiated H2O:pyrimidine ices (Nuevo et al., 2009). This suggests that the nature of the photo-products formed in NH3:pyrimidine and H2O:pyrimidine residues is different, since a large fraction of the products present in NH3:pyrimidine samples seem to diffuse through the GC-MS column without any interaction. The chromatogram of the sample irradiated at 18–24 K with the use of the CaF2 filter (Fig. 7a, middle trace) shows even fewer peaks, in agreement with HPLC data for NH3:pyrimidine (Fig. 5a) and H2O:pyrimidine samples (Nuevo et al., 2009). The GC-MS chromatograms of other NH3:pyrimidine mixtures with relative proportions 10:1, 20:1, and 40:1 (not presented here) show similar results.
As for HPLC, only a few photo-products could be identified with GC-MS in NH3:pyrimidine residues (Table 4). Figure 8 shows the GC-MS SICs of the 19–28 K NH3:pyrimidine=40:1 residue for mass-to-charge ratios (m/z) of 152 and 281 amu, which correspond to the masses of amino- and diaminopyrimidine tBDMS derivatives, respectively, and their direct comparison with the corresponding standard SICs for 2- and 4-aminopyrimidines (152 amu), and for 2,4- and 4,5-diaminopyrimidines (281 amu). They clearly show that 4-aminopyrimidine (Rt =15.08 min) is present in this residue and confirm its detection with HPLC (Fig. 5). Other amino- and diaminopyrimidines were not found in any of the NH3:pyrimidine samples, with the exception of 4,5-diaminopyrimidine, which was detected in only one NH3:pyrimidine=20:1 residue, similarly to what was observed in HPLC chromatograms.
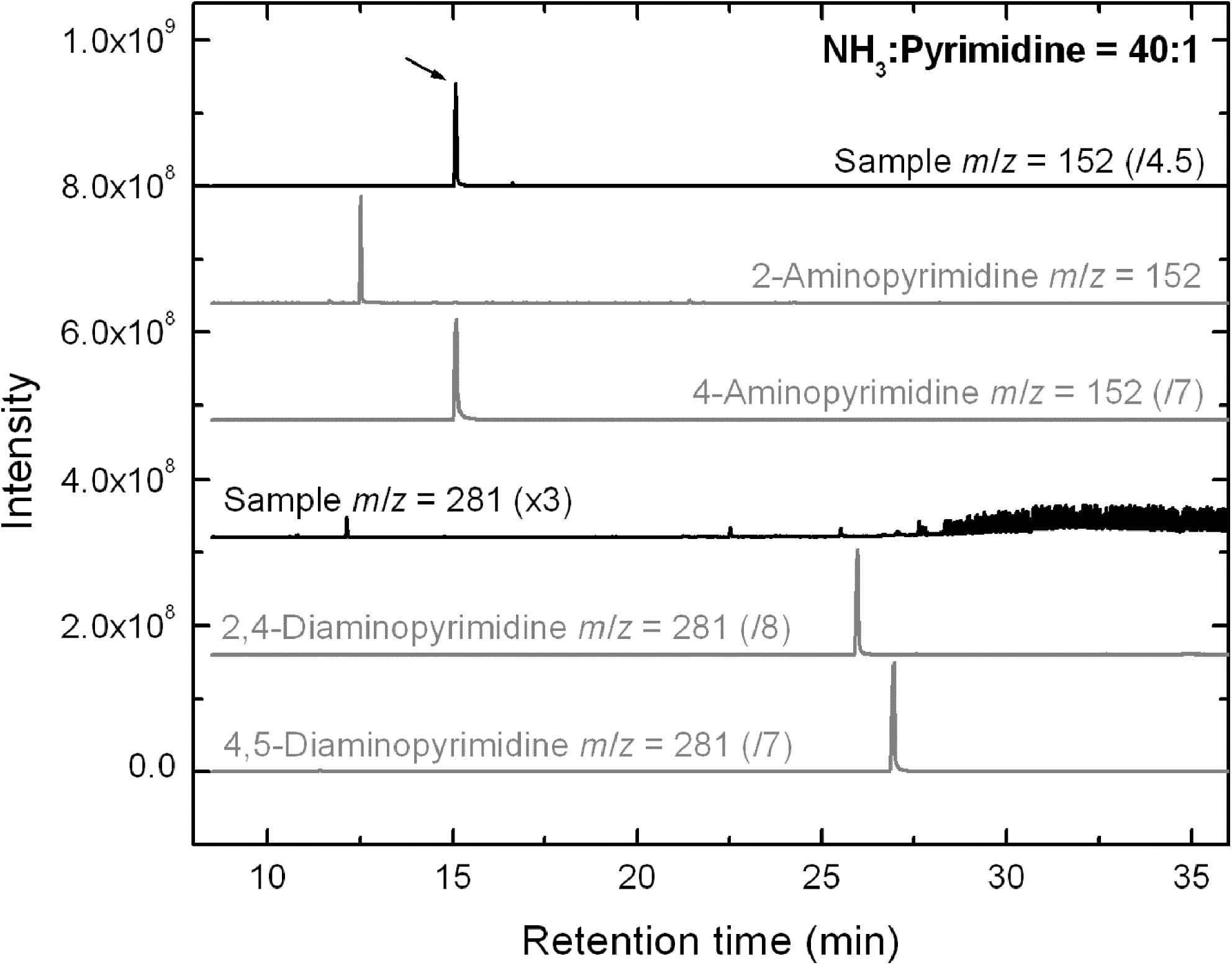
Comparison of the GC-MS single-ion chromatograms (SICs) of the residue produced from an NH3:pyrimidine=40:1 ice mixture irradiated at 19–28 K with aminopyrimidine (m/z=152 amu) and diaminopyrimidine (m/z=281 amu) standards. Only 4-aminopyrimidine was identified in the sample chromatogram (arrow).
