
Abstract
The functional end products of the extant biosynthesis of tetrapyrrole macrocycles in photosynthetic organisms are hydrophobic: chlorophylls and bacteriochlorophylls. A model for the possible prebiogenesis of hydrophobic analogues of nature's photosynthetic pigments was investigated by reaction of acyclic reactants in five media: aqueous solution (pH 7, 60°C, 24 h); aqueous solution containing 0.1 M decanoic acid (which forms a turbid suspension of vesicles); or aqueous solution accompanied by dodecane, mesitylene, or a five-component organic mixture (each of which forms a phase-separated organic layer). The organic mixture was composed of equimolar quantities of decanoic acid, dodecane, mesitylene, naphthalene, and pentyl acetate. The reaction of 1,5-dimethoxy-3-methylpentan-2,4-dione and 1-aminobutan-2-one to give etioporphyrinogens was enhanced in the presence of decanoic acid, affording (following chemical oxidation) etioporphyrins (tetraethyltetramethylporphyrins) in yields of 1.4–10.8% across the concentration range of 3.75–120 mM. The yield of etioporphyrins was greater in the presence of the five-component organic mixture (6.6% at 120 mM) versus that with dodecane or mesitylene (2.1% or 2.9%, respectively). The reaction in aqueous solution with no added oil-slick constituents resulted in phase separation—where the organic reactants themselves form an upper organic layer—and the yield of etioporphyrins was 0.5–2.6%. Analogous reactions leading to uroporphyrins (hydrophilic, eight carboxylic acids) or coproporphyrins (four carboxylic acids) were unaffected by the presence of decanoic acid or dodecane, and all yields were at most ∼2% or ∼8%, respectively. Taken together, the results indicate a facile means for the formation of highly hydrophobic constituents of potential value for prebiotic photosynthesis. Key Words: Origin of life—Prebiotic—Oil slick—Porphyrinogen—Porphyrin—Pyrrole—Partition. Astrobiology 12, 1055–1068.
1. Introduction
T
A variety of mechanisms have been proffered for formation of a primordial oil slick. In a predominantly methane-nitrogen atmosphere, solar ultraviolet radiation would yield methyl and methylene radicals which upon combination form heavier hydrocarbons. The latter could accumulate to give an oil slick from 1 to 10 m thick (Lasaga et al., 1971; Cleaves and Miller, 1998; Nilson, 2002). An oil slick also could be produced from carbon-rich meteorites containing a variety of extraterrestrial organic molecules, including low-molecular-weight hydrocarbons (Cleaves and Miller, 1998; Sephton, 2002). In this regard, short-chain monocarboxylic acids (Lawless and Yuen, 1979; Huang et al., 2005) and aromatic molecules such as alkylbenzenes and naphthalenes (Komiya and Shimoyama, 1996; Sephton, 2002) have been found in organic material extracted from the Murchison carbonaceous meteorite.
The presence of a primordial oil slick could not only result in accumulation of hydrophobic molecules but also provide a cloistered environment for their formation and subsequent transformation. Such hydrophobic compounds also would be expected to localize in surfactant assemblies including lipid bilayers. The lipid bilayer is characterized by a profound change in static dielectric constant in traversing from the bulk aqueous medium (∼78) to the core of the membrane (∼2.1) (Tien, 1974; Coster, 2003). Indeed, the pigments of modern photosynthesis, which are embedded in membrane-spanning proteins, typically are quite hydrophobic. Examples from photosynthetic bacteria include bacteriochlorophylls and carotenes (Scheer, 2003, 2006). The structures of bacteriochlorophyll a and β-carotene are shown in Scheme 1.

Hydrophobic, functional constituents of extant photosynthesis.
Proposals for tetrapyrrole pigments in early photosynthesis have chiefly focused on macrocycles that resemble those produced via the modern biosynthetic pathway or are derivatives thereof. Examples include the following: • Uroporphyrins, of which uroporphyrin III is derived from the biosynthetic intermediate uroporphyrinogen III. Each uroporphyrin is water-soluble due to the presence of the eight carboxylic acid substituents. Indeed, uroporphyrins and uroporphyrinogens are envisaged as functioning as photocatalysts in aqueous solution (Mercer-Smith and Mauzerall, 1984; Mercer-Smith et al., 1985). • Coproporphyrins, of which coproporphyrin III is the unsaturated analogue of the biosynthetic intermediate coproporphyrinogen III. Coproporphyrins contain only four carboxylic acid groups and are envisaged as functioning as photocatalysts upon complexation with other entities (Mercer-Smith et al., 1985). • Protoporphyrin IX, which is the last common precursor to heme and chlorophylls. Protoporphyrin IX is amphiphilic due to the presence of two carboxylic acid substituents on one edge of the molecule, and is envisaged as functioning in membrane assemblies (Olson and Pierson, 1987; Sivash et al., 1991). • Octaethylporphyrin, an abiotic compound that is quite hydrophobic. Octaethylporphyrin has been investigated in a variety of membrane-based photochemical processes, and the magnesium chelate thereof has been shown to enable light-driven transmembrane proton pumping (Sun and Mauzerall, 1996; Mauzerall and Sun, 2003).
While the photofunctional properties of tetrapyrrole macrocycles would be manifested in a photic world, the intrinsic structural features of tetrapyrroles might prove beneficial in other ways. Indeed, hydrophobic aromatic macrocycles have been proposed to have played a variety of roles in the origin of life, including as scaffolding for organization of nucleic acids (Hud and Anet, 2000) and for assembly of functional containers (Ehrenfreund et al., 2006).
