
Abstract
Human mesenchymal stem cells (hMSCs) are one of the important factors that regulate bone anabolism. Osteoporosis resulting from microgravity during spaceflight may possibly be due to a decrease in osteogenesis mediated by hMSCs. This speculation should be verified through culture and osteogenic induction of hMSCs in a microgravity environment during spaceflight. Control of CO2 is a key component in current experimental protocols for growth, survival, and proliferation of in vitro cultured cells. However, carrying CO2 tanks on a spaceflight and devoting space/mass allowances for classical CO2 control protocols make experimentation on culture and osteogenesis difficult during most missions. Therefore, an experimental culture and osteogenic medium was developed through modifying the components of buffer salts in conventional culture medium. This experimental medium was used to culture and induce hMSCs under CO2-independent conditions. The results showed that culture and induction of hMSCs with conventional culture medium and conventional osteogenic medium under CO2-independent conditions resulted in an increase of pH in medium. The proliferation of hMSCs was also inhibited. hMSCs cultured with experimental culture medium under CO2-independent conditions showed a proliferation potential that was the same as those cultured with conventional culture medium under CO2-dependent conditions. The experimental osteogenic medium could promote hMSCs to differentiate into osteoblast-like cells under CO2-independent conditions. Cells induced by this induction system showed high alkaline phosphatase activity. The expression levels of osteogenic genes in cells induced with experimental osteogenic medium under CO2-independent conditions were not significantly different from those cells induced with conventional osteogenic medium under CO2-dependent conditions. These results suggest that the experimental culture and induction system could be used to culture hMSCs and induce the osteogenesis of hMSCs in the atmospheric conditions common to spaceflights without additional CO2. Key Words: hMSCs—CO2-independent culture—Osteogenic differentiation—Proliferation. Astrobiology 13, 370–379.
1. Introduction
M
In vitro culture of cells is a universal technique for cell biology that has been used for more than 100 years and has developed rapidly since the 1950s (Dulbecco, 1952). In vitro culture systems of mammalian cells are composed of conventional medium (including various amino acids, carbohydrates, inorganic salts, vitamins, etc.), 10% serum, 20% O2, 5% CO2, and 37°C constant temperature. CO2 is a key component for growth, survival, and proliferation of in vitro cultured cells. CO2 levels must be in equilibrium with a high concentration of sodium bicarbonate in the conventional medium to compose a buffering system and balance the pH of the medium. Without CO2 in a culture system, sodium bicarbonate in the conventional medium can be resolved into sodium carbonate that may lead to an increase of the pH in conventional medium and thereby result in the death of cells.
The study of cell biology has been extended into various scientific fields such as life science in space. However, it is impracticable to transport CO2 for cell culture into space due to the limited volume of a spacecraft as well as the requirement of safety. In vitro culture of cells in the particular environment of a spacecraft should be performed under CO2-independent conditions. Therefore, it is necessary to design a CO2-independent culture and induction system for hMSCs that is specifically designed for cell experiments during spaceflight. In the present study, we developed an experimental culture and induction system for cells under CO2-independent conditions and used this system to culture hMSCs and induce hMSCs to differentiate into osteoblast-like cells. Then the morphology, proliferation, and osteogenic potential of cells cultured and induced with the experimental culture and induction system were analyzed. Finally, the feasibility of this experimental culture and induction system used for culture and osteogenic induction of hMSCs in a spaceflight environment is discussed.