It is interesting to note that the main photo-product identified in UV-irradiated NH3:pyrimidine ices is 4-aminopyrimidine, that is, a pyrimidine molecule to which the hydrogen atom in position 4 of the pyrimidic ring has been substituted by an amino group (Fig. 1a). This is comparable to what was observed experimentally for H2O:pyrimidine ices, where the most abundant photo-product was found to be 4(3H)-pyrimidone (Nuevo et al., 2009), and supported by quantum ab initio calculations that showed that nucleophilic substitution on position 4 of the pyrimidic ring is favored over the others (Bera et al., 2010).
The presence of 4(3H)-pyrimidone, observed in the HPLC chromatograms of NH3:pyrimidine samples (Fig. 5a), was confirmed by GC-MS analysis for nearly all residues. In addition, very small amounts of uracil and 4,6-dihydroxypyrimidine were also detected. Interestingly, 4(3H)-pyrimidone and doubly oxidized pyrimidines were also detected in the GC-MS chromatograms of dry residues that were extracted from their Al foil with ethyl acetate instead of water (see Section 2.2). This indicates that the oxidation of the NH3:pyrimidine residues is probably mostly due to interactions with traces of residual H2O remaining in the vacuum chamber during photo-irradiation rather than a liquid-phase hydrolysis of the residues.
Other oxygen-bearing molecules formed in H2O:pyrimidine samples, such as hydantoin, urea, and glycine (the smallest proteinic amino acid), were also detected in trace amounts in a few NH3:pyrimidine samples (Table 4), which indicates that the pyrimidic ring can be partially or fully broken upon photo-irradiation, in agreement with the presence of bands assigned to CO, CO2, OCN−, and other small carbon-bearing species in the IR spectra of NH3:pyrimidine samples (Fig. 3). Finally, 1,4,5,6-tetrahydropyrimidine (Rt =14.09 min) and the nucleobase cytosine (Rt =24.53 min) were tentatively detected at trace levels in one NH3:pyrimidine=10:1 sample.
3.3.2. H2O:NH3:Pyrimidine mixtures
The GC-MS total-ion chromatogram of a residue formed from the UV irradiation of an H2O:NH3:pyrimidine=20:2:1 ice mixture at 20–29 K is given in Fig. 7b (top trace) and compared with those of residues formed from the irradiation of a similar mixture at 120 K (top middle trace) and from another similar mixture irradiated at 19–28 K with the use of a CaF2 filter (middle trace). The bottom middle and bottom traces correspond to the TICs of a non-irradiated H2O:NH3:pyrimidine=20:2:1 ice mixture deposited at 22–32 K (blank no UV) and of MTBSTFA (with 1% of tBDMCS) (procedural blank), respectively.
The GC-MS chromatogram of the irradiated 20–29 K H2O:NH3:pyrimidine=20:2:1 sample (top trace) displays a large number of peaks. The GC-MS chromatogram of the non-irradiated H2O:NH3:pyrimidine=20:2:1 sample (Fig. 7b, bottom middle trace) shows only peaks consistent with MTBSTFA by-products (bottom trace).
The TIC of the sample produced from the irradiation of the H2O:NH3:pyrimidine=20:2:1 ice mixture at 120 K (top middle trace) shows a significant number of peaks in addition to those present in the non-irradiated sample and the MTBSTFA. This chromatogram contains, however, a smaller number of peaks than the 20–29 K sample (top trace), as was observed for NH3:pyrimidine=40:1 samples irradiated at both low and higher temperatures (Fig. 7a, top and top middle traces, respectively). This again suggests that the photochemistry of H2O:NH3:pyrimidine ices is different from that of H2O:pyrimidine ices (Nuevo et al., 2009). Finally, the chromatogram of the sample irradiated at 19–28 K with the use of the CaF2 filter (middle trace) shows fewer peaks than the other samples, which indicates again that the formation of photo-products is significantly enhanced when Lyman-α photons are present in the light source. Indeed, a first-order comparison between the GC-MS chromatograms of these two samples shows that singly substituted pyrimidine derivatives such as 4(3H)-pyrimidone and 4-aminopyrimidine are about 5 times less abundant in the experiment in which the CaF2 filter was used. More complex molecules such as uracil were found to be up to 30 times less abundant. This is consistent with the fact that more substitutions are required to make uracil from pyrimidine than to make 4(3H)-pyrimidone, and suggests that the formation of multiple-substitution photo-products from pyrimidine is probably a stepped, multi-photon process.