A key feature of the extant biosynthesis of tetrapyrrole macrocycles is the increasing hydrophobicity in proceeding along the pathway from the starting material δ-aminolevulinic acid (ALA) toward functional end products (Mauzerall, 1976, 1998). This progression is illustrated in Scheme 2. Uroporphyrinogen III contains eight carboxylic acids. Decarboxylation of each acetic acid moiety affords coproporphyrinogen III. Decarboxylation of two propionic acid moieties accompanied by dehydrogenation affords the divinyl-diacid protoporphyrinogen IX. The chlorophylls and bacteriochlorophylls are quite hydrophobic. While the porphyrinogen intermediates of the extant pathway may have provided important functional roles in prebiotic chemistry, only the quite advanced biosynthetic products are employed in extant biochemistry.
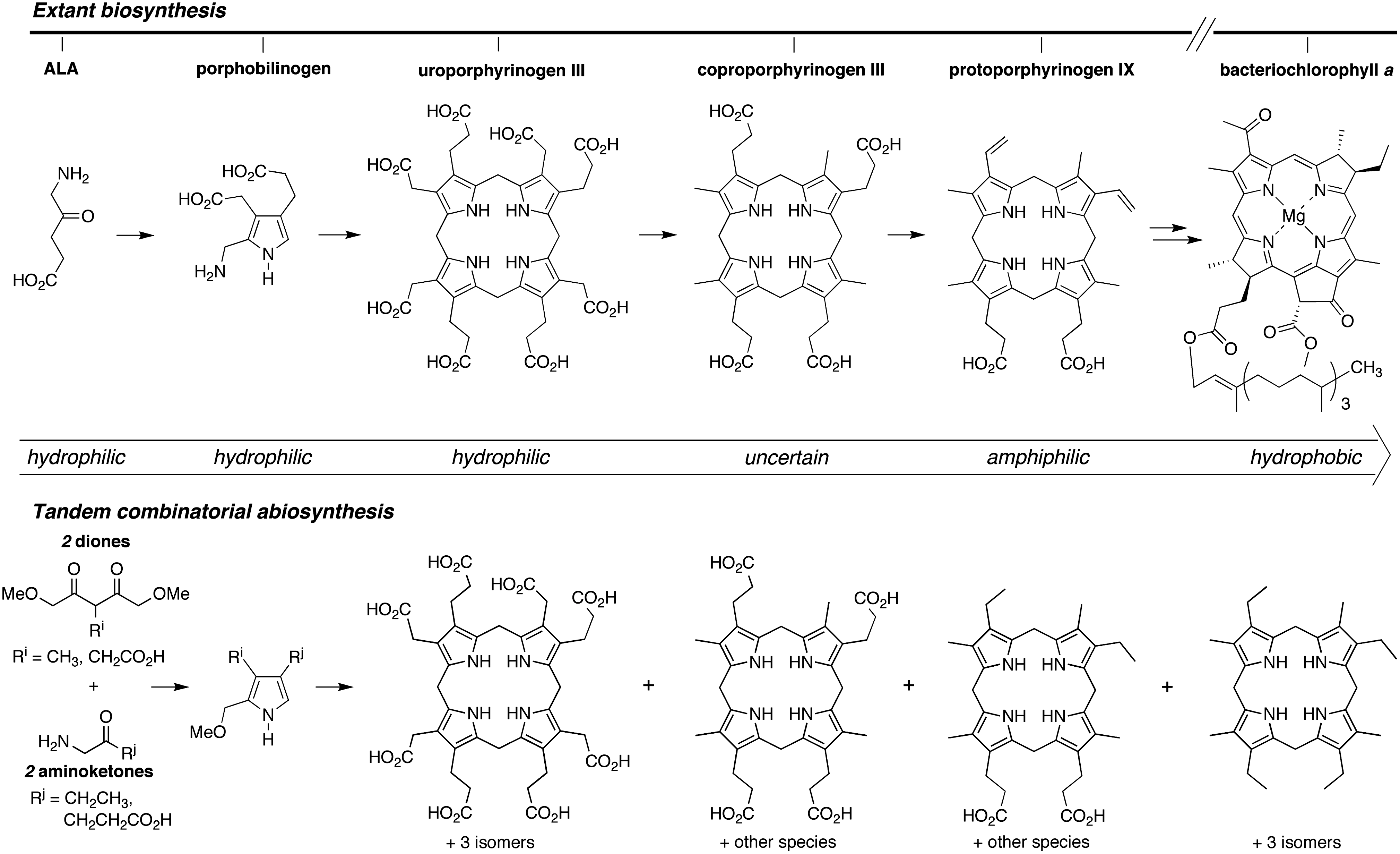
The stepwise extant biosynthetic formation of tetrapyrrole macrocycles is accompanied by a steady increase in hydrophobicity (top). An all-at-once tandem combinatorial (non-enzymic) process affords macrocycles encompassing the entire range of polarity (bottom). Each porphyrinogen can be oxidized to give the corresponding porphyrin (not shown).
