
Abstract
This year marks the 50th anniversary of a proposal by Alex Rich that RNA, as a single biopolymer acting in two capacities, might have supported both genetics and catalysis at the origin of life. We review here both published and previously unreported experimental data that provide new perspectives on this old proposal. The new data include evidence that, in the presence of borate, small amounts of carbohydrates can fix large amounts of formaldehyde that are expected in an environment rich in carbon dioxide. Further, we consider other species, including arsenate, arsenite, phosphite, and germanate, that might replace phosphate as linkers in genetic biopolymers. While linkages involving these oxyanions are judged to be too unstable to support genetics on Earth, we consider the possibility that they might do so in colder semi-aqueous environments more exotic than those found on Earth, where cosolvents such as ammonia might prevent freezing at temperatures well below 273 K. These include the ammonia-water environments that are possibly present at low temperatures beneath the surface of Titan, Saturn's largest moon. Key Words: Astrobiology—Mineral adsorption—Origin of life—RNA world—Titan. Astrobiology 13, 391–403.
1. Introduction: The Status of the “RNA First” Hypothesis
Fifty years ago this year, Alex Rich proposed that RNA might be able to support both genetics (by being copied) and phenotype (by having catalytic activity) (Rich, 1962). Since then, many have suggested that if RNA first did both genetics and catalysis, then the origin of life would be free of the improbability of multiple biopolymers spontaneously emerging together (e.g., DNA and proteins) (Woese, 1967; Crick, 1968). In addition to avoiding this chicken-or-egg problem, an “RNA first” model for life's origins avoids the double improbability that multiple biopolymers would have coincidentally emerged already prepared to interact in an encoder-encoded relationship.
As our understanding of molecular details of the modern terran biosphere improved over the past half century, the RNA-first proposal became increasingly attractive. For example, the structures (White, 1976) and reactivities (Visser and Kellogg, 1978) of RNA cofactors found through terran biology (Fig. 1) argue that modern protein-catalyzed metabolism is a “palimpsest” written over a metabolism that originated when RNA was the only genetically encoded component of biocatalysis (Benner et al., 1989). Further, the RNA components of modern terran ribosomes appear to biosynthesize proteins (Moore and Steitz, 2003; Fox et al., 2012), and the antiquity of this biosynthesis is supported by paleogenetic experiments that resurrected components of 3-billion-year-old translation systems for study in the laboratory (Gaucher et al., 2003). Experiments with alternative forms of DNA (Yang et al., 2011) show that nucleic acids are better catalysts if they carry functional groups (Jäger and Famulok, 2004; Jäger et al., 2005; Hollenstein et al., 2009; Vaught et al., 2010). These functional groups are reminiscent of functional groups added post-transcriptionally to modern ribosomal and transfer RNA.
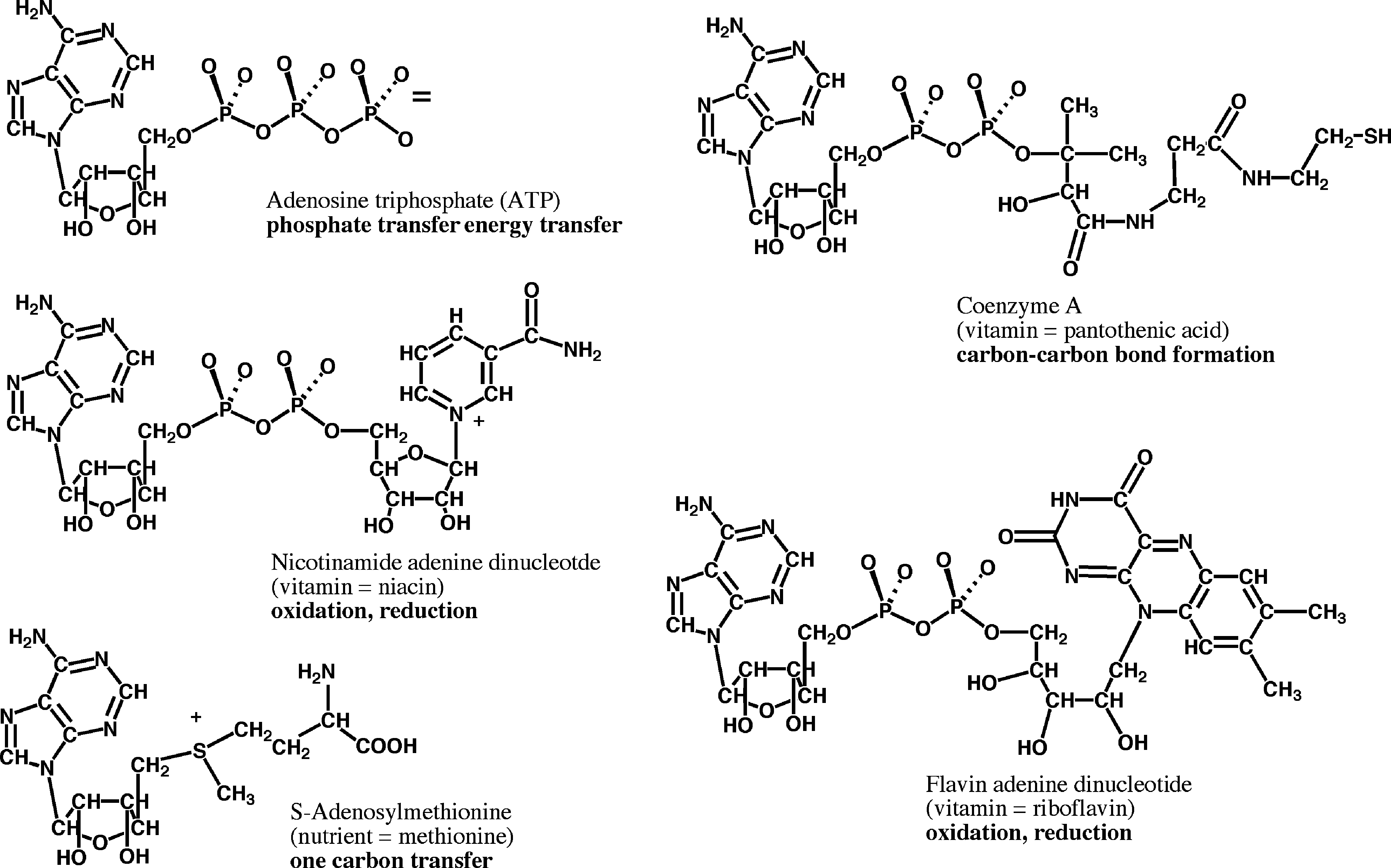
RNA cofactors are universal in terran metabolism, but their RNA components offer no useful reactivity. These are proposed to be vestiges of an episode of life on Earth when RNA was the only encoded component of biological catalysis. The weakest form of the RNA world hypothesis suggests only this episode existed. The “Strong RNA World hypothesis” holds that this episode was the first form of life on Earth. The weak hypothesis has broad support within the community today. The strong hypothesis runs afoul of the failure, so far, to observe the nonbiological synthesis of RNA in geologically plausible models for early Earth.
