
Abstract
A key step in the origin of life is the establishment of autocatalytic cycles controlled by biopolymer catalysts. These catalysts (either ribozymes or proteins) are composed of homochiral monomers. Homochirality in living systems is maintained because biopolymers are asymmetric in their catalysis and synthesize molecules of their own handedness. Asymmetric autocatalysis is also possible with small molecules, as demonstrated by the Soai reaction, but it is rare. As far as we know, single nucleotides and amino acids are not autocatalytic. The observation that organic molecules in meteorites can have an enantiomeric excess of a few percent suggests that the prebiotic mixture may have had a partial chiral bias that was caused by external physical influences. Here, we consider the way that such a partial prebiotic bias would influence the origin of ribozymes in an RNA world scenario. We have previously shown how a transition to a living state can occur in a model for RNA polymerization. Here, we add chirality to the problem by considering simultaneous synthesis and polymerization of left- and right-handed monomers. The two chemical synthesis rates may be equal or unequal, due to physical or chemical effects prior to the origin of life. We determine the stationary states of this reaction system. The nonliving state is racemic, or slightly biased. There are two living states that are almost completely homochiral, whether or not the nonliving state is biased. It is a feature of our model that, for some regions of parameter space, living and nonliving states are both found to be stable under the same conditions. The origin of life therefore involves a stochastic transition between the nonliving and living states. Our model extends previous theories by treating the origin of life and the origin of chirality as aspects of the same model. Key Words: Chirality—RNA world—Prebiotic chemistry—Origin of life. Astrobiology 12, 818–829.
Introduction
S
The key concept is asymmetric autocatalysis: molecules of one enantiomer must selectively catalyze the formation of further molecules of the same enantiomer. In the presence of asymmetric autocatalysis, the racemic mixture (equal concentrations of the two enantiomers) can sometimes be unstable. Any slight asymmetry in concentration of the two enantiomers that may exist initially can be amplified, giving rise to a system with a large excess of one enantiomer over the other. A simple theoretical model illustrating the principle of asymmetric autocatalysis was given by Frank (1953), and this has given rise to a large number of more complex models that will be discussed in more detail below. Experimental examples are much harder to come by. The Soai reaction (Soai et al., 1995, 2001) is the reaction of 5-pyrimidyl alkanol with diisopropyl zinc. This is a well-documented case of a small-molecule chemical system that shows that a large enantiomeric excess (ee) can arise by asymmetric autocatalysis. Although this demonstrates the principle that a homochiral system of small molecules could arise spontaneously, the specific chemical components of the Soai reaction are not directly relevant to prebiotic chemistry. No one has yet found an experimental system involving amino acids, sugars, or nucleotides in which an ee spontaneously arises by autocatalysis, and it would appear that these small biomolecules cannot themselves carry out asymmetric autocatalytic synthesis.
There is an important distinction between small molecules, like amino acids and sugars, and biopolymers, like proteins and nucleic acids. The observation of homochirality of biopolymers in living organisms tells us that asymmetric autocatalytic systems of large biopolymers can maintain high ee. Biochemistry in living organisms relies on specific recognition between biopolymer catalysts and their substrates (either small molecules or other biopolymers). The enzymes or ribozymes in a living system are built from amino acids or nucleotides of a definite handedness, and they would not react with the molecules of the opposite enantiomer. Furthermore, pathways of synthesis of chiral molecules from nonchiral precursors are also controlled by enzymes of one enantiomer, and these can catalyze synthesis of monomers of the same enantiomer in an asymmetric way. Thus, the maintenance of homochirality in a living system appears straightforward because of the high efficiency and specificity of biopolymer catalysts, whereas the origin of homochirality in a nonliving system of small molecules is less easy to understand.
An important part of this puzzle is the observation that amino acids with a relatively high ee of up to 15% have been observed in carbonaceous chondrite meteorites (Cronin and Pizzarello, 1997; Pizzarello et al., 2003, 2008; Glavin and Dworkin, 2009). As these molecules are presumed to be a product of nonliving chemistry, it must either be the case that the amino acids are synthesized by asymmetric autocatalysis involving small molecules or that they are synthesized by a non-autocatalytic process that is asymmetric for some other reason. Among non-autocatalytic sources for an initial asymmetry, one plausible mechanism involves the influence of circularly polarized light. It has been shown that irradiation with circularly polarized light leads to asymmetric photolysis of the two enantiomers, and ees of a few percent have been created by this means (Balavoine et al., 1974; Bonner, 1991 and references therein; Bonner and Bean, 2000). For the Soai reaction, it has been shown that circularly polarized light can provide an initial ee which is subsequently amplified by autocatalysis (Soai et al., 2001; Kawasaki et al., 2004). Furthermore, circularly polarized light has been observed in regions of star and planet formation (Bailey et al., 1998; Bailey, 2001; Fukue et al., 2010). Any chiral bias in the chemistry of the protoplanetary disk arising from asymmetric photolysis by circularly polarized light would be passed on to asteroids and planetesimals that are the parent bodies of meteorites. The direction of the circular polarization depends on the way light from a nearby star is reflected from dust grains in the solar system. Although the orientation could be of either sign, it is likely to be constant over any one solar system; hence, it could provide a planet-wide bias. Another electromagnetic effect that might cause an initial asymmetry is the interaction of spin isomers with large magnetic fields (Popa et al., 2009).
Enantiomorphic crystals of materials such as quartz and sodium chlorate might also provide a possible source of chiral asymmetry. Single grains of these crystals are optically active. The surfaces of such crystals could potentially act as chiral catalysts for organic reactions. Once again, with the Soai reaction, it has been shown that an initial ee induced by chiral sodium chlorate crystals can be amplified by autocatalysis (Sato et al., 2004). However, the sign of the effect would vary between crystals, and there is no net bias favoring one enantiomorph over the whole surface of Earth (Klabunovskii, 2001). Despite this lack of a global biasing mechanism, mineral and/or clay surfaces might provide a platform for asymmetric synthesis of biopolymers (Hazen et al., 2001; Joshi et al., 2011) and thus contribute to an initial local asymmetry that could be amplified by autocatalysis.
It is also possible that crystallization of chiral organic molecules themselves can be important. When a solution of chiral molecules is in equilibrium with chiral crystals, the ee of the molecules in the solution can be higher than that in the crystals (Klussmann et al., 2006). This could provide another means for amplification of the ee, although this mechanism does not amplify the ee unstably from an initial small perturbation, as would be the case with asymmetric autocatalysis. Another crystallization mechanism that does amplify to a high excess from a small perturbation is given by Viedma (2007) and Noorduin et al. (2008). Here, an almost racemic solution of a chiral molecule is in equilibrium with chiral crystals of the two enantiomers. Breakup of the crystals is induced mechanically, and there is a tendency for large crystals to grow at the expense of smaller ones. Racemization of the molecules occurs in solution. The combination of crystal growth, dissolution, and racemization leads to almost complete homochirality.
In any sample of a finite volume with a finite number of molecules, a small random difference in the concentrations of the two enantiomers would arise by chance. If an external bias is to be relevant, it must be larger than this random statistical bias. It has been pointed out that the parity violation of the weak nuclear interaction makes a very slight difference in the thermodynamic properties of the two enantiomers. However, as argued by Bonner (2000), this effect would be much too small to give the observed ees of up to 15% in the meteorite samples, unless an autocatalytic process was able to amplify the excess. If an autocatalytic process was indeed occurring, then a small random bias could be amplified in any case, so a tiny bias arising from parity violation would not be a key aspect of the problem.