Several peaks could be identified in the TICs of the H2O:NH3:pyrimidine samples (Table 4). All amino- and diaminopyrimidine derivatives for which we had standards were detected in most samples. However, 2,4,6-triaminopyrimidine was not detected in any of them. All singly, doubly, and even triply oxidized pyrimidines searched for were also detected in most of the samples. These compounds include the nucleobase uracil (2,4-dihydroxypyrimidine, Rt =20.81 min) and its isomer 4,6-dihydroxypyrimidine (Rt =21.48 min), as shown in the single-ion chromatogram of the 20–29 K sample for m/z=283 amu (Fig. 9a, three bottom traces). Moreover, the nucleobase cytosine (Rt =24.54 min) and its isomer isocytosine (Rt =22.85 min) were also found in the m/z=282 amu SIC of the same sample (Fig. 9a, three top traces), as well as in several other H2O:NH3:pyrimidine samples. 5-Aminouracil, another pyrimidine derivative containing amino and hydroxy/keto groups, was also identified in a few samples, and its isomer 6-aminouracil was tentatively identified (Table 4).
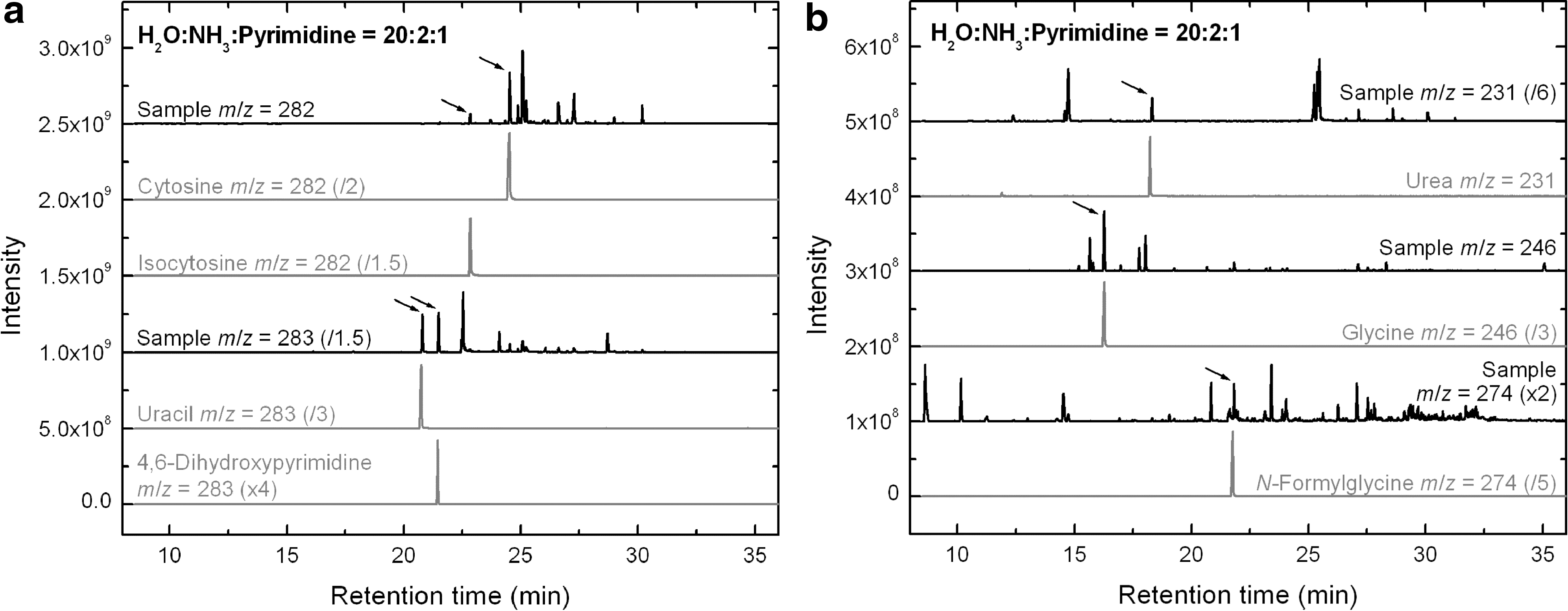
The broad inventory of pyrimidine derivatives identified in H2O:NH3:pyrimidine samples highlights the important role of H2O in the starting mixtures. Indeed, H2O not only contributes to the formation of oxidized species, but it also enhances the formation of non-oxidized compounds, such as amino-bearing pyrimidine derivatives, that were not detected in NH3:pyrimidine samples (Fig. 8, Table 4). These properties of H2O ice will be discussed in Section 4.1.
Additional identified pyrimidine derivatives in the irradiated 20–29 K H2O:NH3:pyrimidine=20:2:1 sample, as well as in several others, include 1,4,5,6-tetrahydropyrimidine (Rt =14.14 min) and 2,2’-bipyrimidine (Rt =17.73 min, also identified via HPLC, see Fig. 6a). Several other intense peaks, with masses consistent with the presence of other bipyrimidine isomers, were also identified in GC-MS chromatograms at Rt =13.03 and 16.75 min, although it is impossible to clearly identify them because of the lack of commercially available standards. The presence of several bipyrimidines in residues formed from the UV irradiation of H2O:pyrimidine ice mixtures was predicted from theoretical calculations (Bera et al., 2010).