2. Materials and Methods
2.1. Preparation of hMSCs
Human bone marrow was kindly provided by the First People's Hospital of Zhejiang. Healthy donors, ranging in age from 21–25 years, gave written consent to the use of bone marrow for research purposes according to procedures approved by the Human Experimentation Committee at Zhejiang Public Health Bureau. hMSCs were isolated and cultured following a previously described method with some modifications (Zong et al., 2010). Briefly, hMSCs were cultured in the ordinary medium consisting of alpha minimum essential medium (α-MEM, Gibco-BRL, Hangzhou, China), 10% MSC-qualified fetal bovine serum (FBS, sterile-filtered, Gibco-BRL), 100 U/mL penicillin, and 100 μg/mL streptomycin (Life Technologies, Beijing, China) and were incubated at 37°C in a high-humidity environment containing 5% CO2. Medium was replaced every 3 days. Cells grew to approximately 80% confluence over approximately 8 days. Cells were subsequently harvested with 0.25% trypsin/1 mM EDTA (Life Technologies) and diluted 1:3 for passage. These cells showed the surface antigen phenotypes of hMSCs (positive for CD29 and CD166, negative for CD34, CD45, CD117, and HLA-DR, with about 85–95% in homogeneity) and had the potential to differentiate into osteoblasts and adipocytes (Xiang et al., 2007; Shi et al., 2010). hMSCs at passage 3 were used for all experiments.
2.2. Preparation of experimental culture and osteogenic medium
The conventional culture medium used in the following experiments was L-DMEM (Gibco BRL, Cargo number 11885084), which is composed of various amino acids, vitamins, inorganic salts (including 3.7 g/L sodium bicarbonate), glucose, pyruvic acid, and so on, as can be seen in Table 1. The conventional culture medium was modified by adding 10% FBS, 0.01 μM dexamethasone, 50 μg/mL ascorbic acid, 10 mM sodium β-glycerophosphate (Gibco BRL), 10,000 U/mL penicillin, and 10,000 U/mL streptomycin (Life Technologies) to prepare the conventional osteogenic medium (Huang et al., 2008; Yang et al., 2010).
CM, conventional culture medium; **EM, experimental culture medium.
To prepare the experimental culture medium, 0.41 g/L sodium bicarbonate, 1.5 g/L disodium hydrogen phosphate dodecahydrate, and 0.07 g/L potassium dihydrogen phosphate (Sinopharm Chemical Reagent Co. Ltd., Shanghai, China) were added to the standard L-DMEM without sodium bicarbonate (Gibco BRL, Cargo number 31600034); then the osmotic pressure of the medium was adjusted with 8.17 g/L sodium chloride (Sinopharm Chemical Reagent Co. Ltd.) to 280–320 mOSM. The components of the experimental culture medium prepared as above are listed in Table 1. The pH of the experimental culture medium was adjusted with NaOH to 7.2 before sterilization by filtration through a 0.22 μm filter. Finally, the experimental osteogenic medium was prepared by adding 10% FBS, 0.01 μM dexamethasone, 50 μg/mL ascorbic acid, 10 mM sodium β-glycerophosphate, 10,000 U/mL penicillin, and 10,000 U/mL streptomycin to the experimental culture medium.
2.3. Culture and osteogenic induction of hMSCs
hMSCs cultured for 2 passages at 37°C in a high-humidity environment containing 5% CO2 were collected and prepared for culture in osteogenic induction experiments. All experiments were designed as six systems: For system A, the experimental culture medium was used to culture hMSCs at 37°C under high-humidity and CO2-independent conditions; for system B, the experimental osteogenic medium was used to induce osteogenesis of hMSCs at 37°C under high-humidity and CO2-independent conditions; for system C, the conventional culture medium was used to culture hMSCs at 37°C in a high-humidity environment containing 5% CO2; for system D, the conventional osteogenic medium was used to induce osteogenesis of hMSCs at 37°C in a high-humidity environment containing 5% CO2; for system E, the conventional culture medium was used to culture hMSCs at 37°C under high-humidity and CO2-independent conditions; for system F, the conventional osteogenic medium was used to induce osteogenesis of hMSCs at 37°C under high-humidity and CO2-independent conditions. hMSCs collected from passage 2 were seeded into six-well plates and cultured to approximately 60–70% confluence before the experimental culture or osteogenic induction was started. The medium was replaced every 2 days. hMSCs were cultured or induced and then harvested for analysis of proliferation or osteogenic potentials at various time points.
