
Abstract
Universal and species-specific quantitative polymerase chain reaction–based methods were employed to compare the effectiveness of four distinct materials used to collect biological samples from metal surfaces. Known cell densities of a model microbial community (MMC) were deposited onto metal surfaces and subsequently collected with cotton and nylon-flocked swabs for small surface areas and biological sampling kits (BiSKits) and polyester wipes for large surface areas. Ribosomal RNA gene-based quantitative PCR (qPCR) analyses revealed that cotton swabs were superior to nylon-flocked swabs for recovering nucleic acids (i.e., DNA) from small surface areas. Similarly, BiSKits outperformed polyester wipes for sampling large surface areas. Species-specific qPCR results show a differential recovery of rRNA genes of the various MMC constituents, seemingly dependent on the type of sampling device employed. Both cotton swabs and BiSKits recovered the rDNA of all nine of the MMC constituent microbes assayed, whereas nylon-flocked swabs and polyester wipes recovered the rDNA of only six and four of these MMC strains, respectively. The findings of this study demonstrate the importance and proficiency of molecular techniques in gauging the effectiveness and efficiency of various modes of biological sample collection from metal surfaces. Key Words: Sampling—Recovery—Biomolecules—Clean room—DNA. Astrobiology 13, 189–202.
1. Introduction
Most studies that address the microbiological sampling of surfaces are limited to cultivation-based approaches, such as the bacterial spore assay (Kirschner and Puleo, 1979) or microscopy-based methods (Brown et al., 2007). Due to the noncultivable nature of most microbial species (Vesley et al., 1966; Kaprelyants et al., 1993; Amann et al., 1995; Kell et al., 1998; Oliver, 2010), culture-based assays often result in only partial identification of the microbial population present. This limitation is somewhat overcome by using rRNA gene cloning-based approaches, which require syntheses of clone libraries and subsequent sequencing of each distinct clone type (La Duc et al., 2009a).
The days of cultivation-dependent assays, light microscopy, and phenotypic microbial classification are drawing to a close and yielding to the ever-evolving fields of molecular biology (Tsourkas and Bao, 2003; Brodie et al., 2007; DeSantis et al., 2007; Cerqueira et al., 2008; Shendure and Ji, 2008; Yoo and Lee, 2008; Yergeau et al., 2009; Vaishampayan et al., 2010; Dwivedi and Jaykus, 2011). Whole-genome sequencing and a multitude of rDNA sequence-based classification schemes (Hugenholtz, 2002; Wilson et al., 2002; Breitkopf et al., 2005; Vaishampayan et al., 2010), along with significant advances in the specificity and sensitivity of molecular technologies, are being reported on a seemingly daily basis (Buttner et al., 2004a; Holman et al., 2009; Girones et al., 2010; Grindberg et al., 2011). All the while an efficient and reproducible means of collecting biological materials (e.g., cells, nucleic acids) from low-biomass surfaces and aqueous samples remains elusive. The low-biomass nature of spacecraft sample surfaces yields an average of ≤105 16S rRNA genes per square meter sampled (Cooper et al., 2011). To achieve the greatest total yield, while inferring the most accurate spectrum of the microbial diversity present from samples low in biomass, methods of collection and processing must place particular emphasis on the purification of highly informative biomolecules (e.g., DNA).
Since the Viking missions to Mars in the 1970s, NASA has adhered to standard procedures for the microbial examination of spacecraft hardware (NASA, 2010), wherein cotton swabs (and polyester wipes for larger surface areas) are the standard device for collecting samples from surfaces (Kirschner and Puleo, 1979). Recently, culture-based studies have demonstrated that cotton swabs and biological sampling kits (BiSKits) yield the highest recovery of bacterial endospores from small (Rose et al., 2004) and large (Buttner et al., 2004a, 2004b; Da Silva et al., 2011) surface areas, respectively. However, Probst et al. (2010) concluded that nylon-flocked swabs outperformed cotton swabs in collecting Bacillus spp. endospores from stainless steel and other surfaces. Similarly, a more efficient recovery of cultivable contaminants from pharmaceutically relevant surfaces was reported with nylon-flocked swabs (Dalmaso et al., 2008).
To date, there are very few published in-depth assessments of strategies for collecting biological samples from low-biomass surfaces. In this study, experiments were designed and executed to (a) test the effectiveness of several sampling devices in recovering cells and naked nucleic acids from surfaces and (b) assess the differential recovery of rDNA of members of a mixed microbial consortium via species-specific quantitative polymerase chain reaction (qPCR).
2. Materials and Methods
2.1. Model microbial community
The model microbial community (MMC) used in this study comprised 11 distinct microbial species (Kwan et al., 2011). These 11 MMC constituents span all three domains of life and comprise a variety of metabolisms, modes of respiration, and cellular morphotypes. Experiments were devised to test the performance of four sampling devices in recovering the rDNA of nine (Table 1) targeted MMC microbial strains. Though not considered in the qPCR-based recovery assays, the purified endospores of Bacillus pumilus SAFR-032 and vegetative cells of Aureobasidium pullulans, a fungal strain, were included to elucidate the effect of nonspecific interactions and competitive influence of nontargeted microbial taxa on recovery efficiencies. Most of the MMC cell lines were previously isolated from spacecraft and associated clean room surfaces (La Duc et al., 2003, 2007; Bruckner et al., 2008; Newcombe et al., 2008), and the others were procured from various culture collections. Detailed procedures addressing cultivation conditions, the mixing of the microbial strains (105 cells each), and their storage at −80°C have been previously described (Kwan et al., 2011).
Working concentration for all primers 9 μM.
Reaction mixture (25 μL/sample) consisted of 12.5 μL 2×SYBR Green Supermix (Cat # 25012; Bio-Rad, Hercules, CA), 10.5 μL nuclease free water, 0.5 μL forward primer, 0.5 μL reverse primer, and 1 μL template DNA.
Cycle parameters for two-step amplification with melt curve: initial denaturation (95°C, 3 min), denaturation (95°C, 10 s), annealing+extension (see table), melt (65–95°C, 5 s at 0.5°C increments).
Total cycles per qPCR run: 40.
Equipment: Bio-Rad CFX96.
2.2. Deposition of MMC onto stainless steel
Stainless steel (grade 304-020) coupons (25 cm2) and sheets (2500 cm2) were precision cleaned (Venkateswaran et al., 2004) and remained individually wrapped in spacecraft-qualified packaging (3M Static Shielding Bag SCC 1000; 3M, St. Paul, MN) until use. For deposition, metal coupons were aseptically kept in sterile Petri dishes, and the metal sheets on pre-sterilized trays. Appropriate aliquots of MMC were thawed, pooled, harvested via centrifugation (3000g, 15 min), and resuspended in UltraPure Gibco water (Cat. # 10977, Invitrogen, NY). One-milliliter aliquots of MMC suspension were deposited (in 4×5 rows of 50 μL spots) onto metal surfaces with a multichannel pipettor. Regardless of whether a given surface area was small (25 cm2) or large (2500 cm2), the same cell/spore density of 106 MMC cells was deposited. The deposition of MMC onto the metal surfaces was performed in a biological safety cabinet that had been previously exposed to UV (254 nm) for 1 h. Once inoculated with MMC, the metal coupons and sheets were allowed to air-dry in the biohood for 24 h at room temperature. Witness plate negative controls were included in the biohood to monitor experimental artifacts resulting from fallout contamination.