Finally, the UV irradiation of H2O:NH3:pyrimidine ices also leads to the formation of a suite of small aliphatic compounds of astrobiological and prebiotic interest such as urea (Rt =18.32 min), glycine (the smallest proteinic amino acid, Rt =16.27 min), and N-formylglycine (a non-proteinic amino acid, Rt =21.83 min). These are seen on the SICs of the 20–29 K H2O:NH3:pyrimidine=20:2:1 sample for m/z=231, 246, and 274 amu, respectively (Fig. 9b). Additionally, traces of alanine, the second smallest proteinic amino acid, were detected in the single-ion chromatograms (m/z=260 amu) of two other samples of compositions 20:2:1 and 20:1:1 at Rt =15.75 min, although it was found to coelute with another unidentified compound.
4. Discussion and Astrobiological Implications
4.1. The role(s) of H2O
Our results highlight the important role of H2O, both in the efficiency of formation and the distribution of pyrimidine photo-products. As mentioned in Sections 3.2 and 3.3, H2O is involved in the formation of photo-products at several independent steps: as a matrix with catalytic properties, as a reactant, and as a solvent when extracting the final residues from their substrate.
In the ice at low temperature, H2O is efficiently photo-dissociated by UV photons (mainly Lyman α) due to its low dissociation energy of 5.1 eV (Woon, 2002), and releases H atoms and OH radicals that can readily react with other species. OH radicals can be efficiently added to pyrimidine in ice mixtures (Nuevo et al., 2009; Bera et al., 2010; Figs. 6b and 9a) to form oxidized derivatives such as 4(3H)-pyrimidone, uracil, and other isomers. The interaction between H atoms and pyrimidine is not as efficient but still observed via the presence of 1,4,5,6-tetrahydropyrimidine in a few samples (Table 4). It has to be noted that while radical species are mainly formed in the ices at (very) low temperature, the majority of radical-radical and radical-neutral reactions take place during warm-up when species become more mobile in the ice matrix and can find each other to interact (Bernstein et al., 1995). In addition, the presence of oxygen-bearing functional groups in samples formed from NH3:pyrimidine (Figs. 3 and 5b; Sections 3.2 and 3.3) clearly indicates that even trace amounts of residual H2O in the chamber are efficiently incorporated into the photochemistry taking place during those experiments.
H2O also plays the role of a third body in the absorption of excess energy from exothermic reactions. As shown by quantum calculations, H2O in these ices assists proton abstraction from intermediate species such that it stabilizes the formation of the final products (Bera et al., 2010). Thus, the presence of surrounding H2O molecules allows the formation of many oxidized species that might not otherwise be thermodynamically favored. In our experiments, these properties of H2O ice are highlighted by the fact that the formation of amino-bearing species from pyrimidine and NH3 appears to be enhanced by the presence of H2O in the starting mixture, since amino- and diaminopyrimidine compounds that were not detected in NH3:pyrimidine samples are present in several H2O:NH3:pyrimidine samples (Table 4).
H2O (liquid) is also used as a solvent to extract final residues from Al foils at room temperature, and it is reasonable to assume that dissolution in water could also affect the chemical composition of the samples. In particular, hydrolysis of organic residues consisting of macromolecular materials will break up polymers into their monomers and oxidize reduced functional groups such as nitriles. To assess whether such an effect occurs with our samples, several residues formed from the UV irradiation of NH3:pyrimidine and H2O:NH3:pyrimidine mixtures were extracted with ethyl acetate instead of H2O (Section 2.3) before analysis with GC-MS. The chromatograms obtained for NH3:pyrimidine residues extracted with ethyl acetate (not shown) are comparable to those extracted with water, within the variation range observed in individual samples, and all of them show the presence of oxidized pyrimidines. Moreover, cytosine is known to be easily hydrolyzed into uracil at room temperature (Ferris et al., 1968; Shapiro, 1999), so the presence of cytosine in all H2O:NH3:pyrimidine residues, even after dissolution in water for several days, indicates that hydrolysis in our samples is limited. These results suggest that liquid H2O has no strong hydrolyzing effect on the residues and that the presence of oxidized products in H2O-free samples is mainly due to reactions of pyrimidine with trace amounts of residual H2O in the vacuum chamber, as suggested by IR spectroscopy (top traces of Figs. 3 and 4).
However, the effects of long-term water solvation have not been studied in this work, and the chemical composition of residues may possibly be altered if they are in contact with H2O for extended periods of time. HPLC chromatograms of NH3:pyrimidine and H2O:NH3:pyrimidine residues kept for 160 days have been measured and compared with those measured immediately after they were extracted (Section 3.2). These chromatograms show slight changes in the relative intensities, shifts in retention times, or both, for a few peaks, but they do not indicate any significant increase of oxidized species, which confirms that H2O solvent-induced hydrolysis is limited. This suggests that a large fraction of the photo-products in the residues are pyrimidine derivatives present in a free form rather than in a macromolecular structure, unlike what is usually observed for residues formed from the UV irradiation of non-aromatic starting compounds, in which the majority of free organic molecules such as amino acids are detected after hydrolysis (Bernstein et al., 2002a; Muñoz Caro et al., 2002; Nuevo et al., 2008).