It should be borne in mind that current observations of the ee in meteorites are based on rather few samples. There is still insufficient information to say what fraction of carbonaceous meteorites contain molecules with significant ee and why certain molecules have a higher ee than others in the same sample. Also, the more general question of the relevance of the delivery of extraterrestrial organic material to Earth is still not resolved (Whittet, 1997; Pasek and Lauretta, 2008). The quantity of organic molecules arriving from outside may have been small compared to what was already on Earth, or chemical reactions (including racemization) may have occurred on Earth after arrival of the material from outside. In either of these cases, the ee on Earth at the time of the origin of life would not be the same as that observed in the meteorites. Furthermore, observations of chirality in meteorites have focused on amino acids, whereas in the context of the RNA world hypothesis, it would be nucleotides that would be more relevant at the time of the origin of life. Unfortunately, we have no data on the extent of the ee that may have existed in nucleotides prebiotically. Encouragingly, however, it has been shown that sugars synthesized in the presence of chiral amino acids may have an ee transferred to them (Pizzarello and Weber, 2004, 2010; Weber and Pizzarello, 2006). Hein et al. (2011) have also shown that RNA precursor molecules can be enriched to high levels of ee from racemic starting materials provided that amino acid enantiomers with a small initial bias are present in the reaction mixture. These two experiments suggest mechanisms by which chirality may have arisen in the backbone of nucleic acids.
If we accept that the ees of a few percent observed for amino acids in meteorites are indicative of the degree of chiral bias that existed in the monomers from which the first living biopolymers were composed, then there is still the question of how to get from a few percent to the essentially 100% homochiral state observed in life. It seems likely that it is the autocatalytic properties of the biopolymers that create the amplification that leads to a highly biased state. An example of a chirally selective peptide replicator has been studied by Saghatelian et al. (2001). Also in an RNA world scenario, polymerization of nucleotides is likely to be chirally selective, whether this is by a non-enzymatic, template-directed mechanism (Bolli et al., 1997) or due to the action of a polymerase ribozyme that catalyzes primer extension (Johnston et al., 2001).
At this point, the chirality problem links to our previous work on the origin of autocatalytic biopolymers (Wu and Higgs, 2009, 2011). We considered a prebiotic chemical reaction system in which monomers (such as nucleotides) and polymers (such as RNAs) could be synthesized. Our aim was to consider how catalytic biopolymers (ribozymes) could arise in this system. We supposed that only polymers longer than some minimum length, m, could be sufficiently complex to be ribozymes. In the absence of ribozymes, the steady state concentration of RNAs decreases rapidly with length. However, if a small number of ribozymes arises by random chemical synthesis, this can cause the system to switch to a living state in which nucleotide synthesis and/or polymerization is autocatalytic. In the living state, the concentration of RNAs decreases much more slowly with length, and the total concentration of ribozymes is large. Our previous work considered the origin of life but not the origin of homochirality, and we considered only one kind of monomer. Thus, we implicitly assumed that homochirality arose before the origin of life and that a supply of homochiral monomers was present from which the first autocatalytic biopolymers could be formed. If so, the origin of homochirality and the origin of life are separate problems. Previous theories for the origin of chirality (see the following section) typically assume that this is the case and do not deal with the origin of life. However, the discussion above suggests that the two problems may be at least partially linked. This leads us to consider a range of possible scenarios linking the origin of homochirality and the origin of life. These are summarized in Fig. 1.
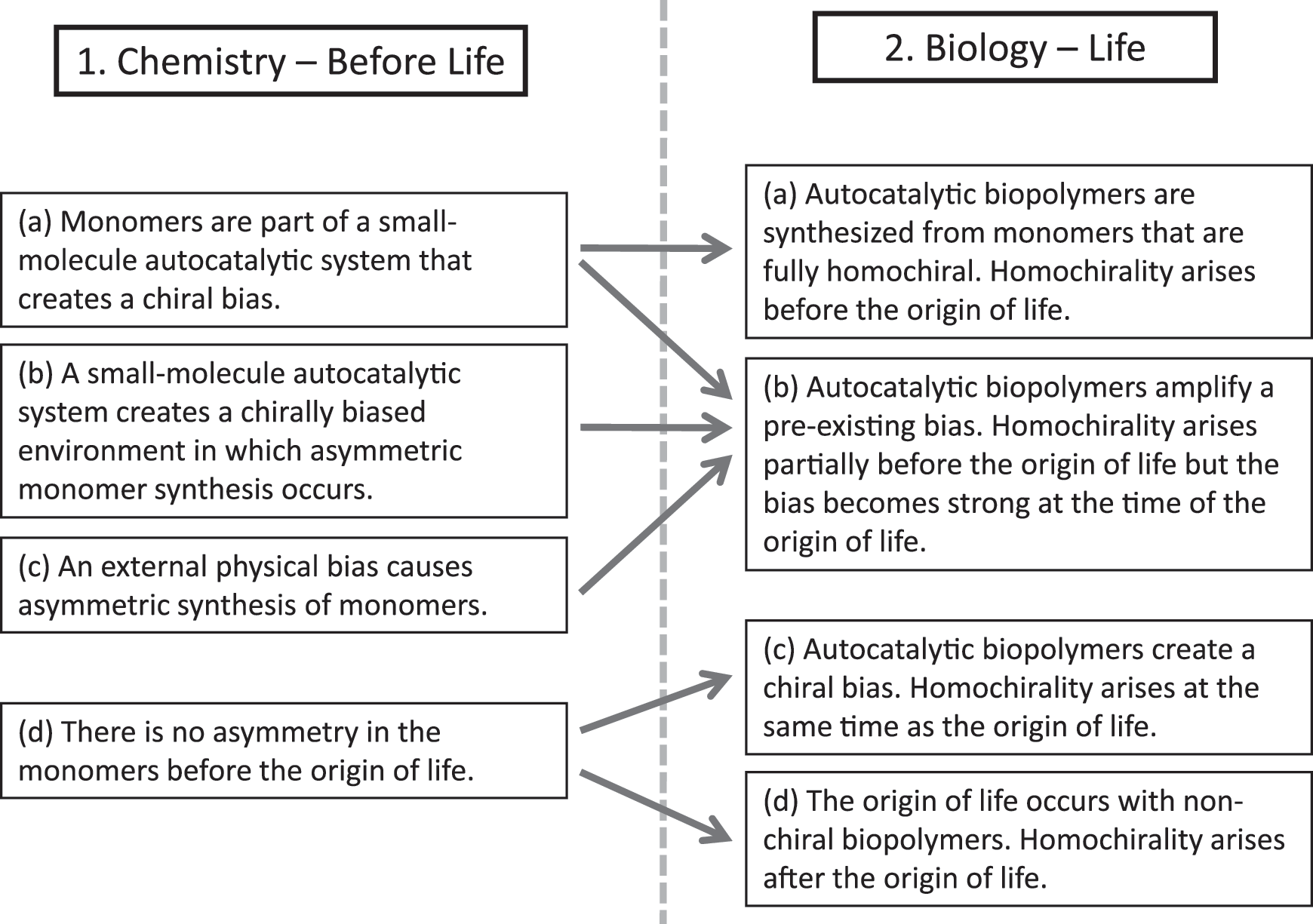
Scenarios relating the origin of chirality to the origin of life.