As part of a program to develop a chemical model for the possible prebiogenesis of tetrapyrrole macrocycles, we have been investigating reactions of relatively simple acyclic compounds that proceed via pyrroles to give porphyrinogens (Lindsey et al., 2009, 2011). Recently, we examined the three pairwise reactions of
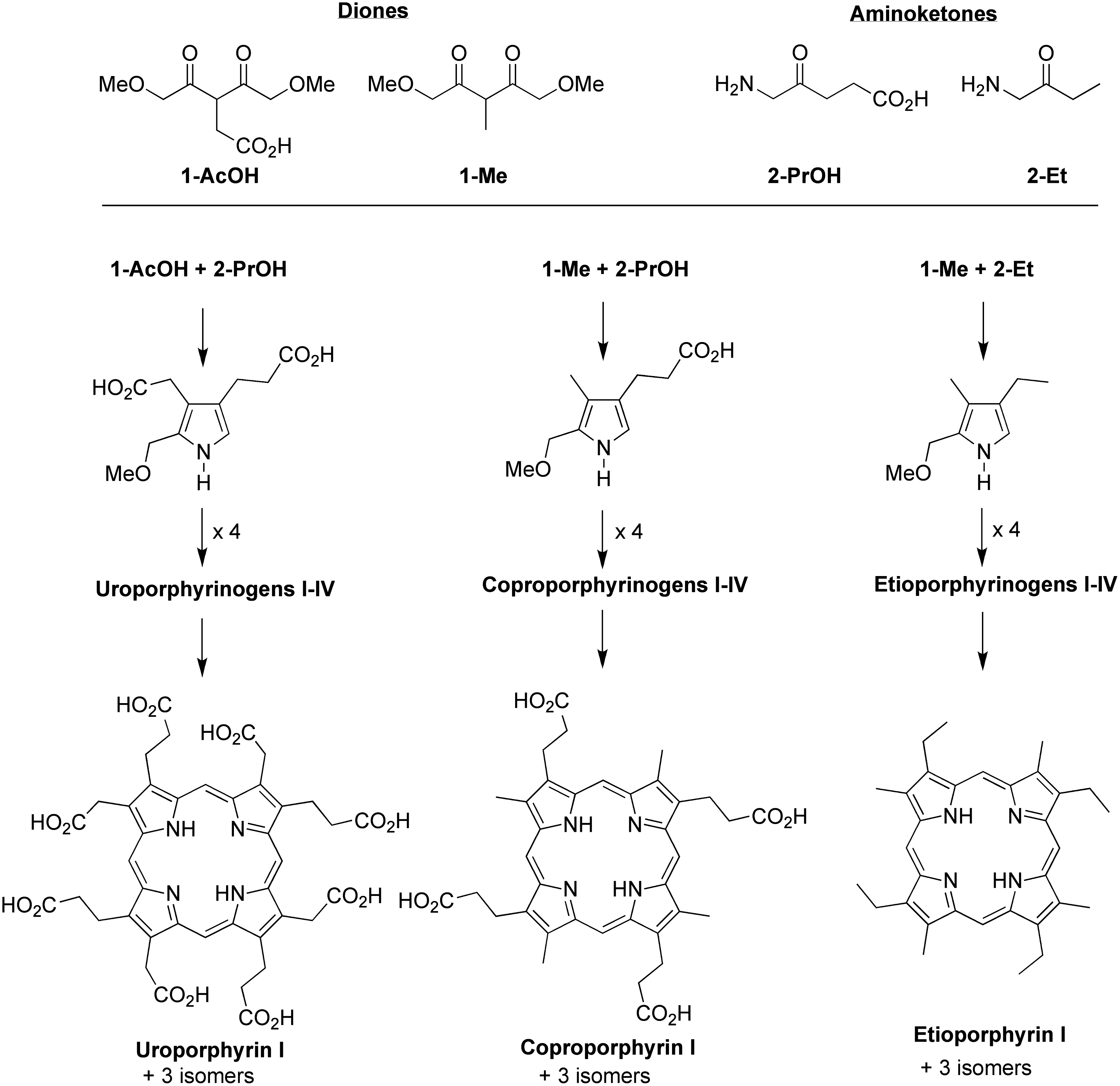
Three pairwise reactions of one dione and one aminoketone (
A combinatorial process afforded a repertoire of molecules spanning the range of polarity, from uroporphyrinogens to etioporphyrinogens (Scheme 2). The compounds formed predominantly contained 2–6 carboxylic acid units, with relatively little of the porphyrins with extreme hydrophilicity (i.e., uroporphyrinogens) or hydrophobicity (i.e., etioporphyrinogens). Indeed, virtual library analysis (Taniguchi and Lindsey, 2012a, 2012b) indicated the fractional amount of such polar extremes was expected to be 0.0039 (of the total macrocycles) for each (Taniguchi et al., 2011). Thus, although the combinatorial process provided a route to hydrophobic molecules such as etioporphyrins, the expected quantity of such porphyrins would be minute. Otherwise, plausibly prebiotic pathways have heretofore not been demonstrated that directly afford tetrapyrrole analogues of the modern functional constituents of photosynthesis.
In this paper, the three pairwise reactions were carried out in aqueous solution at pH 7 in the presence or absence of vesicles composed of decanoic acid, and the presence or absence of the aliphatic hydrocarbon dodecane (which gave phase separation). In addition, the reaction forming etioporphyrinogens was examined in a biphasic medium composed of an aqueous solution at pH 7 and the aromatic hydrocarbon mesitylene (1,3,5-trimethylbenzene), as well as in a biphasic medium composed of an aqueous solution at pH 7 and a mixture of hydrocarbons (dodecane, decanoic acid, mesitylene, naphthalene, and pentyl acetate; in equimolar quantities). Our interest herein is to study the ability of an oil slick, formed by low-molecular-weight hydrocarbons, to accumulate and/or facilitate the formation of prebiotic compounds, and thereby provide suitable conditions for the development and origin of life.
2. Methods
2.1. Samples and solutions
δ-Aminolevulinic acid (ALA=
Stock solutions of
2.2. Preparation of decanoic acid vesicles
Decanoic acid (431 mg) was added to 4.5 mL of deionized water. Aqueous NaOH (280 μL, 10 M) was added to the solution in small aliquots, vortexing between additions, until the acid was completely dissolved (pH ∼11). Aqueous HCl (140 μL, 10 M) was then added in small aliquots to the transparent suspension, vortexing between additions, until pH 7 was reached. The volume of the resulting opalescent suspension was adjusted to 5 mL for a final decanoic acid concentration of 0.5 M.