4.2. Mechanisms of formation of nucleobases and other compounds
Although the formation of photo-products from the UV irradiation of NH3:pyrimidine ices did not appear to be an efficient process (Figs. 5, 7, and 8), a large number of pyrimidic and non-pyrimidic species were found in H2O:NH3:pyrimidine samples. These photo-products include the nucleobases uracil and cytosine (Fig. 9a), as well as other compounds of prebiotic and biological importance, such as urea and the amino acids glycine (proteinic) and N-formylglycine (non-proteinic) (Fig. 9b).
The mechanisms of formation for most of these molecules are not well known, but a few assumptions based on HPLC and GC-MS results (Section 3), as well as previous experimental (Nuevo et al., 2009) and theoretical (Bera et al., 2010) studies on the UV irradiation of H2O:pyrimidine mixtures, can give us a general idea of the chemical pathways that lead to their formation.
It is reasonable to assume that the formation of uracil in H2O:NH3:pyrimidine samples follows a similar pathway to the one deduced from H2O:pyrimidine studies, since H2O is the dominant component of the ices in both cases. In this mechanism, the first step is the addition of an OH group to the position 4 of the pyrimidic ring to form 4(3H)-pyrimidone, which was found to be the most abundant oxidized pyrimidine in all H2O:pyrimidine (Nuevo et al., 2009) and H2O:NH3:pyrimidine (Fig. 6; Sections 3.2 and 3.3) samples, as well as the most stable singly oxidized pyrimidine expected to be formed from pyrimidine in a pure H2O ice (Bera et al., 2010). The second step is the addition of another OH group to 4(3H)-pyrimidone on position 2 of the ring to form uracil, expected to be the most stable doubly oxidized pyrimidine derivative when formed in H2O ice (Bera et al., 2010). 2-Hydroxypyrimidine and 4,6-dihydroxypyrimidine, isomers of 4(3H)-pyrimidone and uracil, respectively, are also observed in H2O:NH3:pyrimidine samples, though with smaller abundances. The same formation process as for H2O:pyrimidine samples (Nuevo et al., 2009) is expected for the formation of uracil in other H2O-containing ices, but competing pathways may exist. Further theoretical work is necessary to identify and evaluate those competing mechanisms for the formation of uracil and its isomers in the presence of NH3.
The formation of cytosine in our samples is not well understood, but it probably follows a similar pattern, that is, a two-step addition of NH2 and OH groups to pyrimidine. According to what we know for the formation of uracil from H2O:pyrimidine ices (Nuevo et al., 2009; Bera et al., 2010) and our experimental results for NH3:pyrimidine and H2O:NH3:pyrimidine mixtures, it seems that the addition of nucleophilic groups (NH2, OH) to the position 4 of pyrimidine is favored over the others, which is supported by the presence of 4(3H)-pyrimidone and 4-aminopyrimidine with higher abundances (Section 3). Therefore, it can be reasonably assumed that the first step for the formation mechanism of cytosine is the addition of an NH2 group to the position 4 of the pyrimidic ring to form 4-aminopyrimidine, which was found abundantly in all NH3:pyrimidine and H2O:NH3:pyrimidine residues (Table 4). Subsequently, an OH group can be added to 4-aminopyrimidine on position 2 to form cytosine. Similarly, isocytosine (Fig. 1b) is probably formed via a two-step mechanism, that is, via the addition of an OH group to pyrimidine on position 4 to form 4(3H)-pyrimidone, followed by the addition of an NH2 group to the position 2 of the ring. These proposed mechanisms for the formation of uracil, cytosine, and their isomers detected in H2O:NH3:pyrimidine residues are summarized in Fig. 10.