Boxes 1(a–d) consider the states of chirality that could have existed before the origin of life, and boxes 2(a–d) consider the states of chirality that could have arisen when life originated. In 1(a), it is supposed that an asymmetric autocatalysis process arises that incorporates the monomers themselves. As this is autocatalytic, it has the potential to create a high ee so that when biopolymer catalysts arise [state 2(a)], they are formed from a completely homochiral supply of monomers. In this scenario, the problem of homochirality is fully solved at the small-molecule stage, prior to the origin of life. On the other hand, if the small-molecule system only creates a moderate ee, biopolymer catalysts will arise within a partially biased mixture of monomers of the two enantiomers. As the biopolymers are likely to be more efficient chiral catalysts, the ee will be amplified when life arises [state 2(b)]. Thus, the origin of homochirality is partially before the origin of life, and the process is completed at the time of the origin of life.
In 1(b), it is supposed that a system of asymmetric autocatalysis arises with small molecules (such as the Soai reaction), but the monomers are not part of this. In this situation, monomer synthesis may be asymmetric because it occurs in a chirally biased environment, that is, chirality may be transferred from the autocatalytic small molecule to the monomer. This relieves the monomer from the dual responsibility of having to act as a chiral autocatalyst when present as a monomer as well as a functional component of a biopolymer catalyst when it is polymerized. For example, ribonucleotides can do the latter, but not the former, as far as we know.
In 1(c), we suppose that some physical influence, such as asymmetric photolysis or the presence of chiral crystals, causes monomer synthesis to be asymmetric. In both 1(b) and 1(c), monomer synthesis is not autocatalytic; therefore it seems likely that these states would only be partially chirally biased. These states would thus lead on to 2(b), in which full homochirality arises only when life arises.
In 1(d), it is supposed that there is no ee at the monomer level. This would be the case if the extraterrestrial supply of chiral molecules is too small to influence the overall chirality on Earth or if any chirality that does exist in the prebiotic mixture is not transferred to monomers from which the biopolymer catalysts are composed. In this case, the biopolymers arise from a racemic mixture, and it is the origin of the autocatalytic process of biopolymer synthesis that creates the instability that leads to an asymmetric state [2(c)]. Homochirality thus arises at the same time as the origin of life in this case.
In principle, state 1(d) could also lead to a state 2(d) in which life arises without chirality. In 2(d), there could be biopolymers composed either of achiral monomers or of racemic mixtures of chiral monomers. Both of these seem unlikely, but we have included this case as it is at least a logical possibility. In this scenario, life would arise before homochirality. Homochiral biopolymers would take over from achiral or racemic biopolymers at some point after the origin of life. Bonner (1991, 1995) reviewed some early proposals of this kind, which he termed biotic theories, because life would be required for the origin of homochirality. However, he effectively rejected these in favor of abiotic theories, in which chirality is required for the origin of life. The scenarios we show in Fig. 1 are an updated and more detailed version of the distinction between abiotic and biotic theories made by Bonner. While we have difficulty in imagining a living system of achiral or racemic biopolymers, as in 2(d), we think that it is quite likely that the problem of the origin of homochirality is at least partly biological, as in 2(b), and we would argue that the possibility of simultaneous origin of life and homochirality, as in 2(c), should at least be considered seriously. The principal aim of this paper is to develop a theory with which to investigate cases 2(b) and 2(c).
In the discussion of Fig. 1, we adopted the viewpoint that the origin of life means the origin of an autocatalytic system of information-carrying biopolymers, such as RNAs. However, if one takes a metabolism-first viewpoint, then it is possible that an autocatalytic system of small molecules may have existed before the origin of biopolymers (Shapiro, 2006) or that compositional inheritance may have existed before information-carrying biopolymers, as in the lipid world theories (Segre et al., 2000; Wu and Higgs, 2008). From this viewpoint, states 1(a–d) might already be classed as living, and states 1(a–d) and 2(a–d) would represent the situations before and after the origin of biopolymers, rather than before and after the origin of life per se. For concreteness in the rest of the paper, however, we will stick to our original viewpoint, and we will describe a system as living only if there are autocatalytic biopolymer catalysts present.
Previous Autocatalytic Theories
Before presenting the model used in this paper, it is useful to discuss the models for asymmetric autocatalysis that have been proposed previously, beginning with the simple case studied by Frank (1953). In Frank's model, L and D monomers are autocatalysts for their own synthesis, and they also inhibit one another by the formation of LD dimers that are nonreactive. This cross inhibition is important because if a slight excess of one enantiomer arises, formation of the LD dimer is a greater penalty for the rarer enantiomer, and this amplifies the ee and makes the symmetric state unstable. Variations of this model have been considered in which LL and DD homodimers form in addition to LD heterodimers and in which dimer formation is reversible (Ribo and Hochberg, 2008; Hochberg, 2009), and the conditions for the instability of the symmetric case have been worked out in detail.
Models have also been studied in which the monomers are not directly autocatalytic, but the homodimers (LL and DD) are catalysts for synthesis of monomers of the same enantiomer (Blackmond, 2004; Islas et al., 2005). Hence there is still asymmetric autocatalysis of monomer synthesis via the effect of the dimers. If dimers are the catalysts, then cross inhibition is less important. For example, if heterodimers and homodimers form at the same rate, there is no instability in models with monomer catalysts, whereas there is still an instability when dimers are catalysts. For this reason, Blackmond (2004) argued that dimer catalysts were essential to interpret the Soai reaction. However, Islas et al. (2005) found that the results of the Soai reaction were still consistent with monomer catalysis. A slightly different mechanism that works at the dimer level is the epimerization model of Plasson et al. (2004, 2007) in which LD dimers are converted to LL or DD, which creates an instability without requiring autocatalysis in monomer synthesis.
A next step toward modeling chiral asymmetry in biopolymers is to allow L and D monomers to make polymers of all lengths and to assume that only long polymers are catalysts. In the model of Sandars (2003), homochiral polymers, Ln and Dn , can grow from both ends, but cross inhibition occurs if a monomer of the opposite type is added, such that LnD and DnL can no longer grow from the blocked end. We will use m to denote the length of the polymer catalyst. Homochiral polymers, Lm and Dm , are assumed to be catalysts for synthesis of monomers of their own type. Saito and Hyuga (2005) described a polymerization model in which only pure homochiral polymers can form and cross reactions do not occur, although only the dimer catalyst case (m=2) was considered in detail. Brandenburg et al. (2005a, 2005b) extended the Sandars (2003) model and considered a case where dissociation of polymers into smaller sequences was permitted as well as polymer growth. Gleiser and Walker (2008) considered a variant that explicitly included monomer synthesis, treated polymers of all lengths as catalysts, and avoided putting in a maximum length of polymers. All these models share the implicit assumption that homochirality must arise prior to the origin of life.