2.3. Reactions
The reactions were carried out at 0.5 mL scale for 24 h at 60°C and pH 7, following experimental procedures described previously (Lindsey et al., 2011; Taniguchi et al., 2012). The 0.5 mL reaction mixtures were prepared by addition of aqueous-based constituents to give a final volume of 0.4 mL, and the volume of the organic “oil slick” constituent was 0.1 mL. The aqueous-based constituents comprised aliquots of
The organic “oil slick” constituent comprised 100 μL of one of the following: decanoic acid (0.5 M), dodecane, mesitylene, or a five-component mixture of organic constituents (decanoic acid, dodecane, mesitylene, naphthalene, and pentyl acetate; each in equimolar quantities).
The samples were placed in an enclosed reaction chamber for investigating multiple reactions in parallel, which was evacuated under reduced pressure and then flushed with argon. The cycle was repeated three times. A slow flow of argon was passed continuously through the reaction chamber. None of the reactions was stirred. After 24 h, the crude reaction mixtures were sampled immediately (to determine the porphyrin yield) and subsequently were frozen.
2.4. Porphyrin yields
Porphyrin yield determinations were performed by oxidation of crude reaction mixtures with I2 directly in a cuvette, as described previously (Lindsey et al., 2011; Taniguchi et al., 2012).
(a) The procedure for uroporphyrin and coproporphyrin was as follows. A 50 μL aliquot of a reaction sample (30 mM of each reactant) was transferred to a dilution vial containing 150 μL of water (1/4 dilution). The solution was mixed briefly, and then 50 μL of this diluted sample was transferred to a cuvette containing 2.6 mL of 0.1 M aqueous HCl. Then the cuvette was treated with 56 μL of I2 (50 mM) in ethanol, followed by 56 μL of Na2S2O3 (0.2 M) in water. The absorption spectrum was acquired. The Soret band was very sharp, as expected in aqueous acid, with a full width at half-maximum (fwhm) of 11 nm (see Figs. S1 and S2 in the Supplementary Material, available online at
(b) The procedure for etioporphyrin was as follows. The crude reaction mixture was diluted with water (1 mL), extracted with cyclohexanone (1 mL), and the organic layer was separated. For reaction at 30, 60, or 120 mM, an aliquot of 50, 25, or 12.5 μL, respectively, of the organic layer was transferred to a cuvette containing 2.6 mL of dimethylsulfoxide (DMSO). Then the cuvette was treated with 56 μL of 50 mM I2 in ethanol, followed by 56 μL of Na2S2O3 (0.2 M) in water. The absorption spectrum was acquired. The Soret band was quite broad (fwhm=41 nm), characteristic of octaalkylporphyrins in neutral aqueous solution or organic media (see Figs. S1 and S2 in the Supplementary Material). Accordingly, the yield was determined by using a molar absorption coefficient of 160,000 M −1 cm−1 at the Soret band maximum (Rimington, 1960). For reaction at 3.75, 7.5, or 15 mM, a 50 μL aliquot of the organic phase was transferred to a cuvette containing 2.0 mL of DMSO. The remainder of the procedure was identical.
2.5. Fluorescence emission analyses
For analysis of etioporphyrin by fluorescence spectroscopy, 50 μL of the reaction mixture (120 mM, pH 7, 60°C, 24 h, and 0.1 M decanoic acid) was treated with 56 μL of 50 mM I2 in ethanol followed by 56 μL of 0.2 M Na2S2O3 (in water). The resulting oxidized reaction mixture was treated with brine and extracted with cyclohexanone (Rimington and Benson, 1967). The organic phase was concentrated upon application of a stream of argon. The resulting crude porphyrin was dissolved in 1-propanol. Fluorescence measurements were performed as described previously (Soares et al., 2012).
2.6. Identification of porphyrin isomers by 1H NMR spectroscopy
For analysis of etioporphyrin isomers, the crude porphyrin extract and authentic samples of etioporphyrin I and etioporphyrin III were examined by proton nuclear magnetic resonance (1H NMR) spectroscopy. The crude porphyrin extract was obtained by treatment of the oxidized reaction mixture (120 mM, pH 7, 60°C, 24 h, and 0.1 M decanoic acid) with brine, extraction with cyclohexanone (Rimington and Benson, 1967), and concentration of the organic phase. The 1H NMR spectra were acquired in CS2 containing tetramethylsilane and benzene-d6 as reference and lock. Carbon disulfide is a very attractive solvent for 1H NMR spectroscopy in this case due to the high solubility afforded hydrophobic tetrapyrrole macrocycles, absence of protons, low viscosity, and low boiling point (Lee et al., 1995; Bothner-By et al., 1996).
2.7. Mass spectrometry measurements
For analysis of etioporphyrin by mass spectrometry, an aliquot (100 μL) of the crude mixture obtained from reaction of
3. Results
3.1. Three porphyrins of distinct polarity
Each of the reactions (
The chemical structures of all the hydrocarbons are depicted in Scheme 4. In each case, the aqueous solution contained potassium phosphate buffer (0.5 M, pH 7), and the reactions were allowed to proceed under anaerobic conditions for 24 h at 60°C. In each case, the aminoketone and the dione were employed in equimolar quantities. The static dielectric constant of each neat compound (in liquid form) is as follows: dodecane (2.002 at 30°C), mesitylene (2.279 at 20°C), naphthalene (2.54 at 80°C), pentyl acetate (4.75 at 20°C) (Riddick and Bunger, 1970). A value for decanoic acid was not identified. The polarity of these bulk solvents thus is comparable to that of the inner, hydrocarbon region of a lipid bilayer. The solubility of water in each is as follows: dodecane (65 ppm at 25°C), mesitylene (0.0291 wt % at 20°C), and pentyl acetate (1.15 wt % at 20°C) (Riddick and Bunger, 1970). Values for decanoic acid and naphthalene were not identified.