2.3. Sampling materials
The sampling materials evaluated in this study were cotton swabs (Puritan Medical Products Cat. # 806-WCL), nylon-flocked swabs (Copan Diagnostics Inc., Cat. # 552C-US), Biological Sampling Kits (BiSKit; Quicksilver Analytics Inc., Cat. # BIS-40001A), and ITW Alpha polyester wipes (Texwipe, Cat. # TX1004). The cotton swabs, nylon-flocked swabs, and BiSKits come pre-sterilized and individually packaged by the manufacturer. Prior to use, polyester wipes were aseptically folded, rolled, and placed in 50 mL conical tubes containing 5 mL of sterile DNA-free Ultraclean phosphate-buffered saline (PBS; Mo Bio, 1X, Cat. # 17330-500) and then autoclaved (15 Pa, N/m2 or kg·m−1·s−2, 121°C) for 30 min. The two swab types were employed to collect samples from small surface areas (25 cm2), and BiSKits and wipes from large surface areas (2500 cm2).
2.4. Sample collection
All sample collection and sample processing procedures (Supplementary Fig. S1; Supplementary Data are available online at
2.5. Sample processing
2.5.1. Sample concentration
Immediately following sample collection, cotton, nylon-flocked swabs, and polyester wipes were placed in sterile 50 mL conical tubes containing 15 mL of PBS. Each device was agitated via vortexing at maximum speed for 5 s and sonicated (2 min, 19–27 kHz) in a degassed (5 min) water bath at room temperature (∼23°C), after which the liquid was decanted into a 50 kD molecular weight cutoff Amicon filtration device (Millipore, Catalog # UFC905024, Chico, CA). After repeating this step a total of three times—samples were also extracted a total of three times from BiSKit devices, as instructed by the manufacturer, with the use of 15 mL of PBS each time—the microbes and biomolecules released from the sampling devices were concentrated via centrifugation with the Amicon filtration devices (5000×g, 4 min). The resulting ∼500 μL sample concentrates were readjusted to 1 mL by using the flow-through liquid from each respective sample, and subsequently subjected to downstream molecular processes.
2.5.2. Sample homogenization
A Fastprep-24 bead-beating instrument (MP Biomedical, Santa Ana, CA) was used to mechanically agitate half the volume of each sample (500 μL). The instrument was run at a speed of 5 m/s for 60 s. Processing in this manner helped to ensure that resulting suspensions would contain DNA released from both hardy and labile cell types, as has been previously described (La Duc et al., 2009b). Samples were subsequently centrifuged (5000×g, 60 s), and supernatants were collected and kept on ice.
2.5.3. DNA extraction
For each sample, the 500 μL bead-beaten portion was combined with the 500 μL portion that was not subjected to bead beating, and the entire volume (∼1 mL) was subjected to DNA extraction. DNA was extracted by the automated Maxwell-16 MDx system according to manufacturer's instructions (Promega, Madison, WI). The DNA eluate was collected in 100 μL of UltraPure Gibco water and subsequently aliquoted into four 25 μL samples in cryovials for long-term storage at -80°C.
2.6. Quantitative PCR
2.6.1. SYBR Green qPCR
All rRNA gene copy measurements were determined in triplicate with a CFX-96 thermal cycling instrument (Bio-Rad, CA, USA). Plasmid standards spanning 102 to 108 full-length 16S rRNA gene copies/μL were created by serially diluting Escherichia coli rrn::pCR4-TOPO constructs (pCR4-TOPO cloning vector; Invitrogen, Carlsbad, CA) of predetermined concentration. Universal eubacterial primers [1369F and 1492R (Suzuki et al., 2000)] targeting the 16S rRNA gene were employed for qPCR analysis. Each 25 μL reaction mixture consisted of 12.5 μL of Bio-Rad 2X iQ SYBR Green Supermix, 10.5 μL of UltraPure water (Gibco), 0.5 μL of forward primer 1369F (10 μM), 0.5 μL of reverse primer 1492R (10 μM), and 1 μL of DNA template. Purified standards and UltraPure Gibco water negative controls were included in all qPCR runs. Thermal cycling parameters for universal 16S rrn qPCR were as follows: hold at 95°C for 3 min to achieve initial denaturation, followed by 40 cycles of the following: 10 s hold at 95°C to denature, ramp-down to 55°C for primer annealing, and extension occurring through a 35 s ramp-up to 95°C.
2.6.2. Species-specific qPCR
To discern differences in the recovery of rRNA genes of the various microbes present in the MMC positive control, species-specific (ss) qPCR assays were designed and carried out targeting each MMC constituent. Species-specific primer sequences are provided in Table 1, alongside detailed thermal cycling parameters for each MMC strain. All gene copy quantifications were performed in triplicate according to previously published protocols for qPCR (Suzuki et al., 2000; Kwan et al., 2011). The standards used were serial dilutions of purified, pre-quantified 16S rRNA gene amplicons of the respective MMC target strain. Gibco water processed through the automated DNA extraction system, and qPCR no template controls were included in each run to serve as reagent and reaction negative controls, respectively.
2.7. Statistical analyses
The statistical significance of qPCR measurements amassed over the course of this study was addressed in accordance with specifications of the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (Bustin et al., 2009). In this study, decimally diluted 16S rRNA gene::pCR4-TOPO plasmid standards consistently yielded qPCR reaction efficiencies of 90–104%, slopes between −3.3 and −3.7, and R-squared values in excess of 0.95. Since all samples were analyzed in triplicate, reactions outlying these constraints were omitted from consideration. To elucidate subtle differences in resulting recovery efficiencies from the sampling materials tested (cotton vs. nylon-flocked swabs; BiSKits vs. polyester wipes), statistical analyses were carried out at all stages of analysis with Microsoft Excel. Briefly, basic statistical calculations such as mean, median, standard deviation, standard error of the mean, and percent recovery were charted and compared. Paired student t tests (p≤0.05) were employed to compare the statistical significance of the means.
2.8. Controls
Sterile water, PBS, Maxwell DNA extraction reagent blanks, and qPCR reagent blanks used in sample collection, processing, and analysis were included in all qPCR runs as negative controls. No template controls were included in every qPCR assay as a means of evaluating melt curves and detecting primer-dimer amplification (multiple melt peaks). Also prepared from each sampling device was a sampling device (SD) negative control, in which the device was only pre-moistened with PBS, and a handling control (HC), in which a sampling device was pre-moistened with PBS and exposed to the ambient sampling environment (without any contact with the metal surfaces). The initial washing step performed for BiSKits, aimed at removing any indigenous DNA, was not performed on other sampling devices, as such treatment compromised the integrity of the swabs and wipes. Instead, an unused sterile device from the same lot was included as a negative control.
3. Results
3.1. Indigenous rDNA associated with sampling materials
The total number of contaminant, indigenous 16S rRNA genes associated with each of the sampling devices, and reagents used in sample collection, processing, and analysis are given in Table 2. Also included, as a positive control reference, are the numbers of MMC rRNA genes directly seeded onto the metal surfaces. Indigenous rDNA associated with the various sampling instruments ranged from 1.8×102 to 8.3×103 average gene copies per device, which was ∼3 logs lower than the detected level of average MMC positive control rDNA (3.6×106 gene copies). Only 10% of the sampling blanks examined (inclusive of sampling devices, reagents, and metal surfaces) exhibited indigenous 16S rRNA genes in excess of 103 (data not shown). Negative controls corresponding to the metal surfaces (sans MMC), and the media used to culture the MMC constituents, contained 101 to 102 indigenous rRNA genes (Table 2). The observed range in indigenous rDNA burden for the metal was roughly 2 logs lower than that corresponding to the sampling device blanks, handling controls, and sampling blanks, and approximately 4 logs lower than that corresponding to the MMC positive control. Based on this disparity, it seems reasonable to attribute the source of DNA contamination observed in the sampling blanks to the sampling devices and not the metal surfaces (see Table 2).