We have previously considered models for the origin of autocatalytic biopolymers such as ribozymes (Wu and Higgs, 2009, 2011). The ingredients of our models are similar to those for the polymer models of chirality, although we were interested in the origin of life rather than the origin of homochirality. We considered monomers of a single type, as would be the case if a nonliving process generated fully homochiral monomers prior to the origin of life. Our models consider monomer synthesis and polymerization reactions. Both of these are presumed to occur at a small spontaneous rate in the absence of ribozymes and at a much higher rate when they are catalyzed by ribozymes. An important feature of our models is that there is a clear distinction between states that are living and dead. In the dead state, reactions proceed at the spontaneous rate, and the concentration of ribozymes is negligible. In the living state, the reactions proceed at high rates that are controlled by the ribozymes. The living/dead distinction is not present in previous models for chirality. Therefore, the aim of this paper is to study a model in which transitions between racemic and homochiral states can be studied alongside transitions from nonliving to living states.
Another feature of our ribozyme models is that an autocatalytic set can be created either by polymerase ribozymes that increase the polymerization rate or by nucleotide synthetase ribozymes that increase the rate of monomer synthesis (Wu and Higgs, 2011). Previous models for the origin of homochirality have focused principally on catalysis of monomer synthesis. In contrast, other papers dealing with the prelife to life transition (Ohtsuki and Nowak, 2009; Manapat et al., 2010) have dealt with catalysis of polymerization but ignored the chirality issue.
A Model Describing Both the Origin of Homochirality and the Origin of Life
Here we propose a system of chemical reaction equations that describes the synthesis of monomers of two possible enantiomers, which we denote L and D, from a nonchiral precursor (or “food”) molecule, F. We consider a reactor system with a fixed external concentration, F 0, of precursor and a constant rate of entry uF 0 of precursor molecules. Molecules of all types exit the system at the same rate u. The monomers are synthesized from F with rate constants sL and sD , which are not necessarily equal.
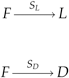
Monomers can polymerize by addition to the end of growing chains. Ln and Dn denote homochiral polymers of length n. Mixed polymers, with at least one monomer of each kind, are denoted Mn . Addition of a monomer of the wrong type to a homochiral polymer creates a mixed polymer. Addition of a monomer of either type to a mixed polymer creates a longer mixed polymer.

We assume that the rate constant for polymerization, r, is equal to the spontaneous rate r
0, which is a constant parameter. However, the synthesis rates are not constant because they depend on the catalyst concentration. The total concentrations of homochiral sequences of at least length m are
We suppose that some fraction of these homochiral polymers can be catalysts. The synthesis rates are the sum of a spontaneous term and a term proportional to CL
and CD
:
The parameter ɛ controls the degree of chiral bias in the chemical system prior to life. If ɛ=1, the chemical system is completely homochiral, and only one type of monomer is synthesized. If ɛ=0, the chemical system is racemic prior to the origin of life. We have set the bias to be in favor of D in Eq. 2 because D riboses arise in nucleotides, but the model is clearly equivalent if the bias goes the other way. The total rate of spontaneous synthesis of the two types of monomer is 2s
0. The parameter k controls the strength of the catalytic effect. The following set of ordinary differential equations (ODEs) can be used to describe the system.
where
Note that Eqs. 8 and 9 are collective equations for homochiral polymers of at least length m, hence Eqs. 6 and 7 are needed only for n<m. Therefore, the total number of independent equations is 2m+2.
Note that, unlike previous models (Sandars, 2003; Brandenburg et al., 2005a, b; Gleiser and Walker, 2008), we assume that mixed polymers can continue to grow and are not inhibited by a single monomer of the opposite sign. We do this for simplicity but also because it is a conservative assumption. The effects of enantiomeric cross inhibition are readily apparent in template-directed polymerization (Joyce et al, 1984); however, it is less clear if this is the case in the absence of a chiral template such as a homochiral polymer or mineral surface. We will show that homochiral living states arise in this model despite the wastage of monomers in nonfunctional mixed polymers. If cross inhibition of polymer growth were treated more explicitly in the model, this would lead to an additional favoring of homochiral living states, because there would be fewer mixed polymers.
Inclusion of the loss term (proportional to -u) in all equations means that polymers escape from the system and do not accumulate indefinitely. The equilibrium concentration inside the system is controlled by a balance between the supply of precursors and the loss rate.
Numerical Methods
Two methods were utilized to find the stationary solutions for the concentrations. The first approach was to integrate Eqs. 3 –10 numerically using a method appropriate for stiff systems of ODEs (Bader and Deuflhard, 1983) until convergence was reached. The second method was to use the NSolve function from Mathematica which is based on solving the eigen system of the right-hand side of the equations (Lichtblau, 2000). This method can find all the fixed points for the ODE system, including both stable and unstable solutions. The stability of the fixed points was determined by analyzing the eigenvalues of the Jacobian matrix of the ODE. For the small m case (m=5) that we consider in detail in this paper, both methods were practical, and we checked that the same results were obtained both ways. For the large m case (m=25), we used only the first method, because it was much faster, although it can find only stable solutions and it requires some prior knowledge of which solutions to expect.
Fully Homochiral Monomer Synthesis (ɛ=1)
Firstly, we consider scenario 1(a)-2(a) in Fig. 1, where the monomer system is fully chiral prior to the origin of life. This means that ɛ=1, and there is only one type of monomer produced, which is D with the sign convention used here. The system can be simplified to the following equation set:
This model is equivalent to the model for the origin of self-replicating biopolymers that we studied previously (Wu and Higgs, 2011). We consider this case briefly here, in order to compare with the case of ɛ<1, where both monomer types are relevant. For the numerical examples in this paper, we set u=1. This sets a timescale to which other reaction rates can be compared. We assume r 0=10 and F 0=10, unless specified otherwise. The two most important rate parameters are the spontaneous monomer synthesis rate s 0 and the catalytic efficiency k, and we consider the effect of varying these parameters in detail. The choice of the minimum catalyst length, m, is also important. We intend that the catalysts should represent ribozymes that are long enough to have a well-defined structure and function. For reasons of computational speed and practicality of finding the numerical solutions, we keep m fairly small and consider the case of m=5 in detail. At the end of the paper, we compare this with a case of longer ribozymes (m=25) and with the special case of dimer catalysts (m=2).