While none of these lines of argument compels an RNA-first model for the origin of life (the “Strong RNA World hypothesis”), other factors might. In particular, catalysis and genetics place contradicting demands on any single molecular system asked to do both. For example, catalytic molecules should fold, to surround a transition state. Genetic molecules should not fold, to allow them to template the synthesis of their complements. Catalytic molecules should have many building blocks, to create versatile catalytic potential. Genetic molecules should have few building blocks, to ensure that they are copied with high fidelity (Szathmary, 1992). Finally, any biopolymer having catalytic potential also has the potential to catalyze its own destruction. A genetic polymer able to evolve to catalyze the hydrolysis of the polymer is more problematic than (for example) a protein able to evolve to become a protease. The second simply destroys the end product of a genetic system; more of that product can later be made. The first, however, destroys the system itself.
RNA appears to be quite unusual among polymeric systems in its ability to strike a balance between the contradicting needs of catalysis and genetics. Indeed, many studies in many laboratories (including our own: Benner et al., 2010) seeking to replace the ribose-phosphate backbone in RNA by alternative backbones that might be more “prebiotic” have shown only how special RNA is in this regard. This supports the RNA-first view.
Balancing this are difficulties in conceiving of routes that might have made RNA prebiotically. Appropriately, Gerald Joyce called RNA “the prebiotic chemists' nightmare” (Joyce and Orgel, 1999). The late Robert Shapiro found RNA so unacceptable as a prebiotic target as to exclude it entirely from any model for the origin of life. Likewise, Stanley Miller, surveying the instability of carbohydrates in water, concluded that “neither ribose nor any other carbohydrate could possibly have been a prebiotic genetic molecule” (Larralde et al., 1995).
Many have attempted to awaken from the RNA nightmare by proposing alternative biomolecules to replace ribose, RNA nucleobases, and/or the RNA phosphate diester linkages, another source of prebiotic difficulty. These have encountered chemical challenges of their own. For example, attempts to create alternative RNA molecules that lack the repeating charge on the backbone phosphates show the need for a repeating backbone charge for a molecule to support Darwinian evolution (Freier and Altmann, 1997). Indeed, a repeating charge might be a universal feature of genetic molecules (Benner and Hutter, 2002).
Likewise, ribose is difficult to replace by more stable carbohydrate analogues (Freier and Altmann, 1997). For example, glycerol (Joyce et al., 1987) can substitute for ribose in the template-directed synthesis of DNA (Heuberger and Switzer, 2006, 2008) but remains problematic as a prebiotic species (Schneider and Benner, 1990). Other alternative carbohydrates (e.g., threose) (Horhota et al., 2005; Ebert et al., 2008) are as unstable as ribose. Still others (e.g., LNA) do not manage the contradicting demands of phenotype and genotype as well as RNA (Wang et al., 2005) and appear to be even more difficult to obtain prebiotically.
For these reasons, efforts have been made to construct “metabolism first” models (Decker, 1979; Copley et al., 2007) for a life that lacks any encoding biopolymer. Here, the hope is that reaction networks might capture enough features of Darwinian evolution to constrain “asphaltization,” recruit resources, and allow the emergence of encoding linear biopolymers in bioenvironments friendlier than a prebiotic soup. As attractive as this concept might be, it remains largely unexplored.
In view of this as the 50th anniversary of the RNA-first proposal, it is timely to review some of the evidence that makes the best case for the concept. The attributes of the proposal, in summary, include (a) the well-known roles of RNA within terran metabolism, (b) the catalytic potential of RNA, (c) the ability of RNA to meet the contradicting demands of genotype and phenotype, and (d) the lack of a competitive model that places metabolism first. Further, we may seek to extend this concept to aqueous environments more exotic than those found on Earth.
2. The “Discontinuous Synthesis Model” for RNA Prebiotic Synthesis
Actually, the prebiotic status of the RNA-first model might not be as problematic as implied above. A “Discontinuous Synthesis Model” has already been adumbrated in the literature that describes how oligomeric RNA might have been formed abiologically on Earth.
First, the atmosphere on early Earth undoubtedly contained CO2, H2O, N2, and some reduced hydrocarbons (e.g., CH4). That atmosphere was undoubtedly exposed to electrical discharge and ultraviolet irradiation. These conditions undoubtedly generated formaldehyde (HCHO) and hydrogen cyanide (HCN) (Cleaves, 2008), which undoubtedly “rained” onto Earth's surface. Because the formaldehyde carbon atom is intrinsically electrophilic (and therefore not prone to form bonds to other formaldehyde carbon atoms), only the Cannizzaro reaction, which converts two HCHO molecules to one molecule of formate (HCOOH) and one molecule of methanol (CH3OH), unproductively prevents higher concentrations. Formaldehyde may have accumulated in primitive oceans and lakes to millimolar concentrations (Pinto et al., 1980; Holland, 1984).
Further, little doubt exists that igneous rocks on Earth's early surface included serpentinizing olivines that generated both reducing power and alkaline environments. As discussed below, HCHO and HCN raining into these aquifers became (respectively) carbohydrates and formamide (Kim et al., 2011) by the formose process (Butlerow, 1861; Breslow, 1959). The alkalinity was undoubtedly eventually neutralized by atmospheric CO2 (Sleep et al., 2011).
Upon heating and/or drying, formamide becomes nucleobases (Saladino et al., 2004; Baross et al., 2007). Further, in some conditions, ribonucleosides are formed from nucleobases and carbohydrates (Fuller et al., 1972; Bean et al., 2007). Finally, in formamide solvents, ribonucleoside phosphates are formed from ribonucleosides and phosphate (Schoffstall, 1976; Schoffstall and Liang, 1985).
The Discontinuous Synthesis Model extends to the formation of oligomeric RNA, for which several reports now exist (Rajamani et al., 2008; Costanzo et al., 2009). Clay-catalyzed processes are reported to convert activated ribonucleotides to generate RNA up to 50 nucleotides in length (Huang and Ferris, 2006; Swadling et al., 2010). Oligomeric RNA molecules have been found to catalyze the synthesis of more RNA (Wochner et al., 2011). Finally, the Joyce laboratory has delivered some compelling multimolecular systems that show several RNA molecules interacting together capable of supporting Darwinian evolution (Lincoln and Joyce, 2009).
Who could ask for more? Each step, from CO2, H2O, and N2 to oligomeric RNA that might support Darwinian evolution, has been illustrated by at least one working example.
3. So Why Is the Discontinuous Synthesis Model Not Compelling “Proof” of the RNA-First Hypothesis within the Bio-Origins Community?