3.2. Sensitivity of ss-qPCR assays
The universal qPCR assay employed in this study to measure total microbial burden was limited in sensitivity to 103 16S rRNA gene copies (Fig. 1B). Upon targeting a distinct MMC constituent with species-specific primers and thermal cycling parameters, the sensitivity of the ss-qPCR assay was pushed down to 102 gene copies (Fig. 1A for B. megaterium). To distinguish background noise from true sample-borne signals, scalability testing was employed by using qPCR and ss-qPCR assays. Corresponding sampling device and reagent negative controls were unaffected, whereas a linear increase in amplification was observed for all real samples (Fig. 1). The rDNA of eight of the nine MMC constituents (exception: M. formicicum) was detected in the sampling blanks (Supplementary Fig. S2A) and handling controls (Supplementary Fig. S2B). With the exception of B. megaterium rDNA in the BiSKit and polyester wipe device controls, none of the sampling material negative controls had rDNA of MMC constituents present in excess of 102 gene copies per sample (Supplementary Fig. S2A). However, attempts at cloning B. megaterium ss-qPCR-amplified rRNA gene fragments failed to yield any pCR4-TOPO plasmid vectors having ligated inserts (data not shown).
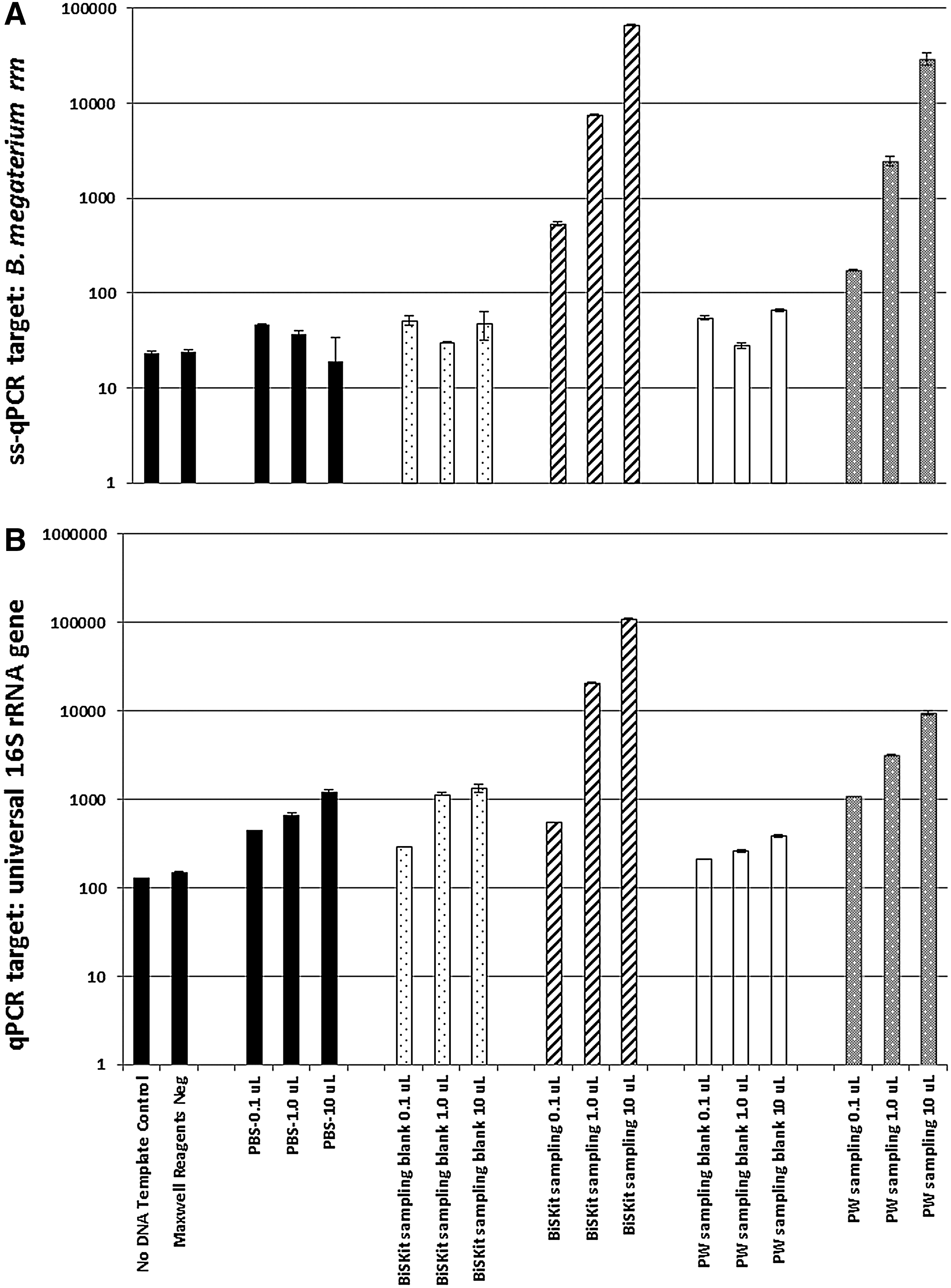
Effect of logarithmic scaling of PCR template volume on sampling device blanks and reagent negative controls versus actual samplings of artificially seeded MMC. (
3.3. Specificity of ss-qPCR assays
The specificity of the various ss-qPCR assays developed in this study was tested by exposing total genomic MMC DNA to qPCR parameters across a matrix of specific primer sets. All nine ss-qPCR protocols developed and employed herein enabled the selective detection and quantitation of their respective strain. As expected, a linear correlation was observed between the decreasing B. megaterium template DNA input and resulting qPCR amplicons (Fig. 2B). A similar pattern of linear regression was noticed for all nine species when their respective rDNA were serially diluted (Supplementary Fig. S3). To examine crosstalk between primers specific for a given MMC constituent and the other eight MMC strains, pairwise ss-qPCR analyses were carried out and compared. For example, when B. megaterium–specific primers were used to test the amplification of rRNA genes from genomic DNA of each of the other eight MMC strains, results demonstrated high primer specificity for B. megaterium (Fig. 2B). The observed M. luteus rDNA yield of 103 rRNA gene copies (Fig. 2B, striped bar) corresponded to a melt-curve profile different from that of B. megaterium, which is indicative of a primer-dimer amplification artifact. The specificity and pairwise selectivity of all nine ss-qPCR primer sets developed and employed in this study are provided in Supplementary Fig. S3. Among the 99 pairwise comparisons (9 ss-qPCR assays×11 MMC constituents) assessed, only 10 reactions gave rise to nonspecific amplification at levels in excess of 103 rRNA genes. Most of these assays yielded melt-curve profiles indicative of primer-dimer artifacts. Finally, the presence of rDNA originating from B. pumilus spores and A. pullulans fungal cells (ss-qPCR data for BP and AP not shown, but both species were used in pairwise assessment, as seen in Fig. S3) exhibited no competitive influence on the amplification plots resulting from targeted MMC constituents.