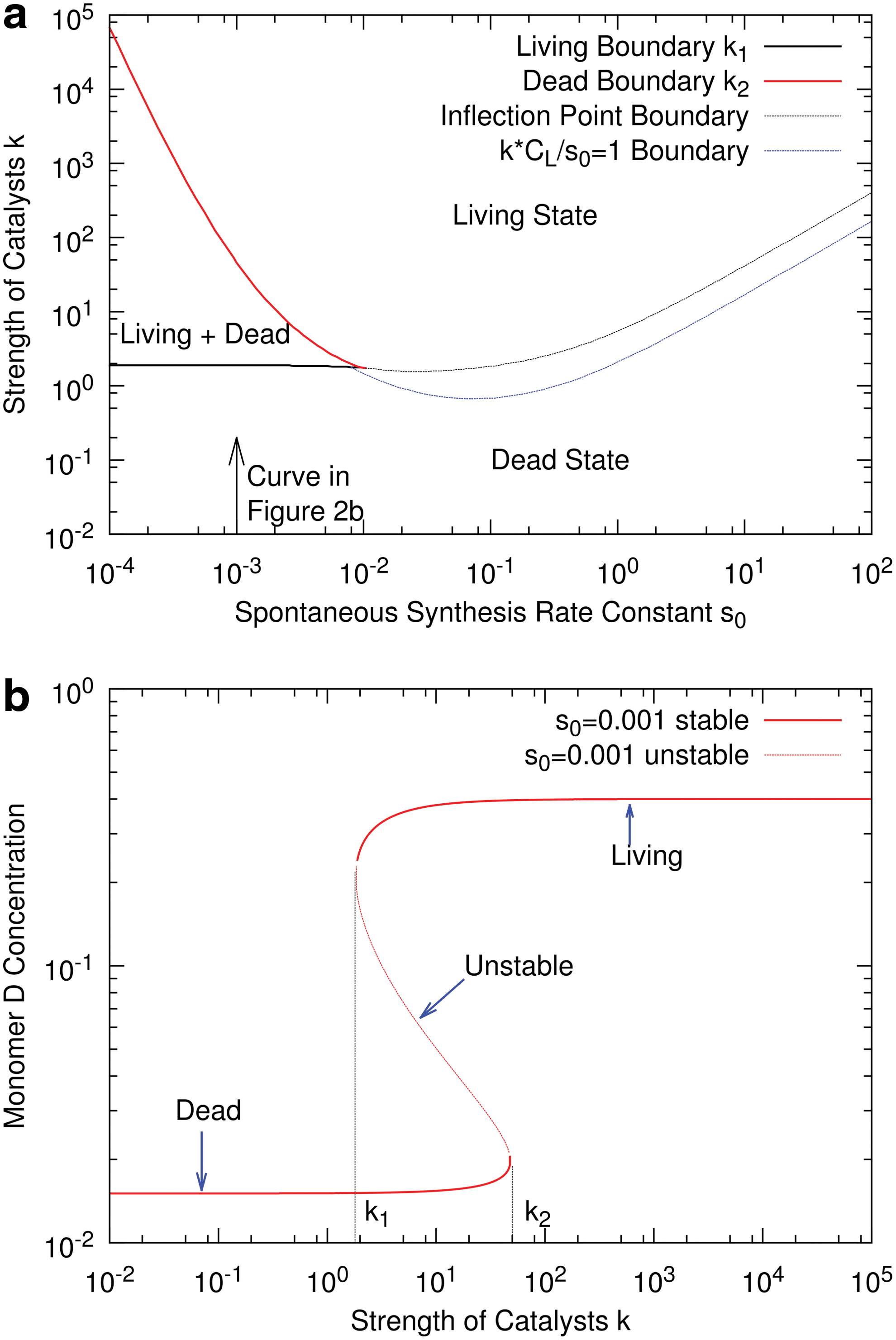
Figure 2a shows the phase diagram of the system in terms of k and s 0. Figure 2b shows the monomer concentration as k is increased, moving vertically along the line indicated by the arrow in Fig. 2a. In the range k 1 <k<k 2, two stable states exist with an unstable state between them. Within this range, the concentrations in Eqs. 12 –15 converge to one or other of the two stable solutions, depending on the starting conditions. Outside this range, only one stable state exists. The ratio kCD/s 0 measures the relative rate of catalyzed to spontaneous monomer synthesis. We call the lower stable state “dead” because the concentration of ribozymes is very small and the catalytic effect of the ribozymes is negligible compared to the spontaneous synthesis rate, that is, kCD/s 0 << 1. We call the upper stable state “living” because it is autocatalytic. The concentration of ribozymes is high, and the monomer synthesis rate is dominated by the ribozymes, not the spontaneous rate, that is, kCD/s 0 >> 1.
(
For larger values of s 0, the two boundaries, k 1 and k 2, meet, and there is only one stable solution for the system in which the monomer concentration increases smoothly with k. In this case, the boundary between the dead state and living states is not clear cut, but we used two ways to define an approximate boundary. The blue dotted line is the point where the ratio kCD/s 0=1, that is, where the catalytic effect of the ribozymes becomes equal to the spontaneous rate (see also Fig. 3).The black dotted line is the point where the monomer concentration increases most rapidly with k (i.e., the point of inflection of the monomer concentration as a function of k). This is a simple way of separating high- and low-concentration regions.
(
Further details of models that consider monomers of a single chirality are given by Wu and Higgs (2009, 2011) and Ohtsuki and Nowak (2009). All these models show regions where both living and dead states are stable, and they have a phase diagram similar to that in Fig. 2.
Symmetric Synthesis of Monomers L and D (ɛ=0)
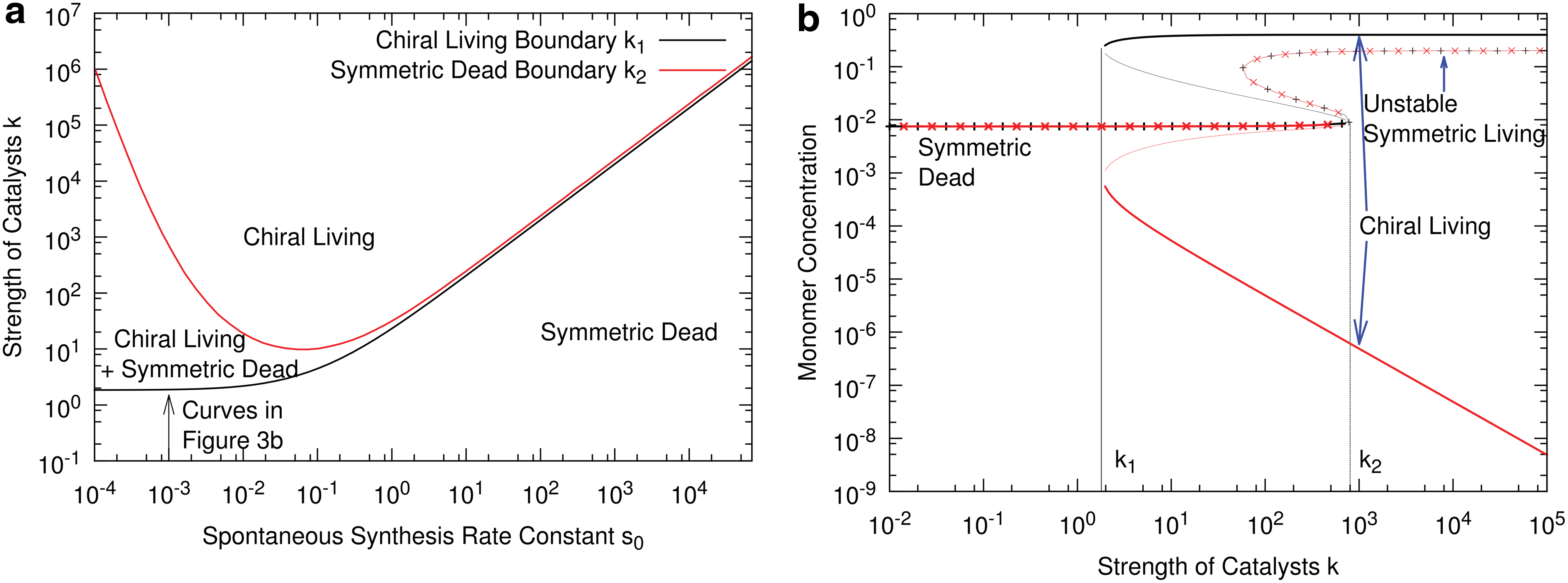
We now consider the scenario 1(d)-2(c) in Fig. 1, where there is no chiral bias in the chemical system prior to the origin of life (ɛ=0), and homochirality arises at the same time as the origin of life. The phase diagram for this case is shown in Fig. 3a, and the solutions for the monomer concentrations as a function of k with s 0=0.001 are shown in Fig. 3b.