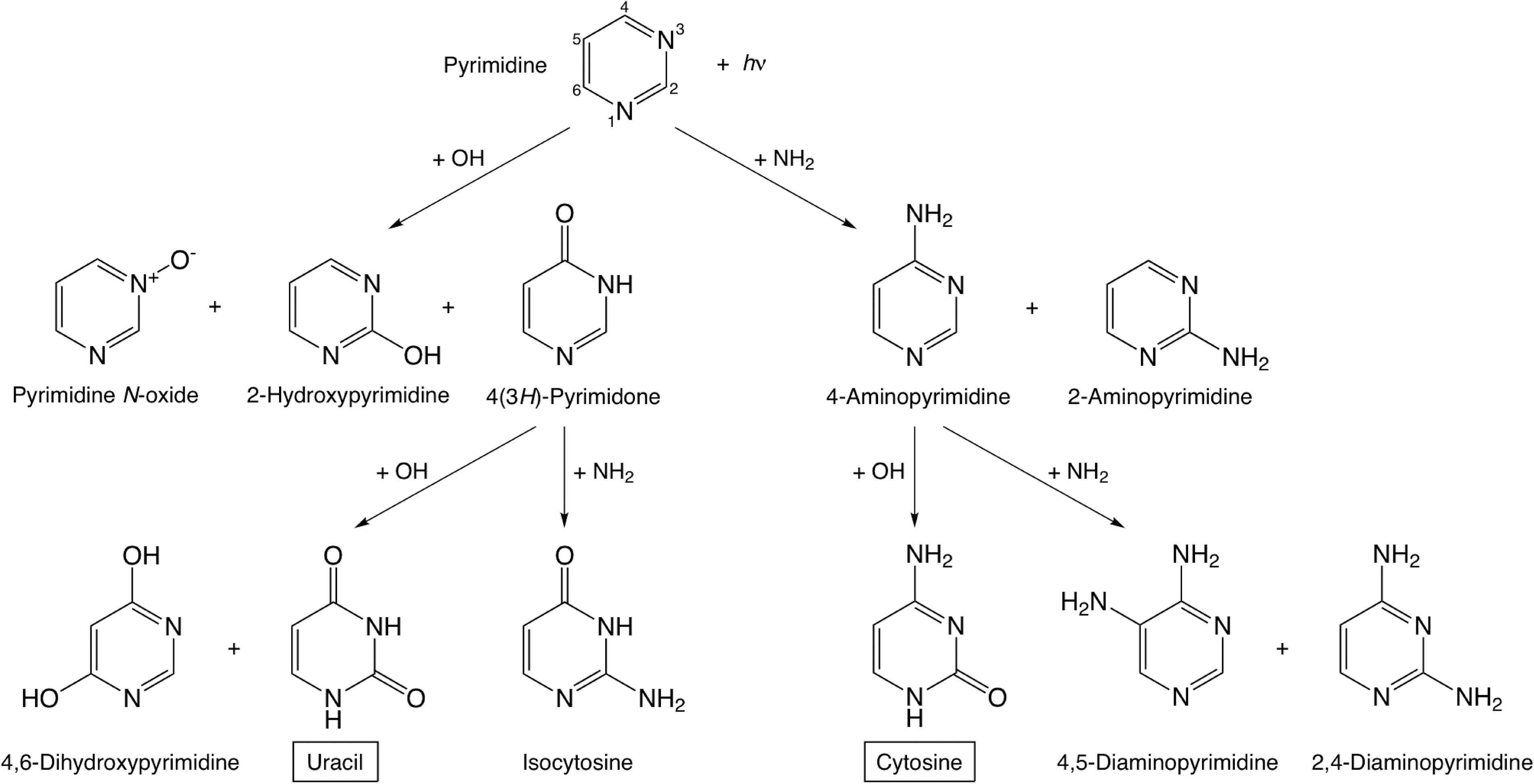
Previous studies have also proposed chemical pathways to form cytosine under prebiotic conditions via hydrolysis from urea (detected in all H2O:NH3:pyrimidine samples) and cyanoacetaldehyde (Shapiro, 1999; Nelson et al., 2001), though such a process may not be efficient at cryogenic temperatures. Under our experimental conditions, these pathways are most probably negligible compared with the formation pathways of cytosine and uracil from the addition of OH and NH2 groups to pyrimidine.
Finally, the formation mechanisms of the small aliphatic compounds such as urea, glycine, and N-formylglycine found in the residues are not well understood. However, IR spectra of irradiated H2O:NH3:pyrimidine mixtures (Fig. 4) show bands assigned to small carbonaceous molecules, including CO2, OCN−, and functional groups such as nitriles and isonitriles, which indicates that a non-negligible fraction of pyrimidine is photo-dissociated upon UV irradiation. Open aromatic rings, in particular radicals, can then easily react with other species such as NH2, OH, and CN groups released from the photo-dissociation of NH3, H2O, and pyrimidine itself, respectively. Subsequent photo-oxidation can then lead to the formation of alcohols, ketones, aldehydes, and carboxylic acids.
4.3. Estimates of quantities and formation yields
We estimated the quantities of the main photo-products detected in NH3:pyrimidine and H2O:NH3:pyrimidine residues based on our GC-MS data. These estimates are based solely on a first-order comparison between the peak area of a given compound in the SIC of a sample and its peak area in the corresponding SIC of a standard. The quantities of uracil, cytosine, and their precursors (Section 4.2) were estimated to be in the 50–500 nmol range in the H2O:NH3:pyrimidine=20:2:1 sample whose GC-MS chromatograms are given in Fig. 9. For comparison, a total of ∼1–10 μmol was estimated for the quantity of all species detected in the sample. Knowing that 0.16 mmol of pyrimidine was deposited on the cold substrate during irradiation, we derived a quantum yield of the order of 10−2 for all detectable pyrimidic photo-products. It must be noted, however, that no specific chromatographic protocol was applied to derive accurate quantities of the standards and samples injected in the GC-MS instrument, so that the uncertainties associated with these quantities are estimated to be around 50%. For this reason, comparison between the quantities of given photo-products is very limited in this study.
Nonetheless, the estimated quantities indicate that 4(3H)-pyrimidone, 4-aminopyrimidine, and 2-aminopyrimidine are among the most abundant pyrimidic photo-products in this H2O:NH3:pyrimidine=20:2:1 sample. Most non-pyrimidic compounds (e.g., glycine and N-formylglycine) were formed in significantly smaller quantities (1–2 orders of magnitude smaller), with the exception of urea, whose yield was estimated to be a few 10−3, which is comparable to that of 4(3H)-pyrimidone and 4-aminopyrimidine.