Organic constituents employed in prototypical oil slicks.
To determine the yield of porphyrin, aliquots from the reaction mixtures affording uroporphyrinogens or coproporphyrinogens were diluted in 0.1 M HCl and treated with an ethanolic solution of I2. The I2 treatment converts the porphyrinogens to the corresponding porphyrins (Mauzerall and Granick, 1958). The yield of porphyrins was then determined by absorption spectroscopy. For the reactions to give etioporphyrinogens, the hydrophobicity of the macrocycles (i.e., lack of solubility in aqueous HCl) mandated an alternative analysis procedure. The crude reaction mixture was first extracted with cyclohexanone, then an aliquot of the cyclohexanone extract was dissolved in DMSO, and finally the DMSO solution was treated with an ethanolic solution of I2 followed by absorption spectroscopy.
The results for reactions at 30 mM are shown in Table 1. The yields for
The pairwise reactions (30 mM for each reactant) were carried out for 24 h at 60°C in aqueous solution (pH 7) in the presence or absence of organic constituents (“oil slick”).
Etio denotes etioporphyrins from the reaction of
Copro denotes coproporphyrins from the reaction of
Uro denotes uroporphyrins from the reaction of
Porphyrin yields upon chemical oxidation (with I2).
Aqueous solution containing 0.1 M decanoic acid.
Aqueous solution (0.4 mL) and dodecane (0.1 mL).
Aqueous solution (0.4 mL) and mesitylene (0.1 mL).
Aqueous solution (0.4 mL) and the five-component organic mixture (0.1 mL).
Not available.
Photographs of the reaction mixtures are shown in Fig. 1. In each case prior to reaction, the aqueous solution was homogeneous, the decanoic acid–containing mixture was turbid, and the dodecane- or mesitylene-containing mixture was biphasic, with a lower aqueous phase and an upper organic layer. In the case of the five-component organic mixture, several phase-separated organic globules were observed at the top of the aqueous solution.

Photographs of 30 mM reactions of diones and aminoketones before and after 24 h of reaction in various media prior to chemical oxidation with I2. Entry 1: reactions of
The reaction to form etioporphyrinogens proceeded as follows: after 24 h at 60°C, the reaction in aqueous solution resulted in a red droplet on top of the aqueous solution. The reaction in the presence of decanoic acid afforded phase separation, with red color localized predominantly in the upper layer. The reaction in the presence of dodecane or mesitylene afforded a relatively colorless aqueous solution beneath a red organic layer. The reaction in the presence of the five-component organic mixture afforded a single red globule on top of the colorless aqueous solution.
The reactions to form uroporphyrinogens or coproporphyrinogens were quite different. The two reactions were identical in the composition of the mixture upon visual inspection: the reaction in aqueous solution afforded a red homogeneous solution; the reaction in the presence of decanoic acid afforded a red turbid suspension; and the reaction in the presence of dodecane afforded a red aqueous solution beneath a colorless layer of dodecane.
Each reaction begins with colorless acyclic starting materials, and if the corresponding pyrrole, oligopyrromethanes, and porphyrinogen are the sole species formed, the final reaction mixture should be colorless given that each such species absorbs only in the ultraviolet region (Lindsey et al., 1987; Lindsey and Wagner, 1989). Indeed, porphyrinogens (206 nm) (Mauzerall, 1962), ketones (∼280 nm) (Jaffé and Orchin, 1962), and diketones (275 nm) (Morton and Rosney, 1926) are not colored. The broad experience in tetrapyrrole chemistry is that reactions of pyrroles leading to tetrapyrrole macrocycles typically afford colored reaction mixtures; the color stems from diverse non-porphyrin substances and can include some amount of porphyrin (Lindsey et al., 1987, 1994; Lindsey and Wagner, 1989; Li et al., 1997; Lindsey, 2000; Geier and Lindsey, 2001). The non-porphyrin substances include molecules that contain dipyrrin chromophores, which can form via oxidation or tautomerization processes (Lindsey and Wagner, 1989), as well as tripyrrins and bilins (Moss, 1987). The 2e−/2H+ dehydrogenation of a dipyrromethane unit gives the corresponding dipyrrin, whereas the 6e−/6H+ dehydrogenation of a porphyrinogen or bilane gives the corresponding porphyrin or bilin, respectively. The facile dehydrogenation under ostensibly anaerobic conditions reflects the electron-richness of the trialkyl- or tetraalkyl-pyrrole constituents, the deep thermodynamic well presented by the porphyrin relative to the porphyrinogen, and the presence in the reaction mixture of entities that function as mild oxidants, such as ketones and azafulvene moieties. In summary, the color of the reaction mixtures observed herein signals the presence of diverse reaction products, which can include intermediates along the path to tetrapyrrole macrocycles, by-products, and porphyrins.
3.2. Etioporphyrin formation across a wide concentration range
The reaction of Top: Yields as a function of reactant concentration for the reaction of • In aqueous solution, the yield increased from 0.5% to 2.6% over this concentration range. As the concentration increased, the size of a phase-separated red droplet increased, suggesting that the reaction largely occurred in the phase-separated layer (see photos). • In the presence of dodecane or mesitylene, the yield increased from 0.4% to 2.9% over this concentration range. The extent of coloration of the dodecane or mesitylene layer increased with increasing reaction concentration. • In the presence of decanoic acid, however, the yield increased from 1.4% to 10.8% over the 3.75–120 mM concentration range. A droplet was formed at the highest concentration of reactants (120 mM reaction), whereas a precipitate was observed for the 3.75–60 mM reactions. • In the presence of the five-component organic mixture, the phase-separated organic globules coalesced to give one globule, which became increasingly colored as the concentration increased. The yield of the reaction increased from 0.9% to 6.6% over the concentration range examined.