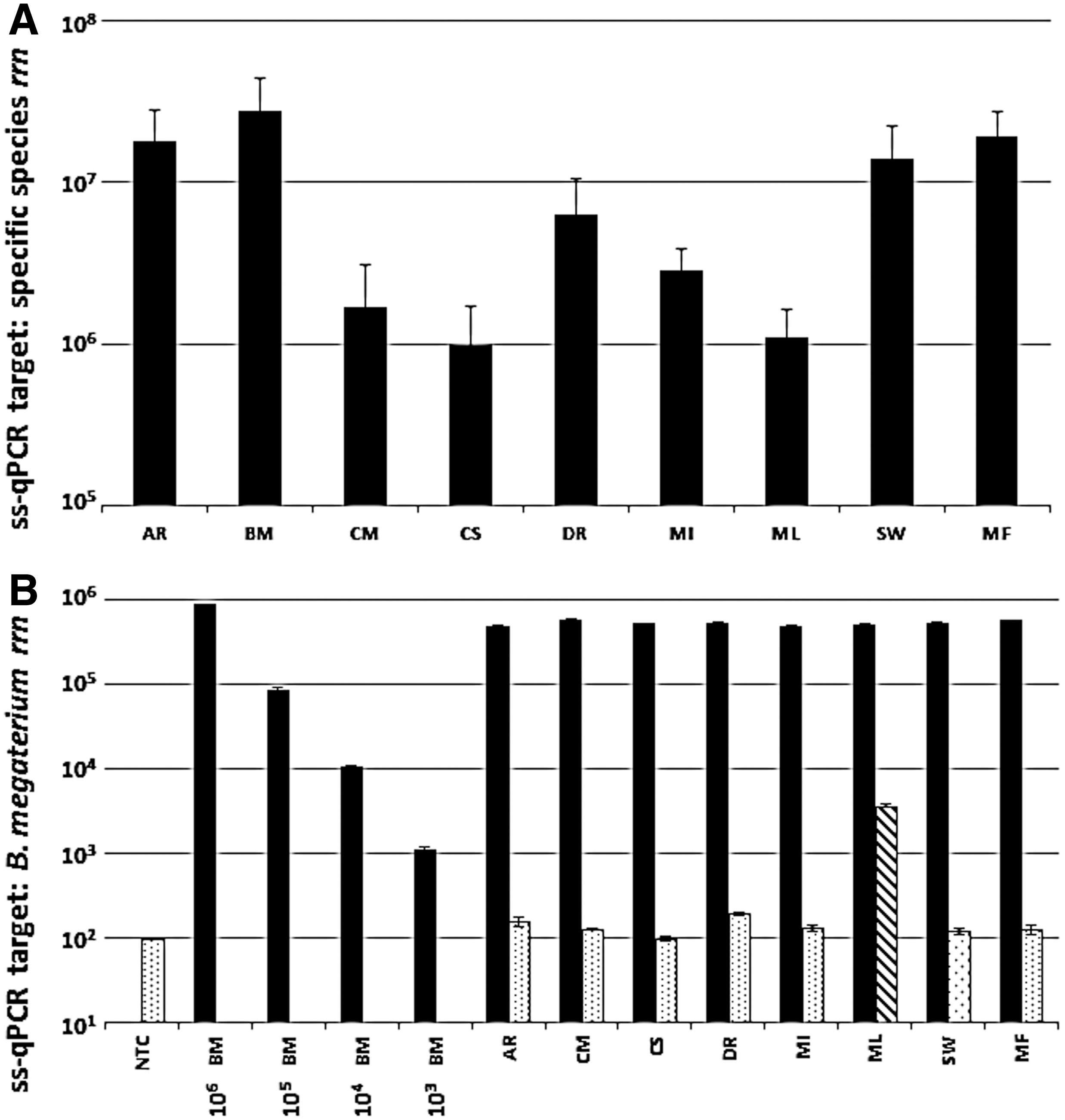
Performance and specificity of ss-qPCR assays developed in this study. (
3.4. Recovery of DNA from metal surfaces
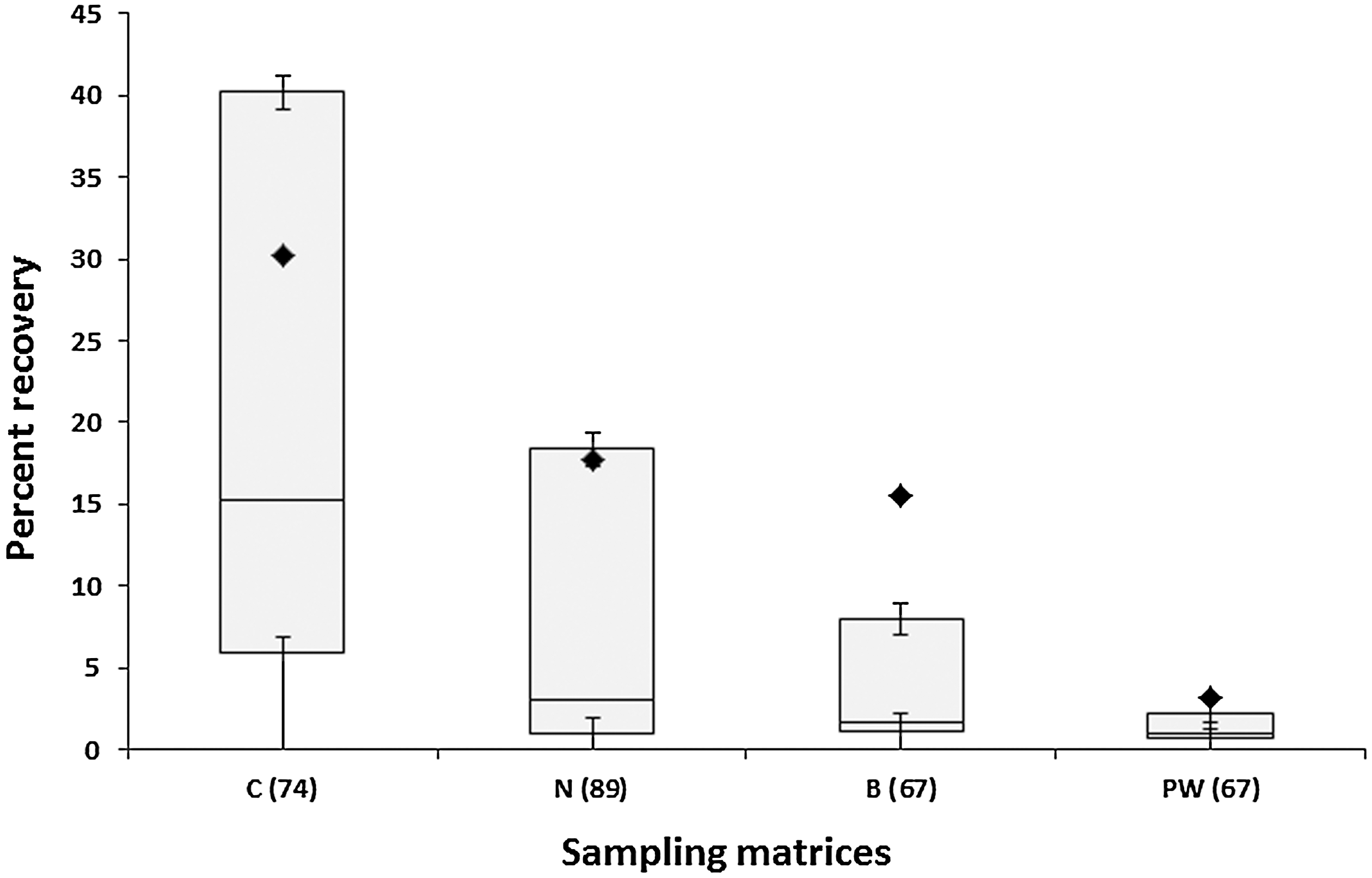
The statistical treatment of 16S rRNA gene fragments (cellular and naked) retrieved from metal surfaces by various sampling materials is shown in Fig. 3 (also see Supplementary Tables 1 and 2). As can be observed in Fig. 3B, cotton swabs exhibited an average yield of 16S rRNA gene copy number (1.2×106), which is nearly twice that observed for nylon-flocked swabs (7.1×105), with Fig. 4 translating this statistic to show a total rDNA recovery efficiency of 30.2% for cotton and 17.7% for nylon. Furthermore, the t test (p-value) analysis in Table 3 revealed cotton swabs collected and released significantly (p<0.05; CI 95%) greater amounts of 16S rDNA than their nylon counterparts. This is validated by Fig. 3B, as no overlap is observed between the total 16S rDNA recovered by cotton (C) and the indigenous DNA present in the device pre-sampling (C-SB). On the contrary, there is some overlap between the minimum gene copy yield recovered by nylon-flocked swabs (N) and the maximum indigenous gene copy present in the device pre-sampling (N-SB). The pie chart in Fig. 5 contributes the overlap observed in Fig. 3B to nylon's overall low efficiency in sample recovery. Specifically, of the 89 nylon-flocked replicates processed, 62 replicates (∼70% of the total nylon swabs) recovered ≤9.99% of the rDNA seeded onto the metal surfaces. Cotton swabs, on the other hand, proved to be much more effective, as 47 (∼64%) of the 74 replicates yielded recovery ≥10% (Fig. 5). In the same fashion as with nylon, the near total overlap observed (Fig. 3C) between the polyester wipe pre-sampling (PW-SB) and the wipe sample replicates (PW) can be attributed to the devices' poor recovery. Of the 67 total wipe replicates, 47 (∼70%) recovered ≤9.99% of the rDNA seeded onto the metal surfaces.