During culture or induction, 2 mL of conditioned medium from each well at time points of 12, 24, 48, and 72 h of culture or induction were collected for measurement of pH with a Delta 320 pH Meter (Mettle Toledo, Hangzhou, China). All pH measurements were run in triplicate.
2.4. Analysis of cell viability and proliferation
Cell proliferation was evaluated by determining the double-stranded DNA content. At various time points, cells cultured or induced were collected and stored in ddH2O at−20°C until the assay was performed. For the DNA content assay, cells were thawed at room temperature and lysed in 1 mL protease K lysis buffer (50 mM Tris/HCl, pH 7.6, 0.1% (v/v) Triton X-100). The lysate was assayed for DNA content by using the fluorescent dye Hoechst 33258. Briefly, the lysate was sonicated on ice for 30 s and vortexed for 5–10 s. After centrifugation, the supernatant was collected. Calf thymus DNA was prepared from 0 to 30 μg/mL for a standard. TNE buffer (10 mM Tris base, 1 mM EDTA, and 200 mM NaCl) was added to each well of a 96-well plate at 50 μL/well. Standards and samples were then added to each well at 50 μL/well in triplicate. Hoechst 33258 dye solution (1 μg/mL) was added to each well at 100 μL/well and allowed to incubate for 10 min in the dark at room temperature. After incubation, fluorescence was read on an Auto Microplate Reader (Infinite M200, Tecan, Austria) with an excitation wavelength of 350 nm and an emission wavelength of 450 nm. Cell number was determined by correlating DNA with a known amount of cells. Samples were run in triplicate and compared against calf thymus DNA standards.
For cell viability assays, MTT (5 mg/mL) was added to the culture at 40 μL/well, followed by incubation at 37°C for 4 h. Following incubation, the medium was aspirated and the cells were lysed with 400 μL dimethyl sulfoxide per well for 10 min. Absorbance of each well was recorded in a microplate reader at 490 nm. Data are presented as means±standard deviation. Three wells were performed per treatment.
2.5. Osteogenic assay of hMSCs
For histochemical staining of alkaline phosphatase (ALP), cells cultured or induced in six-well plates for 14 days were fixed in 10% formalin solution at 4°C overnight and dehydrated in a graded series of ethanol. ALP activity was measured by using modified Gomori staining as described previously with a slight modification (Zhang et al., 1988). Briefly, the cells were kept in the well and incubated at 37°C for 4 h with a 1200 μL mixture of 3% disodium-β-glycerophosphate (2.5 mL), 2% barbital sodium (2.5 mL), 2% CaCl2 (5 mL), 25% MgSO4 (0.5 mL), and distilled water (5 mL), and then rinsed with running tap water for 5 min. Twelve hundred microliters of 2% cobaltous nitrate solution was then added for 3 min, after which the plates were washed with running tap water for 2 min. Twelve hundred microliters of 1% ammonium sulfate was then added for 2 min, and the plates were rinsed again with running tap water for 2 min then dried and photographed.
The ALP enzyme modifies a substrate (disodium phenyl phosphate) to produce a red product (free phenol) that corresponds to the amount of enzyme in solution. For ALP activity assays, cells induced for 14 days were incubated in lysis buffer (0.1% Triton X in PBS) for 30 min in 37°C. The ALP activity of the supernatant was measured with an ALP measurement kit (Jiancheng Biotechnology Institute, Nanjing, China) with disodium phenyl phosphate as a substrate, which could be decomposed into phenol and phosphoric acid by ALP. The solution absorption was read at 520 nm, and the activity of ALP was normalized by using the phenol standards. The ALP activity was expressed as micrograms phenol produced per minute per million cells. Samples were run in triplicate and compared against the phenol standards.