Leaving aside the elusiveness of “proof” for any interesting model in science, many in the bio-origins community do not accept the Discontinuous Synthesis Model because it offers only disconnected steps to create oligomeric RNA and does not proceed from beginning to end without human intervention. Further, various steps in the model suffer from problems, such as: The “asphalt problem.” Compounds like ribose containing C The “water problem.” Although water may be essential for life (Benner, 2010), Cairns-Smith (1982) noted that “all the major biopolymers are metastable in aqueous solution in relation to their (deactivated) monomers.” Even the monomers of RNA have problems, however. In water, deamination reactions convert cytosine to uracil, adenine to hypoxanthine, and guanine to xanthine, in each case destroying information carried by the nucleobase (Fig. 3). The glycosidic bonds holding the nucleobases to carbohydrates are thermodynamically unstable in water. Of course, the phosphodiester bonds that hold nucleosides together are also thermodynamically unstable in water. Indeed, examples of RNA molecules that catalyze the template-directed synthesis of RNA (Wochner et al., 2011) may not be acceptable as a “proof” of the RNA-first hypothesis in part because they work at high concentrations of Mg2+, which in turn catalyzes the hydrolysis of product RNA.
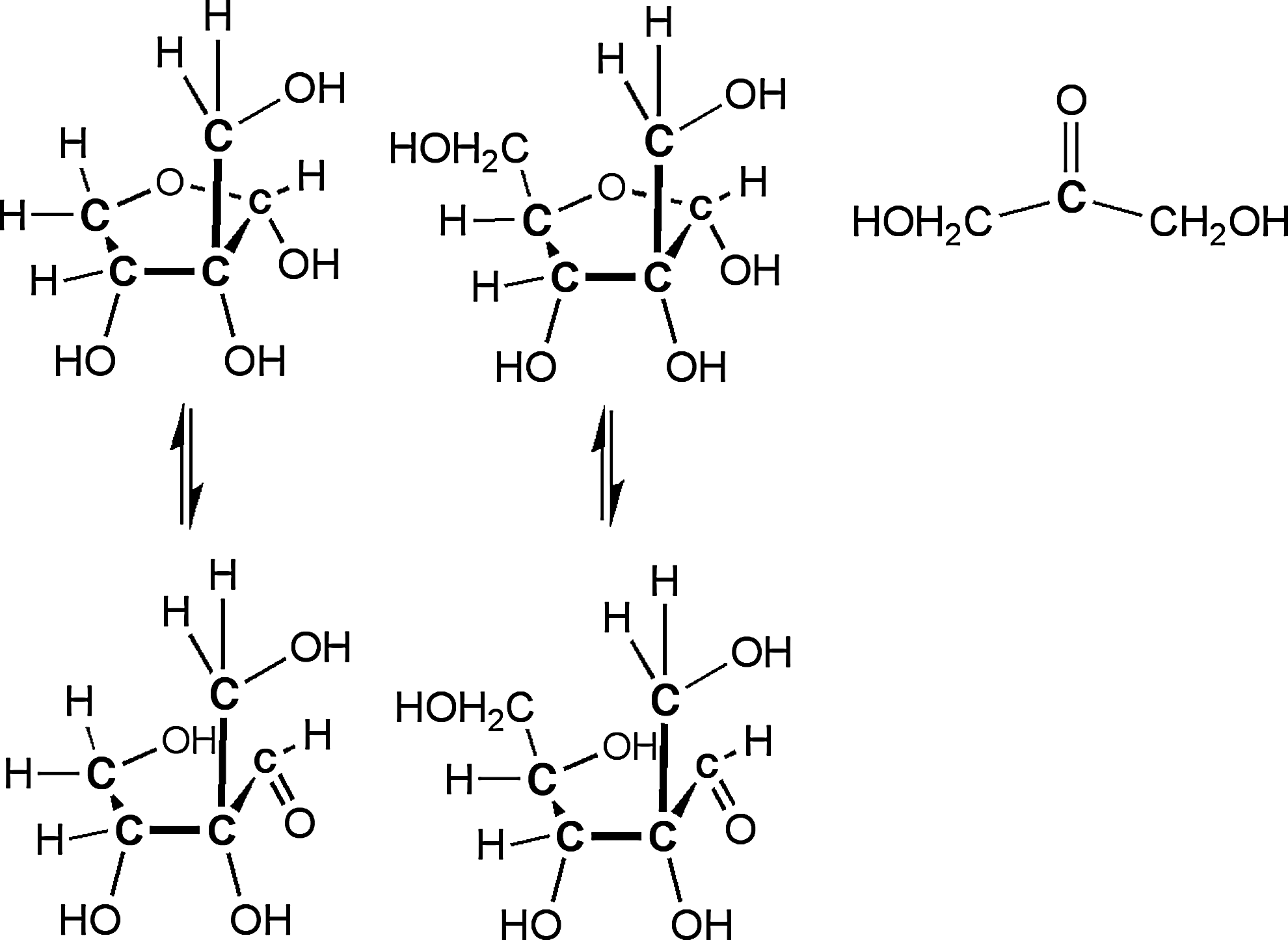
 O (carbonyl) groups are prone to enolize (Fig. 2) and then react as a nucleophilic enol with other carbonyl-containing species in aldol additions, creating molecular complexity. This evolution explains why the formose process is considered inadequate to have delivered carbohydrates to a prebiotic world; the carbohydrates formed are prone to react with each other under the alkalinity of the formose process to give asphaltic messes (Decker et al., 1982). For this reason, some steps in the Discontinuous Synthesis Model involving carbonyl compounds are successful only if reactive compounds are presented in a specific order in large amounts.
O (carbonyl) groups are prone to enolize (Fig. 2) and then react as a nucleophilic enol with other carbonyl-containing species in aldol additions, creating molecular complexity. This evolution explains why the formose process is considered inadequate to have delivered carbohydrates to a prebiotic world; the carbohydrates formed are prone to react with each other under the alkalinity of the formose process to give asphaltic messes (Decker et al., 1982). For this reason, some steps in the Discontinuous Synthesis Model involving carbonyl compounds are successful only if reactive compounds are presented in a specific order in large amounts.

The asphalt problem involves repetition of two reactions, an enolization and an aldol addition.

The water problem arises because ∼ half of the bonds in RNA are unstable thermodynamically with respect to hydrolysis in water. The hydrolytic deamination reactions are shown. The glycosyl bonds holding the nucleobases to the backbone, and the phosphate ester bonds, are also unstable with respect to hydrolysis.
4. Converting Discontinuous Models into Continuous Models for RNA Prebiotic Synthesis
If we accept RNA as our goal, the next step requires us to improve the Discontinuous Synthesis Model by reducing the number of steps requiring human intervention. Let us consider the asphalt problem associated with the prebiotic production of ribose.
One commonly cited prebiotic reaction sequence that converts formaldehyde HCHO into mixtures of carbohydrates is known as the formose process (Butlerow, 1861; Breslow, 1959). The formose process incubates formaldehyde with saturated calcium hydroxide at pH ∼12, normally at elevated temperatures (60–80°C). These high pHs can be provided by the serpentinization of igneous olivine.
Small amounts of glycolaldehyde are also made via electrical discharge through moist CO2 atmospheres (Löb, 1913). Glycolaldehyde helps initiate the formose process (Ricardo et al., 2006; Kim et al., 2011). However, the formose carbohydrates contain carbonyl groups. Therefore, in alkaline environments arising from serpentinizing olivines, absent human intervention, formose carbohydrates react to form “asphalt” (Decker et al., 1982).