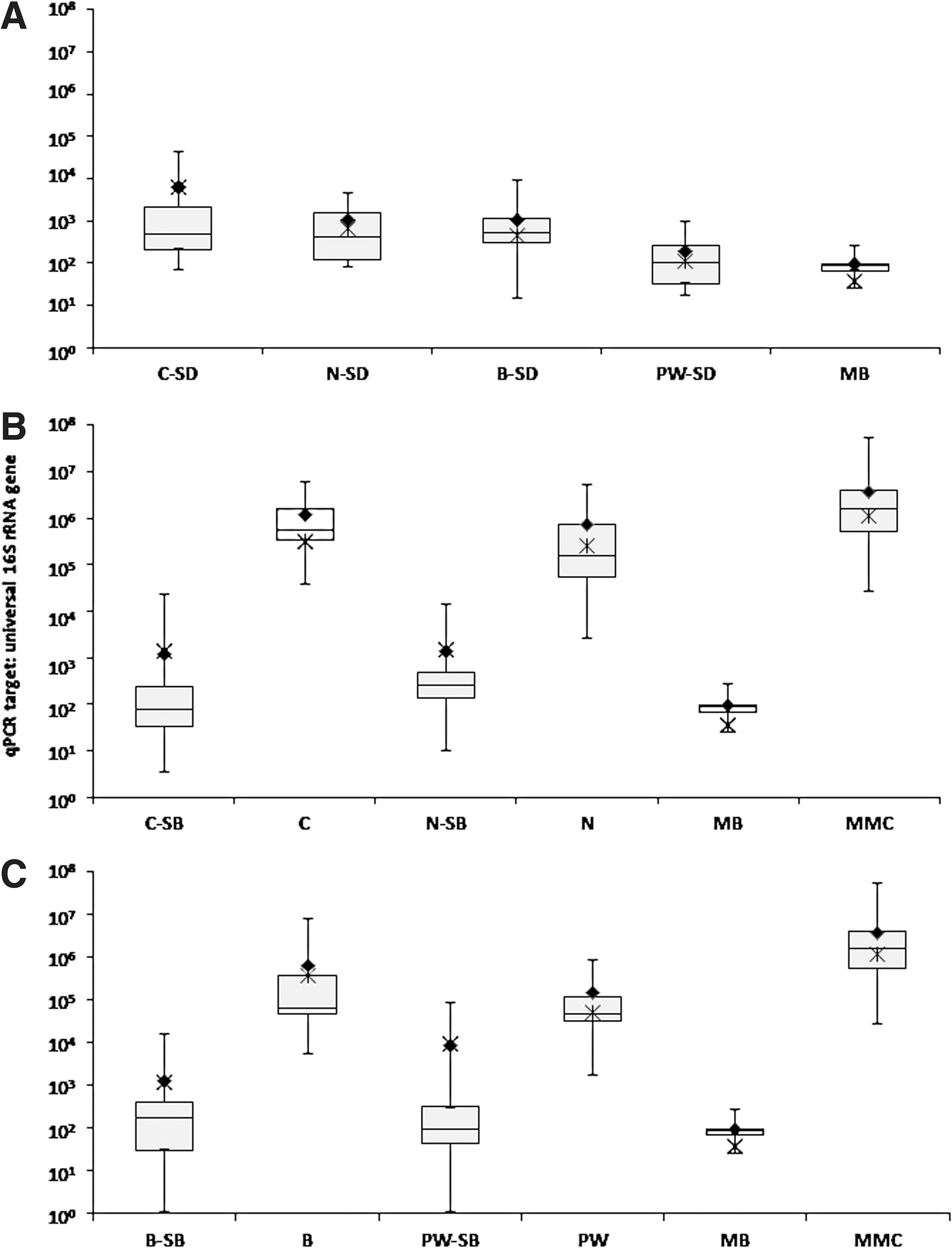
Statistical treatment of 16S rRNA gene fragments (cellular and naked) retrieved from metal surfaces by various sampling materials. (
Performance and recovery efficiency of various sampling devices. Recovered samples were analyzed via universal qPCR targeting 16S rRNA genes, and results were summarized with box plots showing the mean (closed diamonds), median, and upper and lower quartiles for cotton (C) and nylon-flocked (N) swabs, BiSKits (B), and polyester wipes (PW). Error bars depict the standard error of the mean placement of each quartile. The number of replicates performed with each sampling device is provided in parenthesis.