3.3. Characterization of etioporphyrin
The pairwise reactions of

Characterization data for etioporphyrin obtained from the reaction of
A key issue in porphyrin-forming reactions concerns whether the pyrrole self-condensation affords a unique porphyrinogen, or if exchange processes (i.e., scrambling) among pyrromethane intermediates alter the relative orientation of the pyrrole units (Taniguchi and Lindsey, 2012b). The reaction of dione
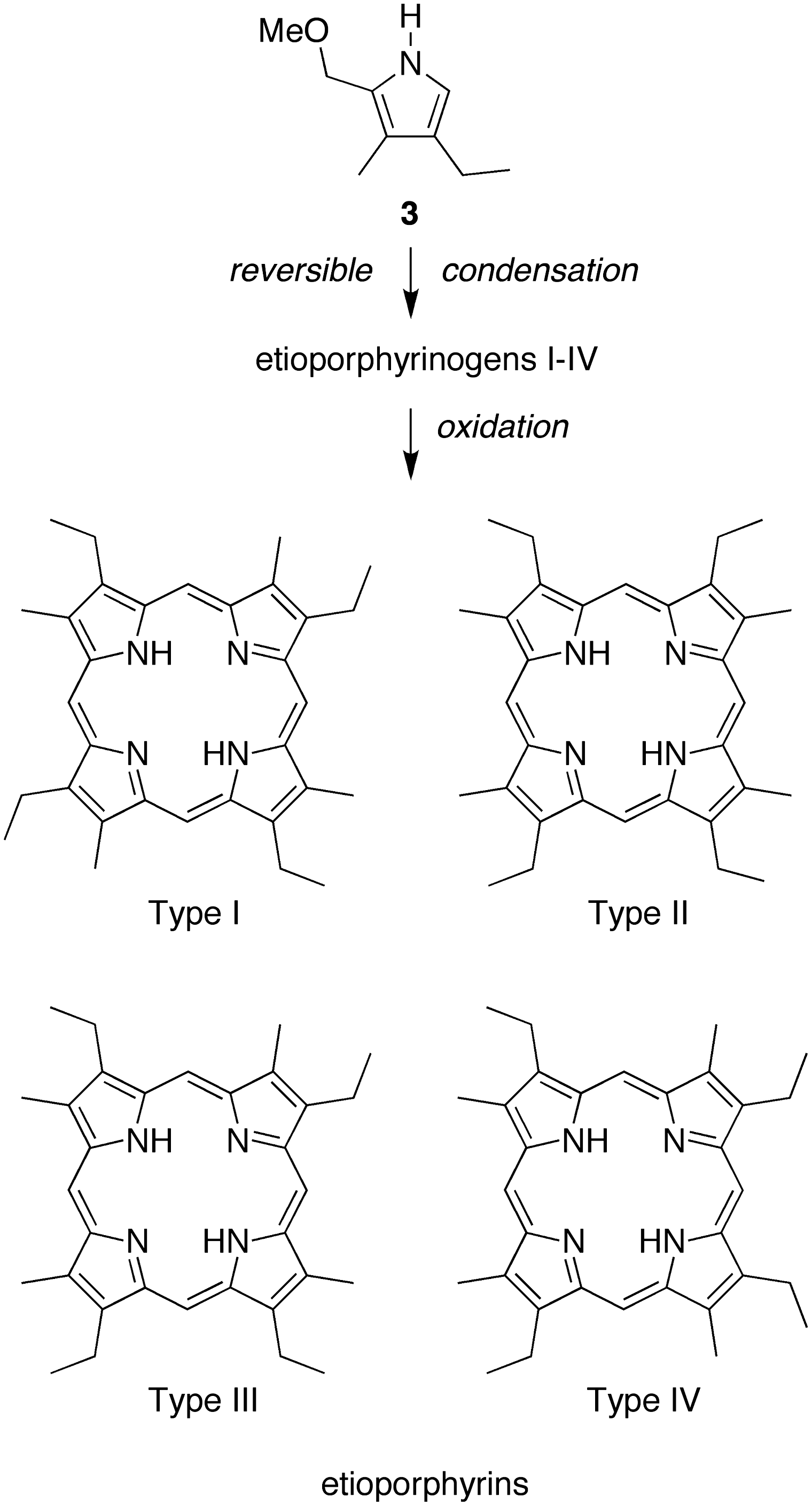
Route to form etioporphyrin isomers I–IV.