2.6. Real-time PCR analysis
Cells cultured or induced for 3, 6, 9, 12, or 14 days were harvested, and total RNA was extracted from cells harvested by using TRIzol (TaKaRa, Dalian, China) according to the manufacturer's instructions. RNA samples were treated with DNase I (Fermentas, Shanghai, China) to remove residual genomic DNA. Total RNA was quantified at an absorbance of 260 nm by a spectrophotometer (Jenway Genova, Hangzhou, China). The RNA samples had an A260:A280 ratio of 2.0 to guarantee high purity. Two micrograms of total RNA from each sample was subjected to first-strand cDNA synthesis by using RevertAid First Strand cDNA synthesis Kit (Fermentas) with oligo d(T). mRNA expression was quantified by the real-time PCR performed in Bio-Rad iCycler 3.0 (Bio-Rad, Shanghai, China) by using Qiagen Green Master Mix. Primer sequences were derived from gene sequences available through GenBank: ALP: forward 5′-ACA AGC ACT CCC ACT TCA TC-3′, reverse 5′-ATT CTG CCT CCT TCC ACC-3′; osteocalcin (OCN): forward 5′-AGC CAC CGA GAC ACC ATG AGA G-3′, reverse 5′-GTG CCT GGA GAG GAG CAG AAC T-3′; collagen Iα (COL Iα): forward 5′-TCA AAG GCA ATG CTC AAA CA-3′, reverse 5′-ACA TCA AGA CAA GAA CGA GGT AG-3′; runt-related transcription factor 2 (Runx2): forward 5′-GTG GAC GAG GCA AGA GTT-3′, reverse 5′-GGT GCA GAG TTC AGG GAG-3′; glyceraldehyde 3-phosphate dehydrogenase (GAPDH): forward 5′-GAA GGT CGG AGT CAA CGG-3′, reverse 5′-GGA AGA TGG TGA TGG GAT T-3′. Quantification of mRNA was performed with the comparative threshold cycle method (ΔΔCt) with GAPDH as the internal reference, and relative gene expression was reported as 2−ΔΔCT.
2.7. Western blot
Cells were homogenized by sonication in RIPA lysis buffer (Beytime Biotech, Wuhan, China) with 1 mM PMSF (Sigma, Shanghai, China) and 0.1% phosphatase inhibitors (Sigma). Cell homogenates were centrifuged at 12,000g for 5 min at 4°C, and the supernatant was collected. An equal volume of 2× sample buffer was added to each sample, after which the samples were boiled for 5 min. Fifteen microliters of lysate was loaded into each lane of an 8% SDS polyacrylamide gel. The proteins in the gel were transferred to a PVDF membrane (Millipore Corporation, Shanghai, China). After blocking with 5% skimmed milk, the membrane was incubated at 4°C overnight with primary antibody against GAPDH (1:2000) (Amyjet Scientific, Wuhan, China), Runx2 (1:800) (Abcam, Beijing, China), ALP (1:10000), OCN (1:1500), and COL Iα (1:1000) (Epitomics, Shanghai, China) followed by three washes with PBST (PBS supplemented 0.1% Tween-20), and 1 h incubation with horseradish peroxidase–conjugated goat anti-rabbit (1:3000; Huaan, Hangzhou, China) at room temperature. Immunoreactive signals were detected with the enhanced chemiluminescent reagent (Tiangen Biotech, Shanghai, China) and quantified in triplicates by normalizing band intensities against the loading controls on scanned films by Image J Software.
2.8. Statistical analysis
All experiments were repeated at least three times, and the representative data were presented as means±standard deviation where indicated. Statistical analysis was performed by factorial analysis of variance (two-way ANOVA), followed by Dunnett's test for comparing treatments with controls. A probability value of less than 0.05 was considered statistically significant: *(P<0.05), **(P<0.01), ***(P<0.001).