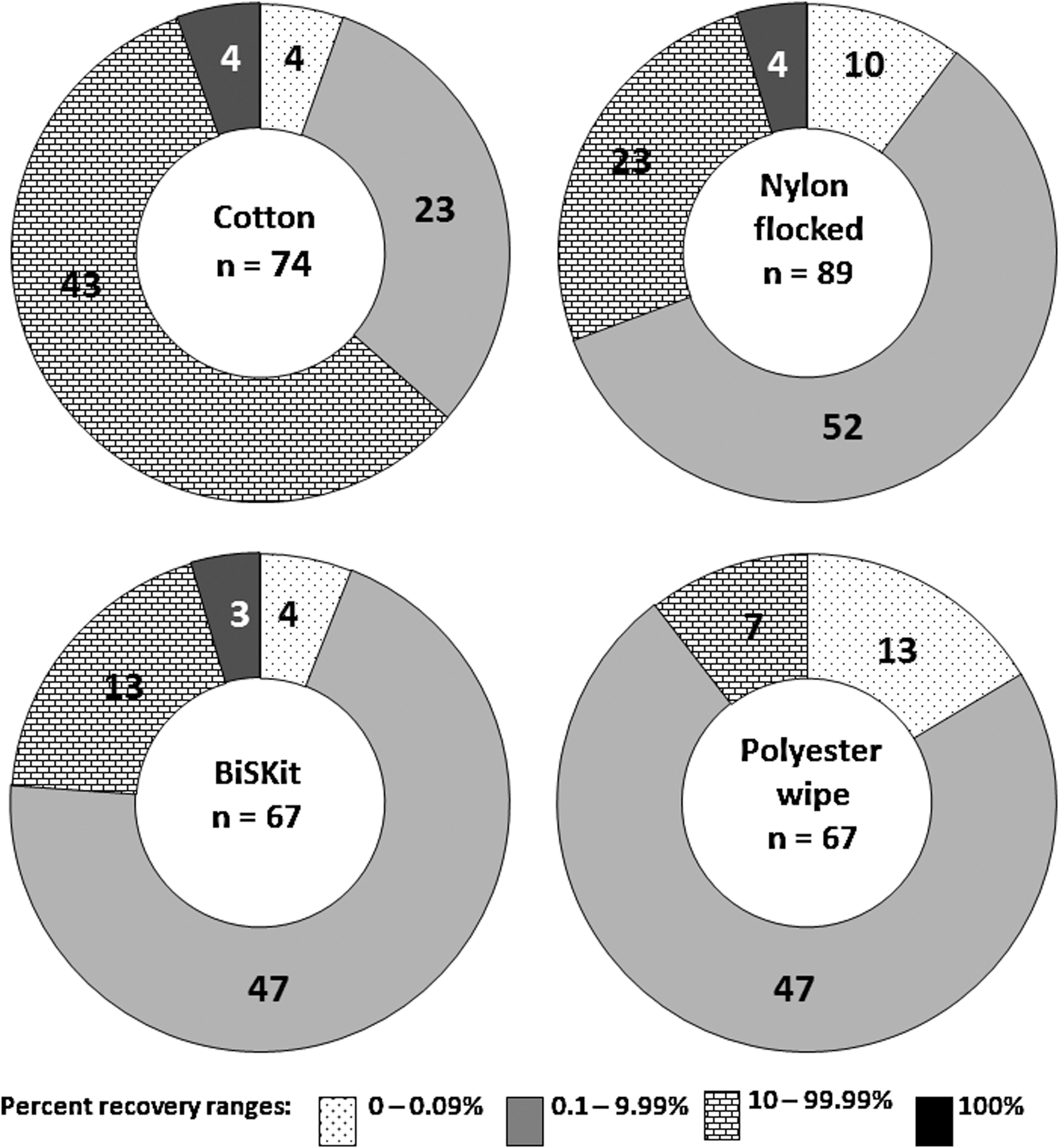
Categorical percent recovery efficiency of various sampling matrices. Numbers on the pie chart represent the distribution of n replicates based on percent recovery range of 0–100%.
Number of replicates are given in parentheses. Each value is the average of three (3) qPCR measurements.
p<0.05 is statistically significant (CI 95%).
Mean total gene copy numbers. Mean values in columns a and b correspond, respectively, to samples a and b of the Sample1 columns.
BiSKit sampling devices were roughly five times more effective in recovering MMC rDNA than polyester wipes (15.5% and 3.2% recovery, respectively; Fig. 4). When BiSKits were used for sample collection, all 13 of the replicates in the 10–99.99% recovery range exhibited >20% recovery, whereas only 2 of the 7 wipes in the same range yielded recovery this high (Fig. 5). Samples collected using BiSKits and polyester wipes were significantly more laden with rDNA than their corresponding sampling blanks (p<0.05; Table 3, Fig. 4). At first glance (Fig. 3C), there appears to be no statistically significant difference between the levels of indigenous rDNA present in the polyester wipe sampling blanks (the positive end of the standard mean of error is 8.3×104 gene copies) and the amount of rDNA actually collected with polyester wipe. However, detailed statistical analyses revealed that the rDNA recovery yield typical of polyester wipes (1.4±2.1×105 gene copies; 3.3%; Fig. 3, Table 3) exceeded the levels of indigenous rDNA burden of the sampling blanks (4.1×101 to 8.3×104 gene copies; Table 3). The levels of indigenous rDNA associated with the sampling device blanks for each of these devices fell below the limits of ss-qPCR detection. The polyester wipe blanks exhibited an average of 1.9×102 indigenous gene copies, compared to BiSKit blanks that averaged 1.1×103 indigenous rDNA genes (Table 3).
To evaluate the direct removal of nucleic acids from metal surfaces, physical [sonication; (Kempf et al., 2005)] and chemical [polyvinyl alcohol spread (PVA); (Tauscher et al., 2006)] techniques were carried out and the results compared. When MMC-seeded metal coupons (25 cm2) were immersed in PBS and subjected to sonication, only 15% of the initial number of rDNA molecules was detected in the resulting solution (data not shown). In contrast, when PVA was applied directly onto the MMC-seeded coupons, roughly 70% of the initial rDNA deposited could be recovered in the resulting PVA layer, upon dissolution (Supplementary Fig. S4). Indeed, scanning electron micrographs of the stainless steel taken after sampling clearly show the presence of whole cells and cellular debris (Supplementary Fig. S5). If the resulting PVA layers are allowed to represent 100% recovery (1.9×106 rRNA gene copies), then device-based recovery was 69.7% for cotton swabs and only 35.4% for nylon-flocked swabs. However, if the original MMC cell suspension in 1 mL of PBS is presumed to represent 100% recovery (3.6×106 rRNA gene copies), as is the case in this study, cotton swabs exhibited 38.5% recovery while nylon-flocked swabs showed only 19.6% recovery. Unfortunately, the clogging of Amicon 50 kDa molecular cutoff filters in downstream processing currently precludes the use of PVA in these studies.
3.5. Differential recovery of MMC rDNA from metal surfaces
The MMC-strain-based recovery efficiencies associated with the various sampling materials tested are provided in Fig. 6. The initial number of rRNA gene copies of each MMC constituent present in 1 mL of MMC suspension (Fig. 2A) was calculated and used as the reference point for determining percent recovery. Cotton swabs enabled the recovery of rDNA from all nine MMC constituent microbes, whereas nylon-flocked swabs failed to recover three of the MMC members, including the archaeaon M. formicicum (Fig. 6A). In addition, cotton recovered considerable levels of rDNA of several MMC constituents that had been totally unaccounted for when sampled via nylon-flocked swabs. Of the nine MMC constituents assayed, cotton recovered at least 25% of the originally deposited rDNA from C. metallidurans (34.5%), C. sporosphaeroides (36.7%), M. luteus (29.1%), S. warneri (31.8%), and M. formicicum (24.8%). In contrast, nylon-flocked swabs recovered the rDNA of S. warneri at 18.6%, and the rDNA of five additional MMC strains at 7% or less. Both swab materials had difficulty recovering the rDNA of D. radiodurans (<1% each).