For k<k 1, there is only one stable state, which is a symmetric dead state, that is, L and D monomer concentrations are equal, the ribozyme concentration is negligible, and the monomer synthesis rate is virtually equal to the spontaneous rate. For k 1<k<k 2, there are two chiral living states in addition to the symmetric dead state. For k>k 2, only the chiral living states exist. The chiral states with L dominant and D dominant are equivalent to one another, and either monomer could become dominant by chance. In the chiral states, the concentration of the dominant enantiomer (black curve in Fig. 3b) is much higher than that of the rare enantiomer (red curve), so the enantiomeric excess is virtually 100%. For example, at k 1=1.97 (close to k 1), the two concentrations are 0.295 and 0.00055, which corresponds to an ee of 99.6%. The ee becomes even larger as k increases.
The chiral states are classed as living, because the concentration of ribozymes of the dominant enantiomer is high, and synthesis of the dominant monomer is predominantly autocatalytic, that is, if D is the dominant monomer, kCD/s 0 >> 1. Figure 3b also shows a symmetric living branch. This corresponds to a situation in which both CL and CD are large and there is no ee in either the monomers or polymers. However, the symmetric living solution is always unstable, so it does not appear in the phase diagram. This is intuitive, because the living state is autocatalytic by definition and the autocatalysis is asymmetric, because only homochiral polymers are catalysts. We know from the many previous models of chirality that the symmetric state is unstable in situations of strong asymmetric autocatalysis. This model therefore illustrates the 1(d)-2(c) scenario, where life and homochirality arise together and it is not possible to have one without the other.
There are some other features of the living and dead states that are worth noting. In the dead state, the precursor concentration, F, is virtually equal to the concentration, F 0, in the surrounding medium (F 0=10 in this example). Monomer synthesis is occurring at a slow spontaneous rate that does not significantly deplete F. In contrast, in the chiral living state, monomer synthesis is occurring at a fast rate that is catalyzed by the ribozymes. This causes a significant depletion of the precursor such that F is approximately inversely proportional to the catalytic efficiency k. This explains why the monomer concentrations do not increase indefinitely, even when k continues to increase. In the dead state, the concentrations of the two homochiral polymers are equal, PL =PD , and both are less than M. Random polymerization leads to a majority of mixed polymers, as we would expect. In the chiral living state, the concentration of homochiral polymers of the dominant monomer is much larger than M, whereas it is much less than M for the rare monomer. Thus, in the living state, most polymers are homochiral. The rare monomer has a very low concentration and does not significantly impede polymerization of the dominant monomer, even though we have assumed a worst-case scenario, where the rates of addition of the two monomers are independent of the previous polymer sequence.
Asymmetric Synthesis of Monomers L and D (0<ɛ<1)
We now consider the situation where monomer synthesis is partially biased and where full homochirality arises with the origin of biopolymer catalysts [scenario 1(b) or 1(c) followed by 2(b)]. For the numerical example, we will set ɛ=0.5 and keep all other parameters as in the previous section. The phase diagram for this case is shown in Fig. 4a, and Fig. 4b shows the monomer concentrations as a function of k for a vertical section through the phase diagram. Regions I–V label parts of parameter space where different combinations of solutions are stable.
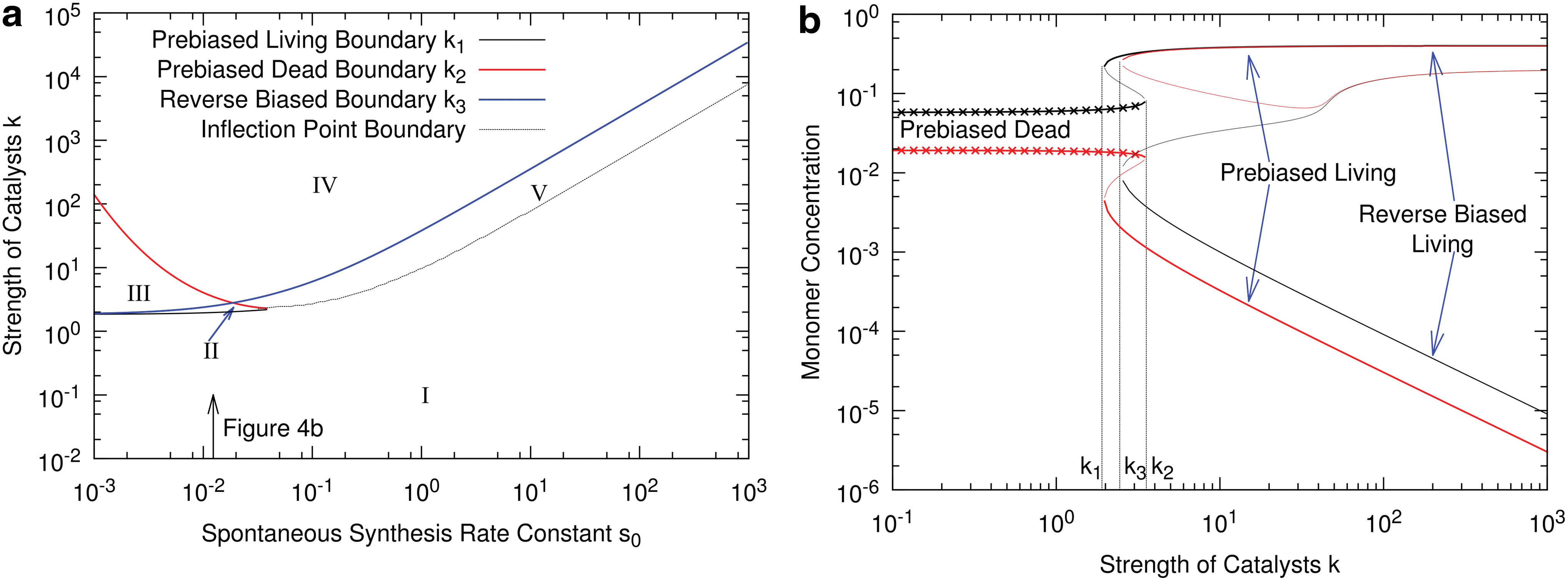
(
In this case, the dead state is “prebiased,” meaning that there is an enantiomeric excess of the monomer whose chemical synthesis rate is higher. A prebiased state would arise if, for example, circularly polarized UV radiation biased one enantiomer over another, or if meteorites seeded the prebiotic Earth with a slight asymmetry of organic precursor molecules as discussed in the introduction. It can be shown from Eqs. 3 –10 that if the catalytic terms kCL and kCD in the synthesis rates are negligible compared to s 0, then the enantiomeric excess of the monomers is equal to the excess in the synthesis rate, that is, (D−L)/(D+L)=ɛ, and D/L=(1+ɛ)/(1−ɛ). In this example with ɛ=0.5, D/L is approximately 3 over the whole range for which the prebiased dead state is stable.