3. Results
3.1. pH dynamics of conditioned medium during culture and osteogenic induction
During culture or osteogenic induction, the conditioned medium was collected after 12, 24, 48, and 72 h, and pH was measured (Fig. 1). The pH of conditioned medium derived from conventional culture medium under CO2-dependent conditions kept a stable level during the culture of cells. The average pH value was 7.39±0.21 (the highest value was 7.61, and the lowest was 7.20). The pH value of conditioned medium after culturing for 12 h was 0.34 higher than the pH value at the starting time point of culture. There was no significant difference between pH values at different time points except for the pH value at the starting time point (P>0.05). However, the pH level of conditioned medium derived from conventional culture medium under CO2-independent conditions increased from 7.20 to 9.08±0.13 after culturing for 12 h and was significantly different from the pH value at the starting time point (P<0.01). The pH value remained above 9.00 until the experiment was ended after 72 h of culture. These results indicate that the culture system without CO2 could result in the alkalization of conventional culture medium, which was unfit for culture of cells. Similar to what occurred under CO2-independent conditions, the experimental culture medium was used to culture hMSCs for 72 h. It was found that the pH level of conditioned medium decreased slightly. The pH value of conditioned medium in this culture system was 7.19±0.07, 7.11±0.09, 7.02±0.11, and 6.94±0.15 at 12, 24, 48, and 72 h, respectively. For osteogenic induction, the pH level of conditioned medium showed a tendency similar to that indicated above. These results suggest that the conditioned medium from experimental culture or osteogenic medium under CO2-independent conditions could acidize gradually. However, this acidification was modest in a short time of culture, such as in 2 days.
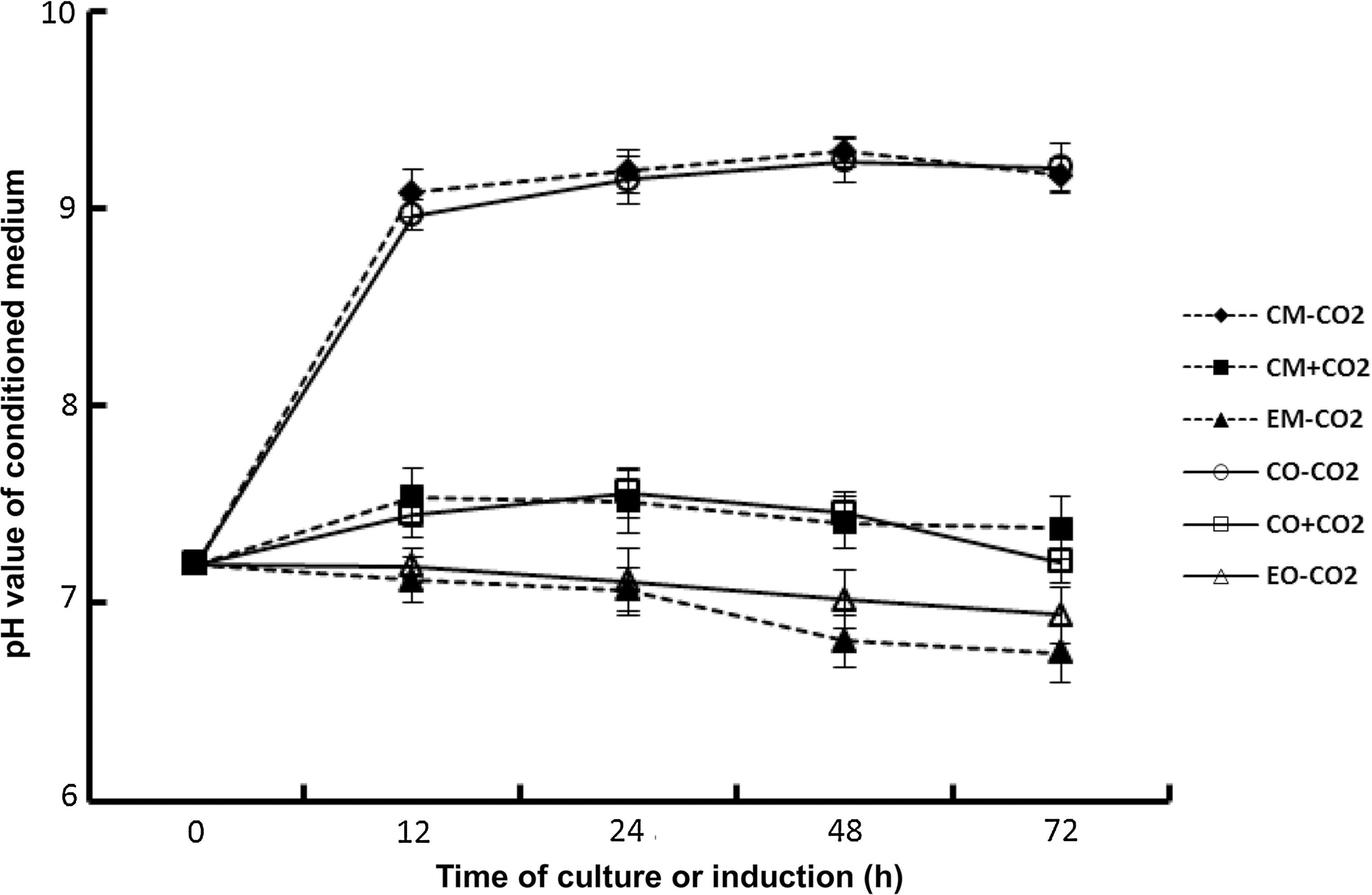