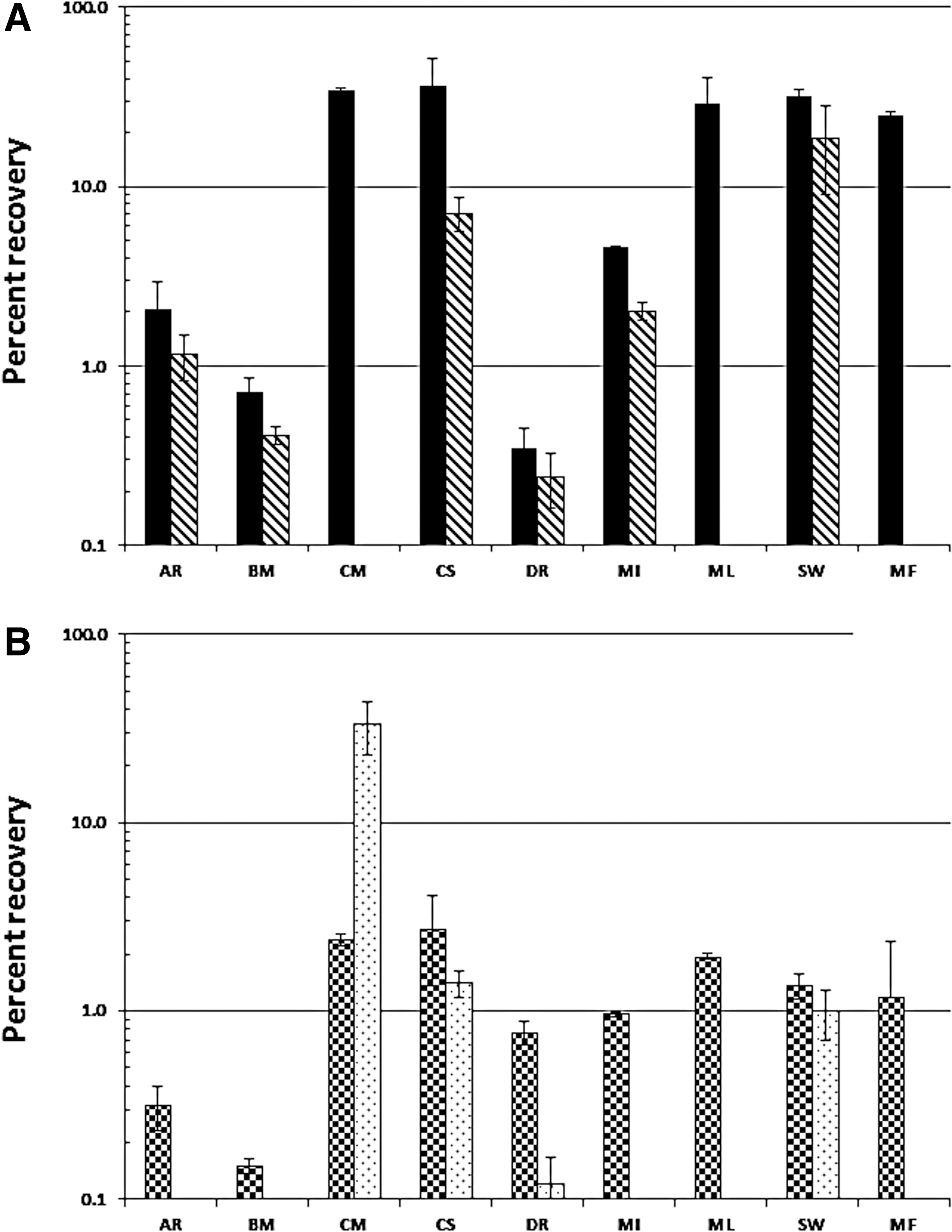
Differential recovery of cells and rDNA of MMC constituents from metal surfaces by various sampling devices. (
As may be expected due to considerable difference in surface area (100-fold), the recovery of MMC rDNA from large surface areas was much lower than that from small surface areas. BiSKit sampling devices consistently collected greater amounts of MMC-borne DNA than polyester wipes (Fig. 6B). Furthermore, BiSKits successfully recovered the rDNA of all nine MMC constituents deposited onto the metal surfaces, whereas polyester wipes recovered only four of the MMC strains. BiSKit samplers were up to 14 times more efficient in collecting individual MMC constituent-borne rDNA than polyester wipes. Of the nine MMC microbial constituents assayed, BiSKits were able to recover at least 1% of the rDNA of C. metallidurans, C. sporosphaeroides, M. luteus, S. warneri, and M. formicicum (Fig. 6B). Coincidentally, these are the same MMC strains recovered in high abundance by cotton swabs (Fig. 6A). In contrast, polyester wipes were limited in recovery to the rDNA of C. metallidurans (33.3%), and three additional MMC members at less than 1.4%.
4. Discussion
The difficulty in accounting for the presence of all the members of a diverse microbial community has been recognized for years (Giovannoni et al., 1990; Pace, 1997; Giovannoni, 2005). This challenge is exacerbated when attempting to analyze the microbial breadth present in samples of extremely low biomass (La Duc et al., 2009b). Several factors contribute to the complexity and difficulty in achieving this objective, perhaps none more so than experimental bias. For any mode of diversity analysis there exist biases (e.g., species high in abundance overshadowing low abundant species) that favor the detection of certain taxa over others, thus skewing the results of comparative quantitative analyses. For the purpose of a genetic census, quantitation is by no means as relevant as presence or absence. However, most molecular approaches are based on some form of universal probe which, in theory, associates with all the cells in a sample irrespective of phylogeny. Unfortunately, many of these techniques are predicated on flawed assumptions. It is presumed that said probe is truly universal and thus associates with the same affinity to every cell present. In practice, however, this is simply not the case. As a finite number of probes are applied to a sample, one can never conclude with confidence that all phylotypes present have been accounted for. There is no molecular safeguard that prevents those phylotypes that happen to have a stronger affinity for the probe from being overrepresented and thus masking the presence of lesser abundant and singleton taxa. Therefore, even if genetic censuses are only interested in qualitative (presence/absence) data, they are constrained to the limitations of these and other experimental biases.
The objective of this study was to determine the most effective sampling device(s) for collecting cells and naked rDNA from low-biomass surfaces. Results demonstrate the intrinsic power of molecular techniques in assessing the differential recovery of rDNA of a mixed microbial consortium deposited and dried on metal surfaces. Measurements of total DNA recovery were directly compared with those corresponding to the specific rRNA gene sequences of each MMC constituent, thereby enabling assessment of differential recovery of the various MMC strains as a function of the device employed for sample collection.
The high throughput and ever-increasing sensitivity of sequence-specific DNA-dependent qPCR methods render them invaluable in detecting both cultivable and noncultivable taxa in samples of extremely low biomass. Recently, molecular techniques were used to evaluate and standardize protocols for the collection and processing of cells and biomolecules from low-biomass surfaces (Kwan et al., 2011). However, these approaches focused solely on the recovery of total nucleic acids. To the best of our knowledge, this report on the use of species-specific PCR-based techniques to assess the differential recovery of various cell lines of a mixed microbial consortium from low-biomass surfaces is the first of its kind.
Prior to testing actual samples, experiments were designed and executed to enhance the current understanding of the limitations and shortfalls associated with the molecular approaches employed. As background noise can have a profound effect on SYBR Green fluorescence detection and thus impair the precision of qPCR reactions (Hilscher et al., 2005; Monis et al., 2005; Lee, 2010), efforts were made to determine whether signals indicative of indigenous DNA contamination associated with negative controls were real or simply an artifact relating to the lower limit of detection. When negative control qPCR template DNA volumes were scaled up in a logarithmic manner, resulting amplification curves overlapped and were not affected by the variability in template DNA volume. This was indicative of nonspecific annealing and extension (e.g., primer-dimers), since amplification plots from sample-borne DNA would be expected to follow a stepwise, log-scale pattern based on the volume of template DNA assayed. It was imperative to demonstrate the non-scalability of qPCR amplification plots resulting from sampling device blanks and reagent negative controls. Without first establishing the origin of qPCR fluorescence signals approaching the lower limits of detection, it would be impossible to set valid threshold limits for sensitivity. This is particularly relevant to investigations centered on low-biomass samples, where it is absolutely critical to discern background noise from faint signals originating from the actual sample.