The yields of the nucleobases uracil and cytosine were found to be ∼10−4, that is, about 10–20 times smaller than the single hydroxy- and aminopyrimidine derivatives, and comparable to what was found for the formation of amino acids from the UV irradiation of simple ice mixtures (Muñoz Caro et al., 2002). Compared with results obtained for H2O:pyrimidine ices (Nuevo et al., 2009), it appears that the formation yields of 4(3H)-pyrimidone and uracil are one order of magnitude larger in H2O:NH3:pyrimidine=20:2:1 samples, which suggests an effect of the presence of NH3 in the starting mixture that has yet to be determined. Such an enhancement in the production of pyrimidine derivatives is in agreement with the photo-irradiation half-lives derived for pyrimidine in H2O:pyrimidine, NH3:pyrimidine, and H2O:NH3:pyrimidine ice mixtures (Table 1), which indicate that pyrimidine can survive UV photons ∼7 times longer in an H2O:NH3 ice matrix than in an H2O ice matrix. Pyrimidine is even more stable upon irradiation when embedded in a pure NH3 ice (Section 3.1), although such an ice composition is clearly less relevant from an astrophysical point of view.
In summary, our results show that, of 5000 molecules of pyrimidine irradiated in our H2O:NH3:pyrimidine=20:2:1 mixture, about 10 will be converted into 4(3H)-pyrimidone and 1 into uracil. The conversion from 4(3H)-pyrimidone into uracil in H2O:NH3:pyrimidine mixtures, assuming that 4(3H)-pyrimidone is the only precursor of uracil (Nuevo et al., 2009; Bera et al., 2010; Fig. 10), is of the same order as what is observed when pyrimidine is mixed with pure H2O, that is, about 10% (Nuevo et al., 2009).
Finally, our estimates also suggest that uracil is the most abundant doubly oxidized pyrimidine produced in these experiments, which is in agreement with theoretical calculations (Bera et al., 2010). Similarly, cytosine was found to be the most abundant isomer among aminohydroxypyrimidines, although no calculations have yet been performed for the formation of cytosine and its isomers from the photo-irradiation of pyrimidine in H2O+NH3 ices.
4.4. Comparison with other laboratory data and meteorites
N-Heterocycles, including puric and pyrimidic nucleobases, have been found in meteorites, particularly in carbonaceous chondrites. Purines are usually found in a broader variety and higher abundances than pyrimidines (van der Velden and Schwartz, 1977; Callahan et al., 2011). Indeed, only a few pyrimidic compounds have been identified in the Murchison, Murray, and Orgueil meteorites among the large inventory of organic molecules present. These compounds include 4-hydroxypyrimidine, tautomer of 4(3H)-pyrimidone (Folsome et al., 1971, 1973; Lawless et al., 1972), and uracil (Stoks and Schwartz, 1979; Martins et al., 2008). Thus, the main pyrimidic compounds seen in meteorites are the same as the main oxidized pyrimidine-based photo-products found in H2O:pyrimidine (Nuevo et al., 2009) and H2O:NH3:pyrimidine (Figs. 6 and 9a) residues.
Compared with Murchison, in which the concentrations of 4(3H)-pyrimidone and uracil were measured to be 6 μg g−1 (Folsome et al., 1971) and 0.03 μg g−1 (Stoks and Schwartz, 1979), respectively, the uracil/4(3H)-pyrimidone ratio derived for our samples (∼10−1) is significantly larger than in meteorites (∼5×10−3). However, it should be kept in mind that pyrimidine has a fairly high concentration in our ices, which results in an efficient formation of uracil via 4(3H)-pyrimidone (Nuevo et al., 2009; Bera et al., 2010; Section 4.2 and Fig. 10). In contrast, in astrophysical environments, the fraction of pyrimidine compared with other dominant ice components should be much lower, so that the amount of uracil formed via this chemical pathway is significantly smaller. Moreover, there is the possibility that the uracil and other pyrimidine derivatives in meteorites formed from non-pyrimidic and even non-cyclic precursors (Ricca et al., 2001).
Cytosine may also be present in meteoritic materials, but it has yet to be detected. This can be partly due to the fact that it is known to be easily hydrolyzed into uracil (Shapiro, 1999; Nelson et al., 2001), which may occur on the meteoritic parent body or during the process of extraction of meteoritic organics. More generally, protocols for the analysis of meteorites with chromatography techniques, including strong acid hydrolysis, may destroy a non-negligible fraction of pyrimidine-based compounds, which are more subject to hydrolysis and degradation than purines. This can explain why several purines, including adenine and xanthine, are detected in the Murchison, Murray, and Orgueil meteorites, whereas pyrimidines are rare (Stoks and Schwartz, 1981; Callahan et al., 2011). In our experiments, extraction of the residues with liquid H2O prior to HPLC and GC-MS constitutes a mild hydrolysis, which does not significantly affect the degradation of pyrimidines and, thus, their detection in the samples.