pH dynamics of conditioned medium during culture and osteogenic induction. The conditioned medium was collected at various given times of culture or osteogenic induction and pH measured with a Delta 320 pH Meter. Samples were run in triplicate. EM-CO2, experimental culture medium under CO2-independent conditions; EO-CO2, experimental osteogenic medium under CO2-independent conditions; CM+CO2, conventional culture medium under CO2-dependent conditions; CO+CO2, conventional osteogenic medium under CO2-dependent conditions; CM-CO2, conventional culture medium under CO2-independent conditions; CO-CO2, conventional osteogenic medium under CO2-independent conditions.
3.2. Morphology and proliferation of hMSCs in the CO2-independent culture and osteogenic induction systems
We used six different culture or osteogenic systems to culture or induce hMSCs (see six groups in Materials and Methods). The morphology of hMSCs cultured with conventional culture medium or induced with conventional osteogenic medium under CO2-independent conditions was changed from a fibrous morphology into a spindle-shaped morphology after 24 h of culture or induction and atrophied gradually even if the medium was replaced every 2 days (Fig. 2A and 2B). However, the morphology of hMSCs cultured with experimental culture medium under CO2-independent conditions kept the fibrous morphology throughout culturing for 4 days, which is similar to what occurred with hMSCs that were cultured with conventional culture medium in an environment with 5% CO2 (Fig. 2C and 2D). For the osteogenic induction, cells induced with experimental osteogenic medium under CO2-independent conditions and conventional osteogenic medium in the environment with 5% kept a fibrous morphology until day 2 and then changed into a spindle-shaped morphology at day 4 (Fig. 2E and 2F). However, no dead cells were detected. As osteogenic induction progresses, the cells in these two systems could differentiate into osteoblast-like cells.