The specificity of ss-qPCR assays was tested by subjecting total genomic MMC DNA to each of the nine primer sets, with thermal cycling parameters specifically designed for each MMC strain. Positive ss-qPCR amplifications were observed for all nine target species assays. The variability observed in the qPCR reaction efficiency, and thus the SYBR Green fluorescence output, may be a consequence of the instrument's application of an estimated baseline, which was calculated from the fluorescence intensity of the first few amplification cycles and applied to the remainder of the reaction. Past reports (Ruijter et al., 2009) have argued that such applications of baseline estimates from early amplification cycles may affect PCR efficiencies and ultimately gene expression quantification. Thus, implementing a “one size fits all” algorithm for determining true baseline values based on early cycle reads will result in variability that is based mainly on statistical errors. Nordgard et al. (2006) evaluated algorithms for estimating optimal starting quantities by limiting the stochastic chemical and physical nature of the first few PCR amplification cycles. They posited that the variability observed in PCR efficiency arises from random errors generated from the noise present at the start of amplification and is thus an artifact of the technology. To minimize the effects of such artifacts, and in accordance with previous reports (Kwan et al., 2011), the authors thoroughly tested each ss-qPCR primer pair and ultimately used only those that yielded highly selective and reproducible amplification. By levying the knowledge base provided by similar published studies (Nadkarni et al., 2002; Ritalahti et al., 2006; Fremaux et al., 2009; Haugland et al., 2010) and subjecting each qPCR assay to rigorous scrutiny and optimization, the authors are confident that resulting ss-qPCR protocols result in highly reproducible, statistically significant reaction efficiencies for each MMC constituent strain. Although qPCR-based rRNA gene quantification is rapidly becoming status quo in the modern-day research laboratory, the bioinformatic infrastructure for processing and analyzing resulting data is still in development. As such, the mathematical algorithms used in these quantitation models must undergo validation to minimize the propagation of error while maintaining precision and increasing reproducibility.
Many factors affect the initial attachment and subsequent retention and/or release of microorganisms to inert substrata (Verran and Whitehead, 2005). Ultimately, the effectiveness and efficiency of sampling devices in recovering biological materials from metal surfaces is dependent on (a) the physical removal of biomaterials from surfaces (Mulligan et al., 2011), (b) the retention versus release of the biomaterials from the sampling matrices (Zain and Bradbury, 1995), (c) the total DNA extraction yield (Barton et al., 2006; Zucol et al., 2006), and (d) the robustness of the analysis techniques (Chandler, 1998; Lee, 2010; Horevaj et al., 2011). To evaluate the direct removal of nucleic acids from metal surfaces, physical [sonication (Kempf et al., 2005)] and chemical [PVA (Tauscher et al., 2006)] techniques were carried out, and results were compared. When MMC-seeded metal coupons (25 cm2) were immersed in PBS and subjected to sonication, only 15% of the initial number of rDNA molecules was detected in the resulting solution. In contrast, when PVA was applied directly onto the MMC-seeded coupons, roughly 70% of the initial rDNA deposited could be recovered into the resulting PVA layer, upon dissolution (Supplementary Fig. S4). Variability in the affinity of stainless steel for microbial cells has been documented, as has an improved release rate upon both physical and electrochemical treatment. Electropolished stainless steel has been shown to harbor significantly fewer bacterial cells than other treated surfaces (Arnold and Bailey, 2000). Hence, the unfinished stainless steel coupons used in this study might have retained a substantial portion (up to 30%) of the initially deposited MMC suspension. To assess the effect of the material properties of the sampling devices tested, MMC suspension was seeded directly onto the cotton and nylon-flocked swab heads, as well as the BiSKit macrofoam. Upon extracting total nucleic acids from volumes expunged from the sampling materials, roughly 10% of the initial rDNA molecules were released and recovered from each of the two swab matrices, whereas BiSKit macrofoam released as high as 50% of the seeded DNA.
In agreement with the findings of previous cultivation-based investigations (Buttner et al., 2004a, 2004b; Rose et al., 2004; Da Silva et al., 2011), this DNA-based study found cotton to be the preferred matrix for collecting samples from small surface areas and macrofoam-based BiSKits the preferred sampling instruments for large surface areas. Polyester wipes were the least efficient of the four devices tested, with percent recovery limited to <10% (Fig. 4). Multiple variables could have contributed to the differences observed in percent recovery and collection efficiency. It seems plausible to speculate that the tightly woven fibers composing the polyester wipes could retain biological materials and thus impede the efficient release of the sampled material from the cloth matrix. Something as seemingly trivial as the pliant nature of the nylon swab shaft, or the very technique in which the liquid nylon is applied to the swab shaft, could have affected the manner in which the samples adhered to the swab head during sampling. Though pertinent, an examination into the cause and effect of these and other subtle nuances was beyond the scope of this study.
The potential impact of this research finding—that BiSKits are superior sampling devices for larger surface areas—could help advance the routine collection of phylogenetic data to a statistically appropriate level to ensure that the microbial diversity of assembly, test, and launch operation environments, and of all NASA spacecraft to be sent to Mars, is reliably assessed. However, the finding of this study only demonstrates the gap-bridging nature of molecular techniques in gauging the efficacy and efficiency of recovery of disparate microbial lineages from metal surfaces. Further BiSKit manufacturing improvements will have to be achieved in order to consider using the currently shed-prone material directly on the spacecraft surfaces. Shed material on the surface of the spacecraft or the ground support equipment used to assemble it is problematic in that it will create increased static or magnetic interference with the microchip devices and tools used to assemble the spacecraft. Additionally, the size of the device (dimensions 6.5″ w×7″ h×2.5″ d) makes it difficult to use on curved surfaces such as that of a spacecraft. On the other hand, the application of the BiSKit device for use in sampling nonspacecraft surfaces (i.e., spacecraft assembly clean rooms and associated surfaces) has proven to be both efficient and promising for use in clean rooms, given that the shedding problem has been adequately addressed.
A recent cultivation-based study (Probst et al., 2010) reported nylon swabs to be far superior to cotton swabs at recovering Bacillus spp. spores from stainless steel and other surfaces. Perhaps these findings are most germane to investigations targeting a single biological entity, such as bacterial endospores or intact cells. To date, however, there are very few published findings (Buttner et al., 2001, 2007; Kwan et al., 2011) on the effectiveness of sampling devices in recovering DNA (both cell/spore-associated and naked) from surfaces, as is reported here. The results of this investigation clearly demonstrate a sampling device-dependent differential recovery of MMC constituent rDNA from metal surfaces. The findings of this study advance the current understanding of the capabilities and limitations of current sampling matrices in collecting biological samples from surfaces.
5. Conclusion
The results of this study underscore the value of universal and ss-qPCR–based methods in studying the collection of biological samples from metal surfaces. This practical application of such innovative molecular tools is in direct response to the National Research Council–Space Studies Board recommendation, which suggests that NASA perform routine collection of phylogenetic data to a statistically appropriate level to ensure that the microbial diversity of assembly, test, and launch operation environments, and of all NASA spacecraft to be sent to Mars, is reliably assessed. The results of extensive ss-qPCR support the conclusion that differential recovery of rRNA genes is largely dependent on the type of sampling device employed. While cotton swabs and BiSKits recovered the rDNA of all nine of the MMC constituent microbes assayed, nylon-flocked swabs and polyester wipes retrieved rDNA of only six and four of the MMC lineages, respectively. Ultimately, the findings of this study demonstrate the prodigious, gap-bridging nature of molecular techniques in gauging the efficacy and efficiency of various modes of biological sample collection (and, in particular, recovery of disparate microbial lineages) from metal surfaces.
Footnotes
Acknowledgments
Part of the research described in this publication was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration. This research was funded by the Mars Program Office. We also appreciate the valuable advice received from J.A. Spry, K. Buxbaum, and C. Conley. Copyright 2011. All rights reserved.
Abbreviations
BiSKit, biological sampling kit; MMC, model microbial community; PBS, phosphate-buffered saline; PVA, polyvinyl alcohol spread; qPCR, quantitative polymerase chain reaction; ss, species-specific.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.