For k>k 1, a prebiased living state becomes stable. This is an autocatalytic state in which there is a high concentration of catalysts of the dominant monomer, and the dominant monomer is the same as that favored by the chemical synthesis. The enantiomeric excess is close to 100% in this case (D >> L) because the dominant monomer is synthesized catalytically and the rare monomer is only synthesized at the spontaneous rate. For k>k 3 a reversed-biased living state becomes stable in which the dominant monomer is the one whose chemical synthesis rate is lower (L in this example). The synthesis rate of L is dominated by the catalytic term kCL , which is much larger than the spontaneous rate of synthesis of D, even though bias in the spontaneous rates favors D. For k>k 2, the dead state becomes unstable. The example of ɛ=0.5 shown here is quite a large bias. This was done in order to make the difference between the prebiased and reverse-biased states clear in the figures. If a smaller ɛ chosen, the difference in concentrations between the prebiased and reversed biased states is smaller.
Dynamical Considerations and the Effect of Changing m
In the previous sections we have considered only the stability of the stationary solutions of the differential equations, and we have not considered the dynamics of how those solutions are reached. We have argued previously (Wu and Higgs, 2009) that the origin of life probably occurred in a region of parameter space where both living and dead states are stable. If the only solution were a dead state, we would not be here, and if the only solution were a living state, we would have rapid spontaneous emergence of life all the time. In reality, the origin of life seems to be difficult but not impossible. We have argued that the origin of life requires a transition from a dead to a living state that occurs due to stochastic fluctuations of concentrations in a finite volume. We demonstrated that this can occur in stochastic dynamical simulations in a model with only one kind of monomer (Wu and Higgs, 2009), similar to the ɛ=1 case in this paper. An infinite deterministic system will remain stable in a dead state forever. In a finite volume, with finite numbers of molecules, concentration fluctuations become significant, and these can cause the system to jump from a dead to a living state.
In the stochastic case, it is necessary to consider the meaning of the catalysis rate k more carefully. If we suppose that all homochiral polymers of length at least m are catalysts, then CD (or CL ) is the concentration of catalysts, and k is the catalytic effect of each catalyst. If we are thinking of a chemical mechanism of catalysis that works with small molecules, then this seems reasonable. For example, if the mechanism is simple enough that dimers are catalysts (m=2), then it seems reasonable to suppose that all dimers are catalysts. However, in our previous papers (Wu and Higgs, 2009, 2011), we were thinking of biological catalysts; that is, the catalysts were ribozymes. In this case, m must be reasonably large, and only a small fraction of sequences of this length will function as ribozymes. If a fraction f of polymers are ribozymes, and the effect of each ribozyme is k 0, then the net effect of the catalysts is k 0 fCD . For deterministic solutions of the differential equations, we can write k=k 0 f; hence, it makes no difference whether we have a large fraction of weak catalysts or a tiny fraction of very effective catalysts. For the stochastic case, these situations are different, that is, both f and k 0 are relevant separately. This has been investigated in our original model (Wu and Higgs, 2009). We have not yet studied the stochastic dynamics of the chiral model. However, it is already possible to draw several conclusions from the deterministic stationary solutions that we have given here.
In most cases, the value k=k 1 at which the living state becomes stable is much smaller than the value k=k 2 at which the dead state becomes unstable. This is particularly true if s 0 is small, as was probably the case for prebiotic synthesis. For example, in Fig. 5, the two boundaries differ by several orders of magnitude when s 0 is small and by a relatively small factor when s 0 is large. The gap between k 1 and k 2 is more pronounced when m is larger. Figure 5a shows the ɛ=0 case with m=25, which may be compared with the ɛ=0, m=5 case in Fig. 3a. For the larger m, there is a wide separation of k 1 and k 2 across the full range of s 0. Thus, given that m may be quite long for functional ribozymes and f is likely to be very small, it would seem much more likely that a real-world situation would fall in the bistable region with k between k 1 and k 2 than in the living-only region with k>k 2. This means that a stochastic transition between dead and living states with parameter values in the bistable region is a probable scenario. In the case where ɛ>0, a slight bias in the spontaneous synthesis is likely to be very relevant for the transition from the dead to the living state. In the dead state, when polymerization is random, the chance of forming homochiral polymers of the dominant monomer may be much higher than for the rare monomer, even if the difference in the monomer concentrations is relatively small. The degree of chiral asymmetry of polymers will increase with increasing chain length (Bolli et al., 1997) due to differential rates of synthesis. It is much more likely that the system will jump to the prebiased living state than the reverse-biased state.
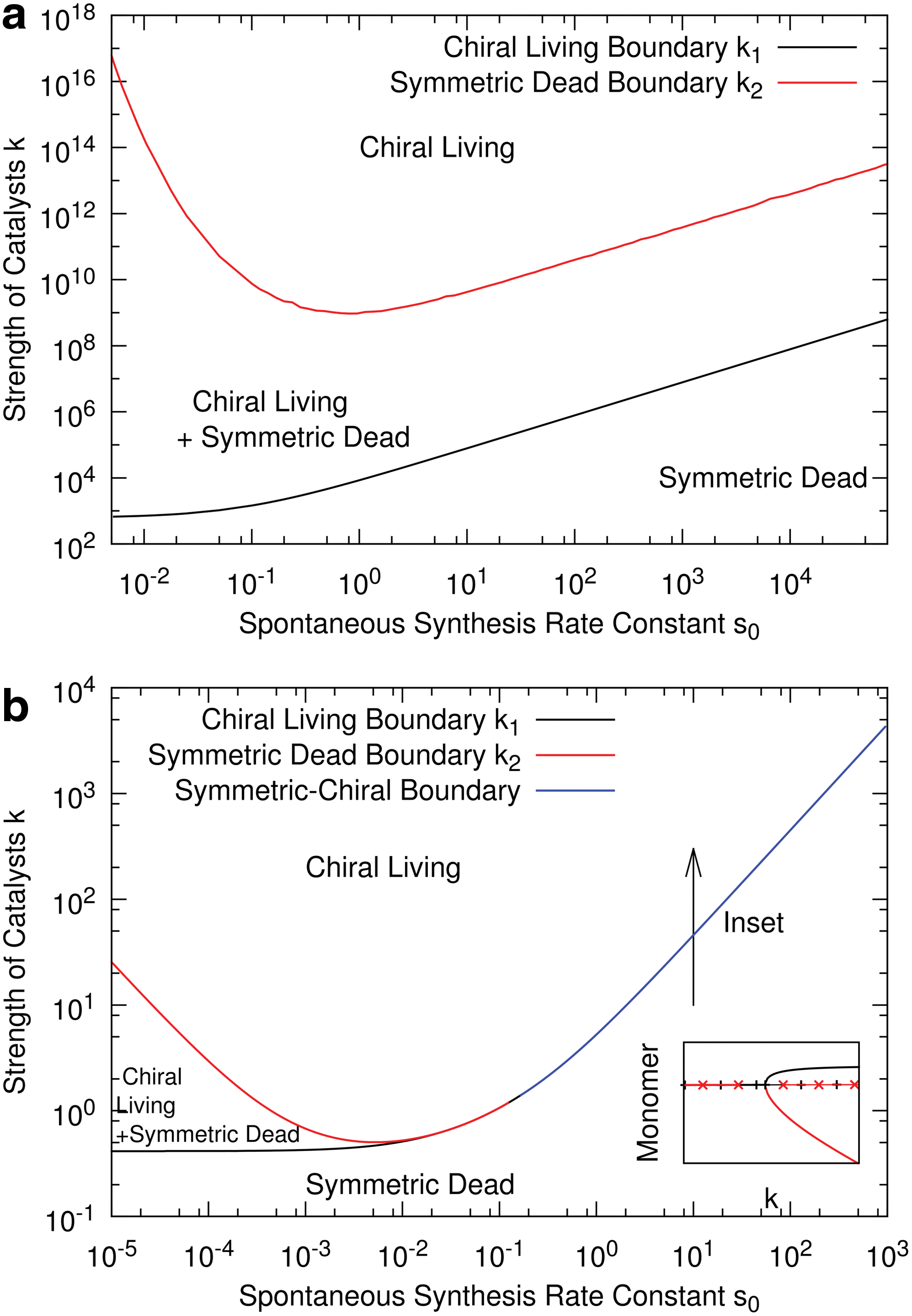
The effect of changing the minimum length m required for catalysis in the case where chemical synthesis is symmetric, ɛ=0. (
For completeness, and for comparison with previous models that deal with dimer catalysts, we will consider the case of ɛ=0 and m=2 in our model. The phase diagram is shown in Fig. 5b. For small s 0 the boundaries k 1 and k 2 for the stability of the chiral living and symmetric dead states are separate, and there is an intermediate region where both types of solution are stable, as in the previous examples. However, for larger s 0 there is a single boundary between the symmetric and chiral states. The monomer concentrations as a function of k go through a forward bifurcation when this boundary is crossed, as shown in the inset to Fig. 5b. Hence there is no region of bistability. This is qualitatively different from the case of m=5 (Fig. 3b), where a backward bifurcation occurs, and there is bistability for k 1<k<k 2. As far as we are aware, this forward bifurcation occurs only when m=2 in our model and not for m>2. As we are particularly interested in the origin of ribozymes, and as these are likely to have m considerably greater than 2, this case of m=2 seems less relevant to scenarios for the origin of life.