A clear distinction between etioporphyrin I and etioporphyrin isomers II–IV is observed upon examination of the resonance of the meso-protons in the 1H NMR spectrum. Etioporphyrin I affords a singlet (10.20 ppm) given that each meso-proton is flanked by one β-methyl group and one β-ethyl group; the molecule exhibits C
4h
symmetry (Taniguchi and Lindsey, 2012b). The spectrum of an authentic sample of etioporphyrin I is shown in Fig. 4A. By contrast, the spectrum of an authentic sample of etioporphyrin III exhibits multiple resonances from the meso-protons (Fig. 4B). The multiple resonances stem from the fact that each meso-proton is unique; the molecule has C
s
symmetry (Taniguchi and Lindsey, 2012b). Samples of etioporphyrin isomers II and IV are not commercially available. The crude etioporphyrin sample obtained upon reaction of

1H NMR spectroscopic data (at room temperature in CS2 containing benzene-d6
and tetramethylsilane) showing the signal(s) from the meso-protons. (
The physicochemical properties of the four etioporphyrin isomers are expected to be essentially identical. Such properties include partitioning into lipid media, absorption spectra, and excited-state characteristics (lifetime, energetics, intersystem crossing yield), all of which together determine photochemical properties in membrane assemblies. The identification of the existence of the etioporphyrin isomers is not important from a physicochemical perspective for etioporphyrin performance in a prebiotic milieu; however, the existence of such isomers reflects the reversibility of latter stages of the reaction, which is profoundly important in determining the number of tetrapyrrole macrocycles formed upon combinatorial reactions (Taniguchi and Lindsey, 2012b; Taniguchi et al., 2012). The mixture of etioporphyrin isomers observed herein indicates that the pyrrole precursor (
A number of reactions have been carried out to prepare etioporphyrins from substituted pyrroles. Such pyrroles (
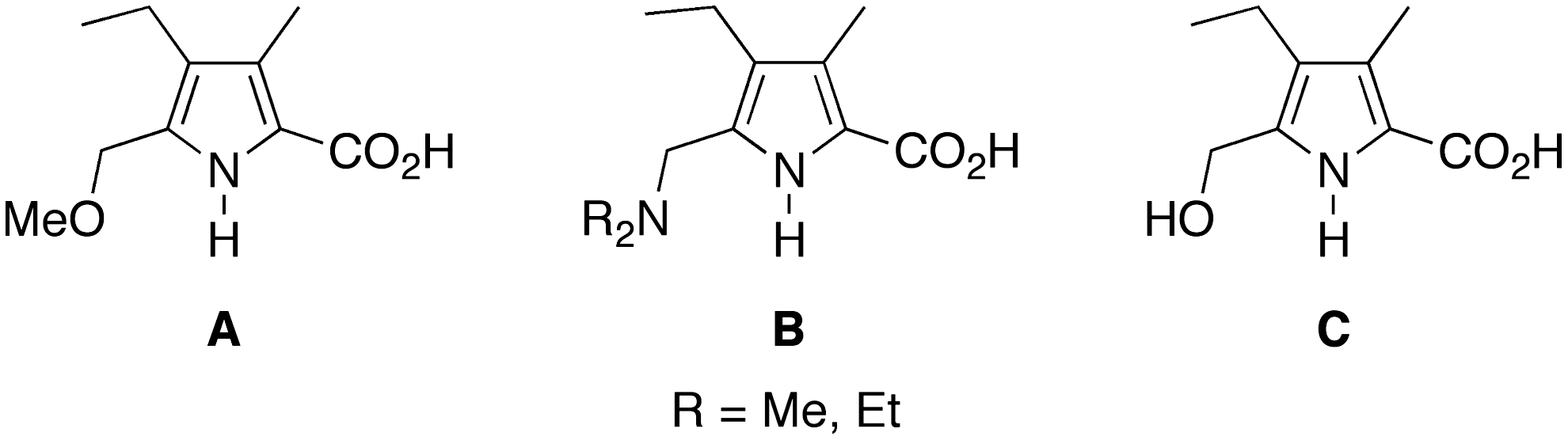
Pyrroles examined in routes to etioporphyrin I and other isomers.
3.4. Examination of the pyrrole-forming step leading to etioporphyrinogen
A key issue for structure-directed chemical processes—where molecular transformations occur due to intrinsic reactivity rather than external action by enzymes or other catalysts—concerns the integrity and yields obtained. In the prior studies of pairwise reactions leading to uroporphyrinogen and to coproporphyrinogen, a pyrrole by-product was formed in each case in addition to the pyrrole leading to the macrocycles (Lindsey et al., 2011). The pyrrole by-product is known as the Fischer-Fink pyrrole. Here, a sample of the reaction of

Competing Knorr and Fischer-Fink pathways to pyrroles.
4. Discussion
The inventory of constituents required for the origin of life remains uncertain. Much emphasis has been placed on the importance of amino acids, nucleotides, and sugars, all of which are soluble in water. More recent attention has been devoted to surfactants such as decanoic acid, which could form primitive vesicles and other assemblies (Apel et al., 2002). By contrast, the origin of hydrophobic compounds has received little attention. The importance of hydrophobic molecules stems in part from their potential function in lipid assemblies, including as photoactive pigments of photosynthesis (Mauzerall and Sun, 2003). The magnesium chelate of octaethylporphyrin, for example, has been shown to give light-driven transmembrane charge transport, which has been proposed as a model for a prebiotic photoreaction system (Sun and Mauzerall, 1996). Octaethylporphyrin is a very close analogue of etioporphyrin. Other structural roles of hydrophobic aromatic hydrocarbons have been proposed (Hud and Anet, 2000; Ehrenfreund et al., 2006).
4.1. Oil slicks and the origin of hydrophobic tetrapyrroles
To investigate reactions in the presence of a model oil slick, several biphasic media were employed here as models for primordial oil slicks on an underlying aqueous solution: a saturated aliphatic hydrocarbon (dodecane); an aromatic hydrocarbon (mesitylene); and a five-component mixture arbitrarily composed of decanoic acid, dodecane, mesitylene, naphthalene, and pentyl acetate. In addition, a monocarboxylic acid (decanoic acid) was employed that forms vesicles. All the reactions studied herein employed an aqueous phase that was buffered with 0.5 M potassium phosphate. Prior studies of analogous reactants in a non-phosphate buffer have also afforded porphyrinogens in yields of as much as 10% (Lindsey et al., 2011).