For formose carbohydrates to avoid this fate, the pH of their environment must drop at least 3 pH units below the pH where they are initially formed. Beneath the CO2-containing atmosphere of early Earth, the pH of serpentinizing aquifers would certainly have dropped, to near neutrality according to some models (Sleep et al., 2011). If the formose carbohydrates could have survived to this point, and if they then had access to amine-containing organic molecules, formose carbohydrates may have formed imines and derivative products, including perhaps amino acids (Weber, 2001). Given strongly dehydrating conditions, they may even have combined with nucleobases to form nucleosides (Fuller et al., 1972; Bean et al., 2007), nucleoside phosphates (Schoffstall, 1976; Schoffstall et al., 1982; Schoffstall and Liang, 1985), and RNA (Huang and Ferris, 2006; Swadling et al., 2010).
However, for us to consider such optimistic scenarios, the formose carbohydrates needed to avoid “asphaltization” in alkaline media long enough to be rescued by a pH drop. Accordingly, Kim et al. (2011) looked for ways for useful carbohydrates (such as ribose and other pentoses) to have accumulated in alkaline environments. They began by considering other minerals in alkaline aquifers surrounding serpentinizing rocks. They noted that the same igneous rocks that carry olivines often also carry igneous borate minerals (e.g., tourmalines). These are also easily weathered to create borate-rich alkaline washes. Death Valley is a modern example of an intermountain valley whose chemistry is dominated by these processes; its alkaline soda lakes have highly concentrated borate in a desert environment conducive to dehydration reactions.
Borate binds to 1,2-diol units in organic molecules (Fig. 4). This is especially true when the two hydroxyl groups are presented on a ring in a cis-conformation preorganized to point in the same direction, toward borate. Many carbohydrates present preorganized hydroxyl groups in their cyclic “hemiacetal” forms (Fig. 4). The borate complex of ribose is especially stable because of a network of hydrogen bonds to the borate complex, a network made possible by its particular stereochemistry (Fig. 4). If equilibration is possible, the borate complexes of ribose and two pentuloses (ribulose and xylulose) are expected to come to dominate all other five-carbon carbohydrates in a mixture at equilibrium.

Borate influences the reactions of formaldehyde and glycolaldehyde to generate pentoses that bond borate in their cyclic forms, which have no electrophilic CO carbon.
But can pentoses be formed in the presence of borate under conditions where borate would stabilize them after they are formed? Our earlier work showed that they could (Ricardo et al., 2004). Glycolaldehyde (
Glyceraldehyde (
Thus, when glycolaldehyde and HCHO are mixed under alkaline conditions in the presence of borate, pentoses (ribose, arabinose, xylose, and lyxose) are formed (Fig. 4). In our proposed path, glycolaldehyde first forms an enediol
Borate moderation of the formose process has a “good news-bad news” aspect. As “good news,” borate manages its asphalt problem: it allows useful carbohydrates to be made and accumulate in a single pot without the need for human intervention. However, borate complexation also prevents the pentoses formed from enolizing as the next step to fix more HCHO. Thus, as “bad news,” borate appears to limit the amount of pentoses that might have accumulated on early Earth to half the amount of glycolaldehyde formed in the atmosphere. This would seem to generate too little ribose to comfortably support nucleoside synthesis and would waste the large amounts of HCHO expected to rain out from prebiotic atmospheres into serpentinizing rocks.
Fortunately, borate acts to mitigate this limitation. For example, relatively weak coordination by borate to the 3,4-diol unit of erythrulose directs its enolization toward the first carbon in the erythrulose chain (“carbon 1”) (Fig. 5). Further, this coordination directs the addition of HCHO to the more hindered carbon of the enediol, leading to a pair of diastereomeric branched pentoses (
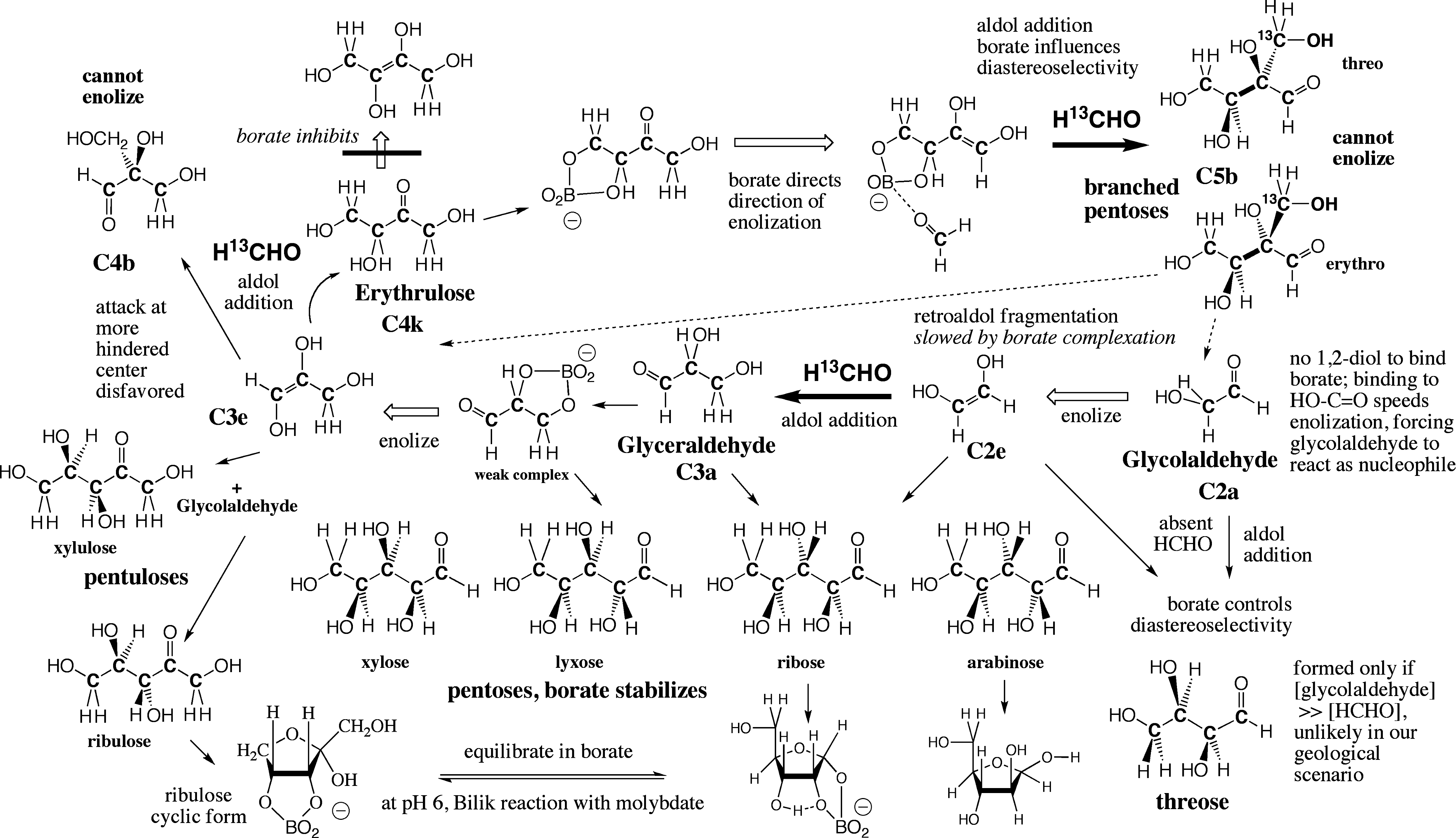
The borate-moderated formose process and the production of pentoses. Features shown include: (a) Borate binds to 1,2-dihydroxy units; shown are weak complexes with glyceraldehyde and erythrulose in their linear forms, modulating their reactivity, and strong complexes to ribose and ribulose in their cyclic forms, which drive their accumulation under formose conditions. (b) The structure of the especially strong borate-ribose complex (bottom). (c) Borate enhances the enolization of glycolaldehyde. (d) Borate coordination to the erythrulose diol drives enolization to the right and induces HCHO to add to the more hindered nucleophilic center to give branched pentoses. (e) The branched pentoses suffer retroaldol fission (dotted arrows) to generate more
Because of their branched structure, the
This analysis of the formose process suggests a conclusion: five-carbon carbohydrates accumulate in borate-moderated formose processes because they are the first species formed by one-carbon addition reactions that can form cyclic hemiacetals that present cis-diols preorganized for borate complexation. In contrast, glycolaldehyde, glyceraldehyde, and erythrulose do not accumulate because they do not present diols preorganized for borate complexation.