The existence of a region of bistability of living and dead states is an important feature that emerges in all the variants of our model that we have considered. The bistable region occurs in the models in our previous papers, which ignore chirality (Wu and Higgs, 2009, 2011), and in the fully homochiral case, ɛ=1 (Fig. 4), in which the origin of homochirality is a separate problem from the origin of life. The models for the prelife-life transition considered by Ohtsuki and Nowak (2009) also have this structure. In contrast, almost all previous models that consider the origin of homochirality have a simple forward bifurcation and no bistable region (Sandars, 2003; Brandenburg et al., 2005a; Gleiser and Walker, 2008; Ribo and Hochberg, 2008). The model of Saito and Hyuga (2005) is an exception in that it is a model for homochirality that does show a bistable region for some parameter values. The origin of homochirality is a symmetry-breaking transition, whereas the origin of life is a transition between dead and living states that differ in concentrations and reaction rates, and it is not directly associated with symmetry breaking. In this paper, we have considered the way these two transitions interact. When chirality is introduced into our origin of life models, the feature of bistability is retained from the nonchiral model, and the instability in the chirality is induced by the transition from dead to living states. This leads to the novel feature of our models that the chiral states arise via a backward bifurcation rather than a forward bifurcation.
The idea of a stochastic transition between dead and living states occurs in the simple abstract model for the origin of life considered by Dyson (1999), and we expanded on this in the context of the RNA world (Wu and Higgs, 2009). This picture has important consequences for the way that homochirality and life will spread spatially across the surface of Earth. Previous spatial treatments of chirality (Brandenburg and Multamaki, 2004; Gleiser and Walker, 2008; Gleiser et al., 2008) dealt with models that have a forward bifurcation. It was supposed that the system begins in a racemic state and that this becomes unstable if the parameters are suddenly quenched into a range where the chiral states are stable. This leads to simultaneous emergence of spatial domains of L and D with a length scale that grow slowly over time until one domain takes over the whole surface. According to the models in this paper, however, the dynamics would be substantially different. If the system begins in a racemic dead state (with ɛ=0) in a parameter range where the dead state is also stable, then the origin of life is initiated by a rare stochastic transition occurring in a localized region. Life would then spread rapidly and deterministically across the surface, because the living state synthesizes more catalysts that diffuse out into neighboring regions and immediately trigger the transition to life in the neighboring regions. The spread of life would take along whichever chirality happened to arise in the initial event. If there were a small pre-existing chemical bias (ɛ>0), then the initial living state would be likely to follow the handedness of the prebias. Whether ɛ were zero or nonzero, it would still be unlikely that simultaneous origins of life would occur in more than one place at the same time. The idea that there were simultaneous competing forms of L and D life on the surface seems improbable, in our view. We intend to investigate the spatial dynamics of our model in detail in the future.
Conclusions
The evidence from analysis of meteorite compositions now seems to indicate fairly conclusively that a modest ee of a few percent can arise in some kinds of organic molecules for nonbiological reasons. Asymmetric photolysis seems to be the most plausible cause for this (as reviewed in the introduction of this paper), although other mechanisms are also possible. Formation of biopolymer catalysts, such as RNAs, or some other nucleic acid analogue, is a key step in the origin of life, in our view. Life must have arisen in an environment in which the chemical synthesis of nucleotides was possible. If the chemical environment was chirally biased to some extent, then it is possible that prebiotic nucleotide synthesis was also chirally biased, that is, there may have been a small but nonzero ɛ. However, the scarcity of examples of asymmetric autocatalytic systems based only on small molecules suggests that it is unlikely that a fully homochiral system could have arisen by prebiotic chemistry at the monomer level. We argue that it is the biopolymers themselves that are the asymmetric autocatalysts. It is the origin of biopolymer catalysts that drives the transition to the living state. As these biopolymer catalysts are chiral, then the same transition drives the system to a homochiral state. If the chemical world were racemic (ɛ=0), then the origin of life and the origin of homochirality would occur together. If there were a partial chiral bias before life (ɛ>0), then the origin of life would complete the transition to homochirality that was begun by chemical forces existing before life.
As discussed in the introduction, the possibility that homochirality emerges with or after the origin of life has largely been dismissed (Bonner, 1991, 1995), and a majority of research has focused on abiotic theories where homochirality is required to emerge prior to the origin of life. Here we have shown that scenarios in which a homochirality emerged with life are serious possibilities. This has implications for astrobiological searches for life on other planets. If the abiotic hypothesis is correct, homochirality is not a valid biomarker, because nonliving chemistries with a high chiral bias might exist that would be false positives for life. Alternatively, if homochirality arises only with life, homochirality may be one of the best biosignatures for detecting life on other planets, being both easily identifiable and characteristic only of living systems.
Footnotes
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada. S.I.W. gratefully acknowledges support from the NASA Astrobiology Institute through the NASA Postdoctoral Fellowship Program.
Abbreviations
ee, enantiomeric excess; ODE, ordinary differential equation.