The reaction of
The reaction of
The reaction to give etioporphyrinogens in the presence of dodecane or mesitylene was not as expected: the yield was no higher than that in aqueous solution alone. The reaction in the five-component organic mixture, however, afforded an increase in yield, reaching 6.6% at the highest concentration (120 mM) examined. It may well be that the phase separation of reactants into dodecane or mesitylene creates a reaction environment that is too hydrophobic. Said differently, the reaction at certain steps is expected to benefit from a polar environment, which can stabilize transition states or intermediates that entail the formation of charged or polar species. Examples of such intermediates include an iminium ion and tertiary alcohol during pyrrole formation (Lindsey et al., 2011), and the 2-azafulvenium ion (Scheme 8) formed by displacement of the pyrrolic α-methoxy group. Reaction of a pyrrolic species with the neutral 2-azafulvene, derived by deprotonation of the 2-azafulvenium ion, may still entail formation of polar species. If so, such steps in the absence of specific catalysts would proceed very slowly in a hydrophobic medium such as dodecane or mesitylene, which have low dielectric constants. Thus, pyrrole formation may proceed in aqueous solution and then partition into the organic layer for subsequent oligomerization, or partitioning may only occur following oligomerization and cyclization. Accordingly, the necessity to stabilize transient polar species may result in reaction occurring largely at the aqueous-organic interface, in which case a thin oil slick containing surfactants may better support reaction chemistry of this type than a thick hydrophobic layer. In this context, it warrants emphasis that the role of organic constituents of an oil slick may be multifold, affording solubility, supporting interfacial reactions, and contributing to phase-separated nanoscale or microscale assemblies.
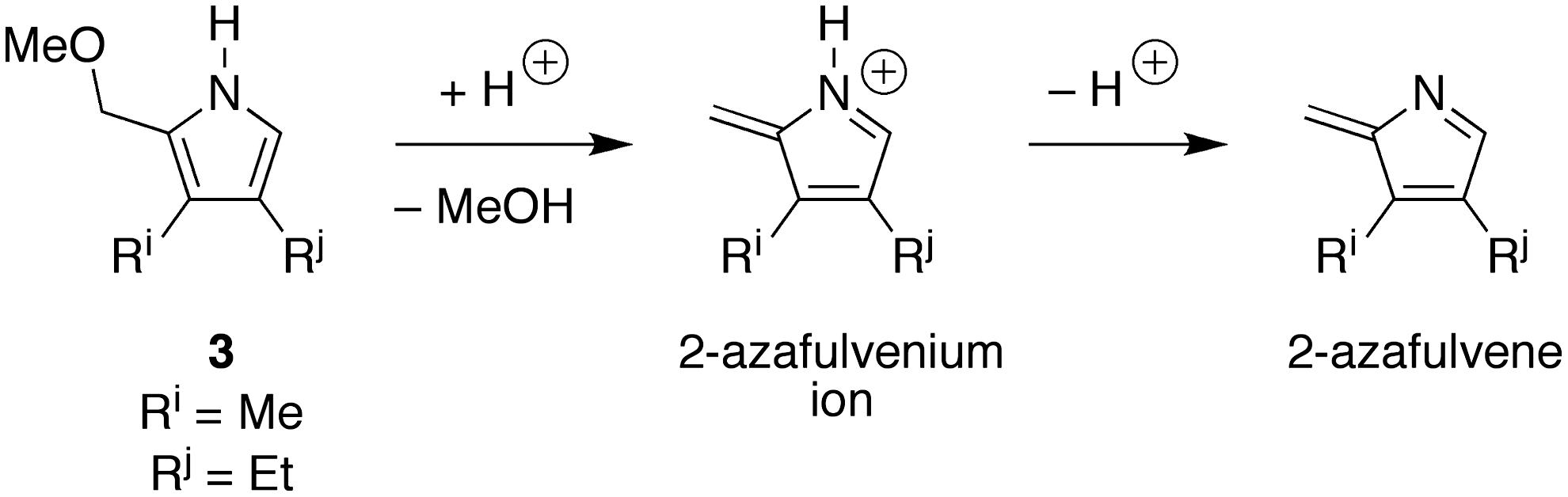
Putative polar or nonpolar reactive species derived from pyrrole
The yields of porphyrins remain <10% in all cases examined. The colored reaction mixture prior to oxidation with I2 in part consists of non-porphyrin species. Moreover, regardless of the ultimate yield of porphyrin, the reactions afford colored products. Oxidation (by chemical or photochemical means) of the crude mixture formed upon condensation of acyclic reactants affords further colored (non-porphyrin) species that absorb across the visible and into the ultraviolet spectral region. The facile formation of colored, non-porphyrin species suggests that such compounds could function as a shield of ultraviolet light (Cleaves and Miller, 1998), thereby protecting biorelevant molecules (e.g., nucleobases) from photodestruction. The distinction between a photoprotective layer and a layer that could impede photosynthesis depends on the wavelength and intensity of absorption; the latter of course depends on concentration. It would appear extremely unlikely that the flux of acyclic precursors (e.g.,
4.2. Origin of precursors
The route described in the chemical model proposed herein relies on a β-diketone (
In this regard, ALA (
The reactants
5. Outlook
The results described herein illustrate a very simple pathway from acyclic reactants (
Footnotes
Acknowledgments
This work was supported by a grant from the NSF Chemistry of Life Processes Program (NSF CHE-0953010). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation.
Author Disclosure Statement
The authors (Ana R. M. Soares, Masahiko Taniguchi, Vanampally Chandrashaker, and Jonathan S. Lindsey) have no conflict of interest or competing financial interests concerning the studies and results reported herein.
Abbreviations
ACP, acyl carrier protein; ALA, δ-aminolevulinic acid; CoA, succinyl-coenzyme A; DMSO, dimethylsulfoxide; ESI-MS, electrospray ionization mass spectrometry; fwhm, full width at half-maximum; 1H NMR, proton nuclear magnetic resonance; TCA, tricarboxylic acid.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.