Given serpentinizing rocks weathering with igneous borates, a CO2 atmosphere, and rain containing abundant prebiotic HCHO and catalytic glycolaldehyde, these results force the conclusion that branched carbohydrates, ribulose, xylulose, and pentoses (including ribose) almost certainly formed on early Earth and accumulated as their borate complexes. Indeed, it is hard to imagine scenarios for early Earth where they did not accumulate.
5. What Other Minerals Might Replace Borate?
As attractive as borate is as a species to solve the asphalt problem in one part of the discontinuous model for the synthesis of RNA, it need not be compelling. Boron's high neutron cross section means that very little boron is synthesized in stars, and boron made rapidly adsorbs a neutron and becomes another element. Thus, boron on Earth came largely through nucleosynthesis outside the Sun in the Solar System by a process known as spallation. Thus, although boron is relatively scarce on Earth compared to its neighbors in the periodic table, it is not absent, making up ∼ 0.3–2.4 ppm of Earth's crust (Chaussidon and Jambon, 1994).
While this is sufficient to make borax and other minerals containing borate rather inexpensive today, this does not mean that borate minerals were present in useful amounts on early Earth. For example, Grew et al. (2011) suggested that tectonics on an early Earth might not have processed Earth's crust sufficiently to have concentrated enough borate in the lithosphere to yield local borate concentrations high enough to be useful at the time when life emerged.
Various facts make this argument less than compelling. Borate forms minerals poorly and is therefore concentrated in residual igneous melts, even in the absence of tectonics. This requires that Earth's inventory of borate, even early in its history, must have come to the surface even without plate tectonics. Borate would have been easily weathered from surface rocks, even if it were scattered. Therefore, borates would have been concentrated in the hydrosphere in any time of Earth's history, provided that a single ocean did not exclude land. 1 Direct evidence for early (if not very early) borate concentration is found in the tourmalines, a borate mineral, found in the 3.8 billion-year-old rocks in Isua, Greenland (Chaussidon and Uitterdijk-Appel, 1997).
The relative scarcity of borate caused us and others (Kim and Benner, 2010; Lambert et al., 2010) to seek alternative elements to see whether they could coordinate diols under conditions where such coordination might stabilize carbohydrates formed by formose processes. Two obvious candidates for mineral stabilization are silicate and aluminate, simply because they are very abundant.
Unfortunately, both silicate and aluminate proved to be relatively impotent, both for guiding the formose process and stabilizing its product carbohydrates (Kim and Benner, 2010; Lambert et al., 2010). This is attributable to the low solubilities of both silicate and aluminate in water except at a very high pH that renders carbohydrate enolization very rapid.
Interestingly, recent geochemical modeling has raised the possibility that strongly alkaline conditions arise in cold aqueous environments, where ammonia (NH3) acts as an antifreeze. Icy satellites of the outer Solar System, such as Titan, may feature such a high-pH water-ammonia subsurface ocean (Marion et al., 2012), beneath the methane oceans. Here silicate and aluminate could be soluble and could stabilize carbohydrates formed in prebiotic synthetic routes on Titan. However, at very high pH in the (still hypothetical) subsurface aqueous ocean on Titan, to compensate for the facile enolization of carbohydrates, any stabilizing effect achieved by complexation of carbohydrates to silicate and aluminate is best reinforced by lower temperatures. In the subsurface on Titan, low temperatures are most likely.
Iron is another candidate. It is likely involved in early protein cofactors (e.g., Burgess, 1990; Beinert, 2000), and it could have played a significant role in prebiotic chemistry (Cleaves et al., 2012). Although ferrous iron destroys nucleic acids in the presence of O2, it may have been a useful cofactor on the surface of an anoxic prebiotic Earth (Athavale et al., 2012). However, at moderate to high pH necessary for the formose process to occur, Fe(OH)2 and Fe(OH)3 form various insoluble oxides. Thus, it is doubtful that iron could have helped overcome the asphalt problem.
Accordingly, we examined a range of other mineral species that might dynamically associate with diols to create stabilized carbohydrates, without regard for their abundance in the crust. Phosphite, vanadate, germanate, arsenate, and arsenite anions all can form complexes with alcohol R-OH units, just like borate. The results are phosphite, vanadate, germanate, arsenate, and arsenite esters that are, at least in part, analogous to borate esters. A lower redox potential favors, of course, phosphite and arsenite over phosphate and arsenate.
To detect complexation, we mixed these anions with various diols (ethylene glycol, glycolaldehyde, glycerol, glyceraldehyde, dihydroxyacetone, sorbitol, cis-1,2-cyclohexanediol, anhydroerythritol, cysteamine, xylose, and ribose) at pH values from 4 to 14. Evidence for complexes was sought by seeking shifts in the nuclear magnetic resonance (NMR) spectra of these diols as the amounts of the anions were increased from 0:1 to 2:1 ester:diol. (See Fig. 6 for further experimental details.)