Non-pyrimidic compounds found in our samples have also previously been detected in organic residues produced from the UV photo-irradiation of astrophysical ice analogues and carbonaceous chondrites. A broad range of amino acids (both proteinic and non-proteinic) have been detected in hydrolyzed organic residues formed from the UV irradiation of different combinations of starting ice components with non-aromatic carbon sources, including H2O, CO, CO2, CH3OH, CH4, NH3, and HCN (Bernstein et al., 2002a; Muñoz Caro et al., 2002; Nuevo et al., 2008). In such residues, the distribution of amino acids formed follows a trend in which their abundance decreases exponentially with their molecular weight (Nuevo et al., 2008). All these amino acids have also been detected in chondritic meteorites (up to 70 in Murchison alone), from which they are released after acid hydrolysis (Kvenvolden et al., 1970; Shock and Schulte, 1990; Cronin and Pizzarello, 1999; Engel and Macko, 1997; Sephton, 2002; Martins et al., 2007). In our H2O:NH3:pyrimidine samples, only glycine, the smallest proteinic amino acid, and one of its non-proteinic derivatives, N-formylglycine, have been detected (Fig. 9b). Alanine, the second smallest proteinic amino acid, is probably also present in a few residues, but only in trace amounts (Table 4). This suggests that aromatic compounds, including pyrimidine, may be an additional source of carbon for the formation of amino acids and other small prebiotic molecules after UV irradiation under astrophysical conditions, although this process is not as efficient as the UV photolysis of carbonaceous species such as CO, CO2, CH3OH, and CH4, which are significantly more abundant in the interstellar medium.
Among other non-aromatic compounds detected, urea, a small molecule of prebiotic importance believed to catalyze amino acid polymerization (Mita et al., 2005), was found in all H2O:NH3:pyrimidine samples with high abundances. The presence of this species has previously been reported in organic residues formed from the UV irradiation of CH3OH:NH3 and H2O:CH3OH:NH3 ice mixtures (Bernstein et al., 2002a; Nuevo et al., 2010; de Marcellus et al., 2011), as well as in the Murchison meteorite (Cooper and Cronin, 1995). Similarly, hydantoin has recently been detected in the residues of UV-irradiated CH3OH:NH3 and H2O:CH3OH:NH3 ices (de Marcellus et al., 2011). It is probably formed from the combination of urea and glycolic acid, as both of these compounds have also been detected in residues (Nuevo et al., 2010). Hydantoin, which has also been detected in the Murchison and Yamato-791198 meteorites (Cooper and Cronin, 1995; Shimoyama and Ogasawara, 2002), may, in the presence of amino acids in an aqueous medium such as primitive oceans, lead to the formation of carbamoyl amino acids and N-carboxyanhydride amino acids, known to be precursors of polypeptides (Commeyras et al., 2004; Danger et al., 2006).
The presence of a large inventory of pyrimidine derivatives, including the nucleobases uracil and cytosine, together with species such as amino acids, urea, and hydantoin in H2O:NH3:pyrimidine samples, supports the idea that molecules of astrobiological importance could have been formed abiotically in space and delivered to the early Earth by comets and asteroids, seeding primitive oceans in which the first prebiotic reactions that led to the emergence of life may have taken place.
5. Conclusions
The UV photo-irradiation of NH3:pyrimidine and H2O:NH3:pyrimidine ice mixtures leads to the formation of a large variety of photo-products, including pyrimidine-based species such as 4(3H)-pyrimidone, 2- and 4-aminopyrimidine, the nucleobases uracil and cytosine, several of their isomers, as well as non-cyclic species such as urea and the amino acids glycine and N-formylglycine.
First-order estimates of the quantities of several photo-products suggest formation yields for uracil and cytosine of the order of 10−4, which is comparable to the formation of amino acids from the UV irradiation of simple astrophysically relevant ices. This work is also in agreement with previous experimental and theoretical studies about the mechanisms of formation of uracil, and proposes similar pathways for the formation of cytosine as well as other OH- and/or NH2-bearing pyrimidines.
Although the presence of pyrimidine in the interstellar medium is still an open question, our experimental results confirm that the photochemistry that takes place in cold astrophysical environments is very rich and leads to the formation of molecules of prebiotic interest under abiotic conditions. These molecules can then be preserved in small bodies like asteroids and comets before being delivered to telluric planets such as the primitive Earth. Such a possibility is consistent with the detection of non-terrestrial uracil and puric nucleobases as well as other astrobiologically interesting molecules in Murchison and other carbonaceous chondrites.
Footnotes
Acknowledgments
This work was supported by NASA grants from the NASA Astrobiology Institute and Origins of Solar Systems programs. M.N., S.N.M., and S.A.S. would like to acknowledge M.G. Martin, J.E. Elsila, and J.P. Dworkin (NASA Goddard) for technical advice on the GC-MS data analysis, R.L. Walker (NASA Ames) for technical support, and C.K. Materese (NASA Ames/SETI) for his help in preparing and running standards for HPLC and GC-MS.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations
GC-MS, gas chromatography coupled with mass spectrometry; HPLC, high-performance liquid chromatography; ISM, interstellar medium; MTBSTFA, N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide; m/z, mass-to-charge ratio; PAHs, polycyclic aromatic hydrocarbons; PANHs, polycyclic aromatic nitrogen heterocycles; SICs, single-ion chromatograms; tBDMCS, tert-butyldimethylchlorosilane; tBDMS, tert-butyldimethylsilyl; TICs, total-ion chromatograms.