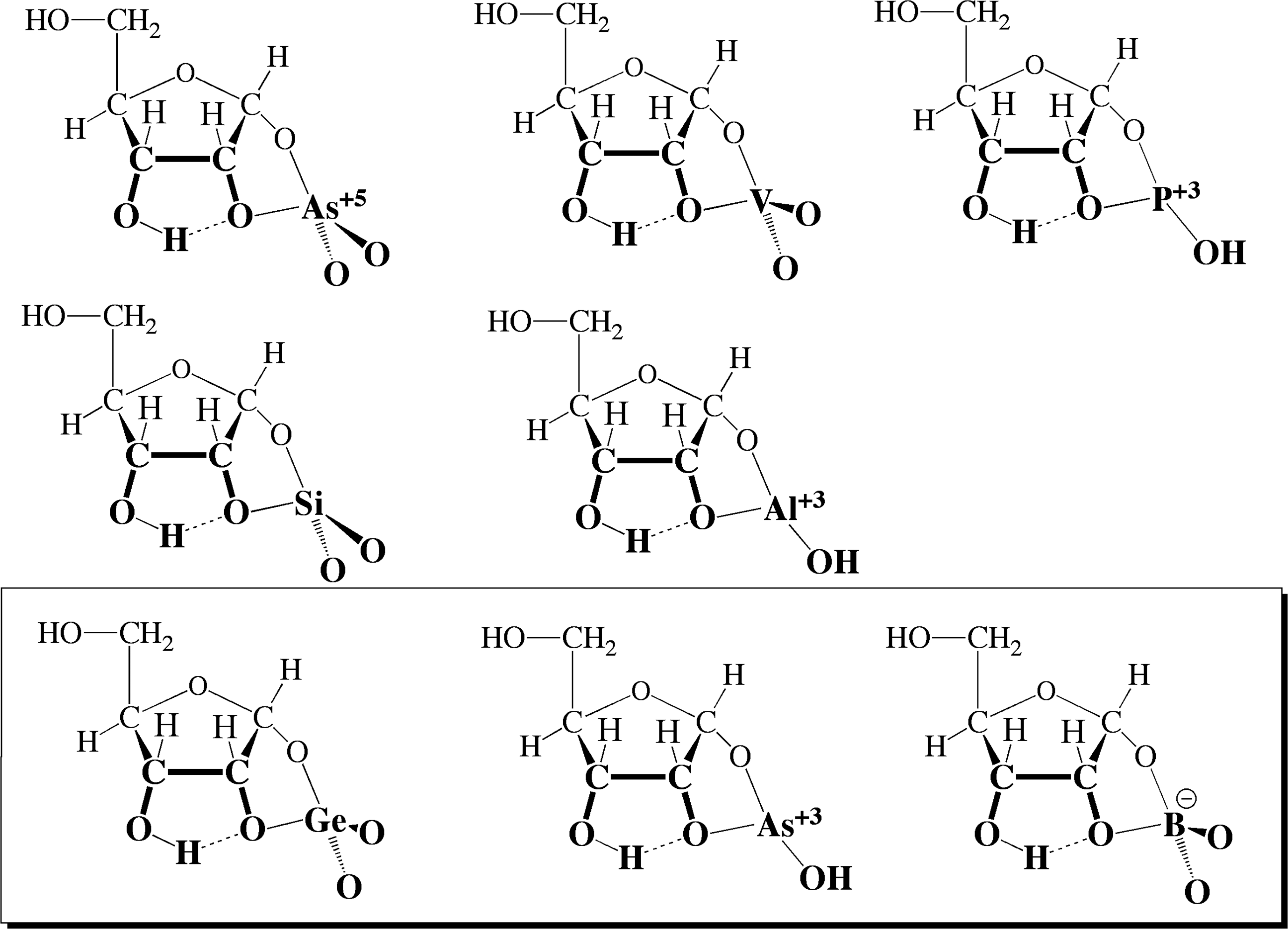
The capacity of esters other than borate (first row: arsenate, vanadate, phosphite; second row: silicate, aluminate; third row: germanate, arsenite, borate) to form complexes with diols (ethylene glycol, glycolaldehyde, glycerol, glyceraldehyde, dihydroxyacetone, sorbitol, cis-1,2-cyclohexanediol, anhydroerythritol, cysteamine, xylose, and ribose, the latter shown in the figure) was investigated. The experiments involved NMR to observe complexation, and pH-metry to observe if (and how many) protons are released upon complex formation. Germanate and arsenite complexes with anhydroerythritol, cysteamine, xylose, and ribose were observed in both NMR and pH experiments, but only at pH>12.5. Data not shown.
Further, upon complexation from many of these species, protons are released. This allows a measured decrease in pH to provide evidence for complex formation. To exploit such measurements, a mixture of anion and diol is titrated and used to confirm NMR observations. Further, the stoichiometry (1:1 or 2:1 diol:ester) of the anion:diol complex was assessed.
Results from these experiments are summarized in Fig. 6. Arsenite and germanate appeared (by NMR) to form stable complexes with ribose. In contrast, arsenate, vanadate, and phosphite did not. However, arsenite and germanate were observed to dissolve in water only at pH >12.5, and their complexes with ribose could not be observed at lower pH. The unavailability of dissolved arsenite and germanate at lower pH makes them unlikely candidates to have guided prebiotic carbohydrate synthesis. Although these solutions were in contact with atmospheric dioxygen in our experiments, the rates of oxidation of arsenite and phosphite are too slow to have altered these conclusions.
6. Obtaining Cycles
These results suggest that borate is unique among the mineral species that might prevent formose sugars in formose environments from becoming asphaltic long enough for them to enter environments where the pH is buffered by atmospheric CO2, where carbonyl compounds enolize only slowly. This returns us to the branched carbohydrate
Lowering the pH will release the borate from
This creates an apparent paradox. High borate is required to stabilize pentoses and guide their formation, in particular, the formation of
To resolve this apparent paradox, we sought to observe cycling at intermediate concentrations of borate. Based on an overall view of the detailed mechanism of the formose process (Kim et al., 2011), we speculated that, so long as formaldehyde is present, any enediol formed by any higher carbohydrate will be trapped by formaldehyde. This, in turn, would ensure that any enediol nucleophile formed via the enolization of a higher carbohydrate would not react with the electrophilic centers of other complex carbohydrates to create asphalt. Ultimately, all carbonyl-containing systems would eventually add HCHO molecules until their branching no longer permits enolization.
The constant delivery of formaldehyde from the atmosphere into an aquifer is entirely reasonable from a prebiotic geological perspective. However, this environment is not easily reproduced in the laboratory. If the experiment adds a large amount of formaldehyde initially at high pH, much of it will be consumed unproductively in the Cannizzaro reaction, as the rate of the Cannizzaro reaction is dependent on the square of the concentration of HCHO. The Cannizzaro reaction lowers the pH of the reaction mixture. If the pH is lowered to prevent the Cannizzaro reaction even at high HCHO concentrations, then the rate of enolization becomes too slow to measure conveniently on a laboratory timescale. 2
Alternatively, to simulate the slow rain-out of formaldehyde on a prebiotic Earth, we can add formaldehyde continuously. This might be done manually or with a chemostat, where the drop in pH due to the Cannizzaro reaction, or the drop in pH due to the complexation of a product carbohydrate and borate, signals the chemostat to add more formaldehyde (and more base).
These considerations suggest a relatively simple experimental setup. We begin at a high pH (ca. 12) so that enolization is rapid. Also rapid at high pH is the Ca2+-catalyzed Bilik reaction (Petrus et al., 2001), which rearranges branched non-enolizable carbohydrates to linear enolizable carbohydrates. Also at high pHs, the retro-aldol fragmentations of non-enolizable carbohydrates are rapid.
In these experiments, formaldehyde was initially present at concentrations low enough that the Cannizzaro reaction is negligible. Further, borate is present initially at modest concentrations; more borate is added over the course of the experiment, simulating the erosion of borate-containing rocks on prebiotic Earth.
Results are collected in Table 1. These experiments started with dihydroxyacetone (DHA,
Experimental Conditions Seeking Continuous Formaldehyde Fixation
In two control experiments, erythro-branched pentose (EBP,  O unit (Fig. 7). Borate complexes both, preventing their rapid retroaldol fragmentation. Thus, in these controls, only the products of retroaldol fragmentation and the Cannizzaro reaction could consume formaldehyde.
O unit (Fig. 7). Borate complexes both, preventing their rapid retroaldol fragmentation. Thus, in these controls, only the products of retroaldol fragmentation and the Cannizzaro reaction could consume formaldehyde.
Starting materials. Left: EBP. Center: hamamelose. Right: DHA. The carbons adjacent to the carbonyl group in EBP and hamamelose carry no acidic hydrogen atom, preventing EBP and hamamelose from enolizing; they can fix formaldehyde only following retroaldol reactions. In contrast, DHA has four acidic hydrogen atoms, enolizes rapidly, and therefore can rapidly fix formaldehyde from solution.
These facts set up the interpretation of the experiment. If formaldehyde is consumed in stoichiometric excess over the amounts of higher carbohydrate added, then we conclude that retroaldol fragmentation has occurred to create a “premetabolic cycle” to fix formaldehyde. In this calculation, we must only account as well for the amount of formaldehyde consumed by the Cannizzaro reaction to give methanol and formate.
The results (Fig. 8) suggest that formaldehyde was consumed in a premetabolic cycle but with different efficiencies. In the hamamelose control, three equivalents of formaldehyde (with respect to hamamelose) were consumed after 90 h. Starting with EBP (
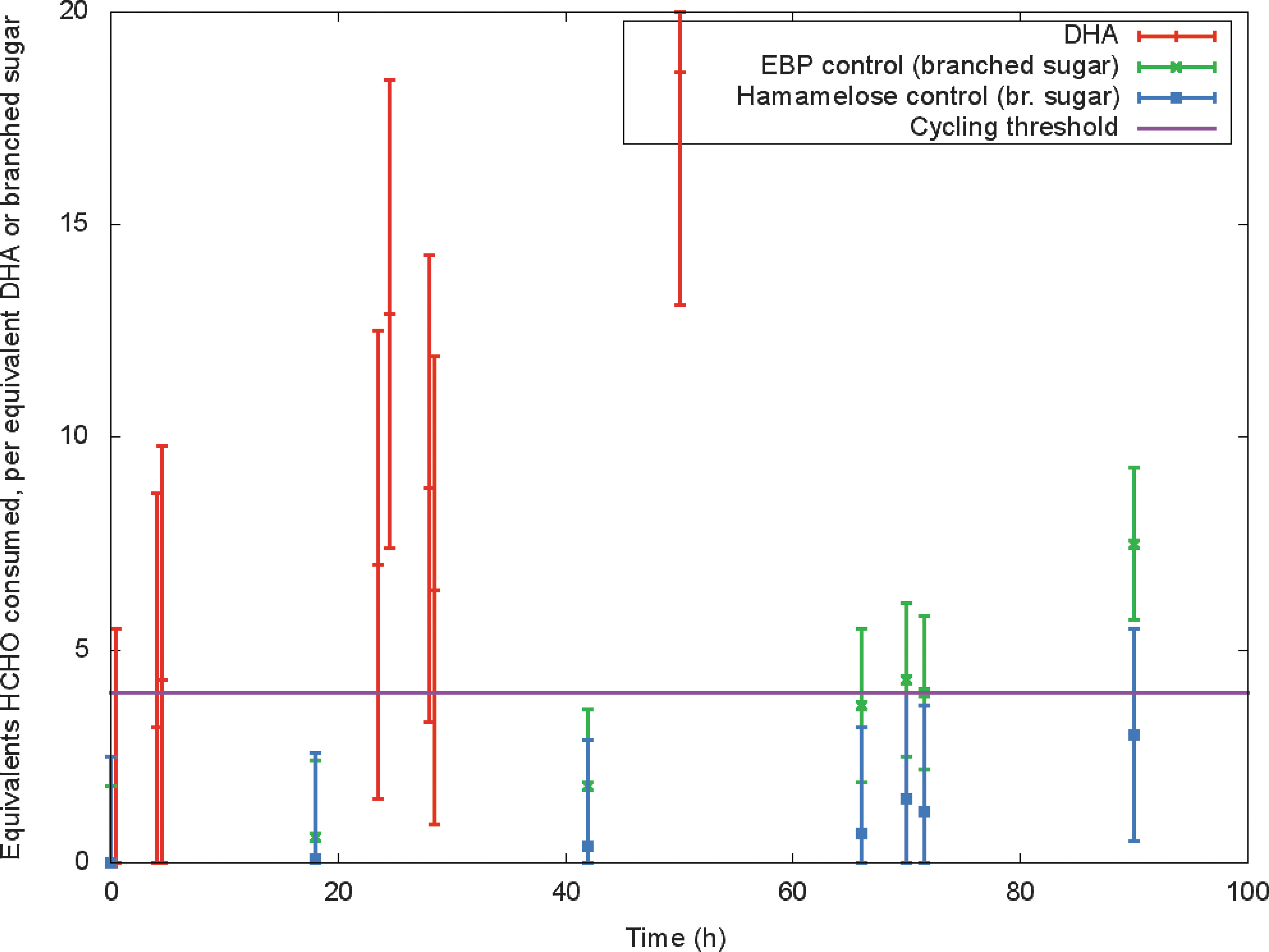
Amounts of formaldehyde consumed in base-catalyzed cycling with catalytic amounts of EBP, hamamelose, and DHA. (Color graphics available online at
If we assume that the formaldehyde (HCHO) consumed in the experiments where
While this conclusion is consistent with cycling, a large experimental error remains due to the background Cannizzaro reaction. For example, the DHA experiment started with 200 mM HCHO instead of the 50 mM HCHO used to initiate the experiments with
7. Simulating Geological Time
These experiments covered 50 h, a long time in the laboratory but a very short time geologically. Further, even with the controlled addition of reaction components for which a chemostat was used, these laboratory experiments do not necessarily reproduce well conditions on prebiotic Earth. Formaldehyde, glycolaldehyde, borate, calcium, and even base are likely to enter a natural aquifer only slowly, because atmospheric processes are slow, and the erosion of rocks is even slower. A good simulation needs a procedure that allows slow addition over weeks, months, or even years of unattended laboratory processing.
One way to simulate this in the laboratory may also resemble how it occurred on early Earth. For example, a “formaldehyde buffer” would be an unreactive species that is in equilibrium with formaldehyde in solution. As the formaldehyde is consumed, the buffering species would decompose to generate more formaldehyde. This would simulate slow addition.
Hydroxymethylsulfonic acid (HMSA) is a compound built from HCHO and sulfurous acid (H2SO3). On prebiotic Earth, sulfurous acid was almost certainly present from the reaction of water with sulfur dioxide, which in turn comes through volcanic discharge. HMSA in water is in equilibrium with formaldehyde and bisulfate
A buffer for the concentration of borate would also be desirable. A borate “buffer” would maintain a concentration of borate high enough to stabilize pentoses but not so high as to prevent cycling to generate multiple molecules of pentose from scarce glycolaldehyde.
Here again, mineral borates have some advantages. An indefinitely large amount of a slightly soluble borate mineral provides an indefinitely large reservoir of borate without having the concentration of dissolved borate be any higher than allowed by the solubility constant of the borate salt. As dissolved borate becomes bound to pentose as it is formed, more borate dissolves from the precipitate to maintain the concentration of free borate constant.
Calcium borate (colemanite) is an example of a salt that does this. Curiously, the sulfite released from HMSA as it dissociates to replenish consumed formaldehyde forms a salt (calcium sulfite) that is less soluble than calcium borate. Thus, if the formose reaction proceeds in solution above colemanite minerals, with formaldehyde released from HMSA to maintain the concentration of formaldehyde, and as the borate is consumed by complexation to carbohydrates as they are formed, the colemanite decomposes to generate more borate, and calcium sulfite precipitates the calcium released. The overall reaction sequence is proposed as follows:
8. Conclusion
It has been a half-century since Alexander Rich initiated the search to identify conditions where components of RNA might have emerged without human intervention. The following inferences have emerged from this search: (a) Asphaltization of pentoses can be avoided for substantial periods of time by complexation to the borate anion. (b) No other mineral anion is as satisfactory as borate to stabilize pentoses across the critical range as pH drops from that delivered by serpentinizing rocks to a pH where enolization of carbonyl compounds is too slow to create significant asphaltization. However, in more exotic environments (such as the hypothetical subsurface aqueous oceans on Titan, at high pH and low temperature created by large amounts of ammonia), silicate might serve as well. (c) Pentoses can be viewed as the natural products of the formose process in the presence of borate because they are the first carbohydrates to be formed in that process that have cyclic forms able to bind borate tightly. (d) Indeed, as long as formaldehyde is present in an aquifer, it prevents asphaltization because any enolized higher carbohydrates react with formaldehyde before they can react with any electrophilic center of any other higher carbohydrate. (e) However, simulating a low but constant concentration of formaldehyde, and a low but constant concentration of borate, is challenging over long periods of time that might reproduce geological time on early Earth. There is no convenient way to maintain high pH at high concentrations of HCHO, as the Cannizzaro reaction lowers both [HCHO] and pH through the formation of methanol and formate. (f) This notwithstanding, imperfect experiments suggest that, if low but constant concentrations of both formaldehyde and borate are arranged, a cycle can be achieved that fixes many molecules of abundant formaldehyde for each molecule of scarce glycolaldehyde.
These facts give further support to the Rich hypothesis. Now, attention must return to the geological environment on early Earth. Of special interest are models that show that the inventory of water on early Earth was sufficiently high as to make dry land scarce (Kirschvink et al., 2006). Should this have been the case, other locales for this process may have been available in the early Solar System, most notably on early Mars.
9. Experimental Section
9.1. Ester-diol complexation experiments
The capacity of esters other than borate (phosphates prepared from phosphoric acid, vanadate prepared from vanadic acid, germanate, sodium arsenite, mono- and dibasic sodium arsenate) to form complexes with diols (ethylene glycol, glycolaldehyde, glycerol, glyceraldehyde, dihydroxyacetone, sorbitol, cis-1,2-cyclohexanediol, anhydroerythritol, cysteamine, xylose, and ribose) was investigated. All reagents were obtained from Sigma-Aldrich. Experiments were executed at room temperature. Proton NMR was used to observe complexation, and pH-metry was used to observe if (and how many) protons were released upon complex formation. Proton NMR spectra were obtained on a Varian Mercury 300 NMR spectrometer from solutions of constant volume (0.6 mL) in D2O (Sigma-Aldrich) with diol concentrations that were 50 mM in all solutions. Complex formation was confirmed by measuring the pH of solutions of constant volume (1 mL) and of concentrations ranging from 250 mM ester:0 mM diol (1:0) to 250 mM ester:250 mM diol (1:1) in H2O.
9.2. Continuous formaldehyde fixation experiments
All reagents were from Sigma-Aldrich unless indicated otherwise. In each experiment, 1 mmol (final concentration 10 mM) of a reactive carbohydrate (EBP, hamamelose, and DHA, respectively) was added to a 100 mL solution containing sodium borate (100 mM boron, obtained from Fisher Scientific), formaldehyde (HCHO, 200 mM in the DHA experiment, 50 mM otherwise), and calcium hydroxide [Ca(OH)2, 10 mM, obtained from Fisher Scientific] in H2O. The DHA experiment was done at 45°C and pH 12.3, maintained by addition of an alkaline feed solution containing 100 mM sodium hydroxide (NaOH, Fisher Scientific), 10 mM Ca(OH)2, and 200 mM HCHO in H2O. The EBP and hamamelose experiments were done at 50°C and pH 9.8, adjusted after 18 h to 12.4 and 12.1, respectively, with concentrated NaOH. The pHs were maintained at 12.4 and 12.1, respectively, by addition of an alkaline feed solution containing 100 mM NaOH, 10 mM Ca(OH)2, and 200 mM HCHO in H2O. HCHO concentrations were monitored by sampling 20 μL of the experiments, diluted 100-fold in H2O (final volume 2 mL) in 15 mL tubes. To these 2 mL, 300 μL of a 5% chromotropic acid solution (prepared from chromotropic acid disodium salt, Sigma-Aldrich) and 3 mL of concentrated sulfuric acid (ACS plus, Fisher Scientific) were added. Following the procedure described by Georghiou and Ho (1989), the tubes were thoroughly mixed by vortexing, sealed with Parafilm, placed in a boiling water bath for 1 h, left to cool, and mixed by vortexing again. Absorbances were measured at 580 nm with the use of 1.0 cm optical glass cells.
Footnotes
Acknowledgments
This work was supported by the NASA Exobiology program (NNX08AO23G) and in part through the NASA Astrobiology Institute (via the Jet Propulsion Laboratory, RSA1371457). M.N. was an intern from the Institut Supérieur de l'Aéronautique et de l'Espace in Toulouse. This work was his master's thesis.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations
DHA, dihydroxyacetone; EBP, erythro-branched pentose; HMSA, hydroxymethylsulfonic acid; NMR, nuclear magnetic resonance.
1
2
We might, of course, avoid these experimental problems by simply assuming that, given geological time, these processes will have occurred. However, this would not create a model that would be widely accepted by many in the community, as it does not generate an observation that can be made today.