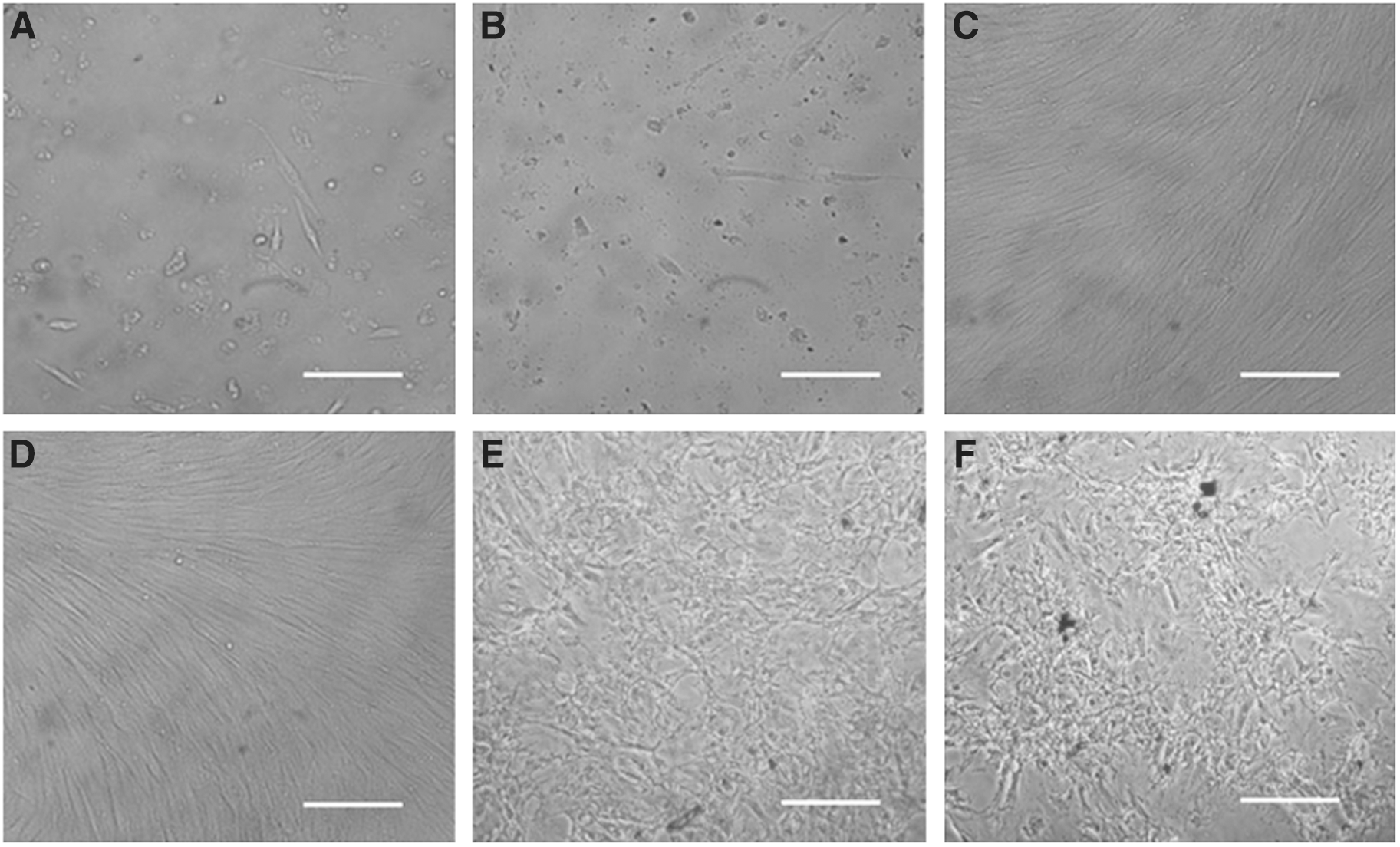
Morphology of cells from CO2-independent culture and osteogenic induction systems. hMSCs were cultured or induced for 4 days in different culture or induction systems and then photographed. (
The growth characteristics of cells were assessed by analyzing the total DNA content at different time points during culture or induction. As shown in Fig. 3A, the densities of cells cultured with conventional culture medium or induced with conventional osteogenic medium under CO2-independent conditions decreased significantly more than 50% after culture or induction for 2 days compared to the initial density of cells. Also, few cells could be detected after culture or induction for 4 days. The density of cells cultured with experimental culture medium under CO2-independent conditions continued to increase at a logarithmic growth rate and reached its peak at day 4. Cell growth in this system was similar to that in the system with conventional culture medium under CO2-dependent conditions. There was no difference in cell densities between these two systems (P>0.05). The density of cells induced with experimental osteogenic medium under CO2-independent conditions and conventional osteogenic medium under CO2-dependent conditions increased slightly and reached its peak at day 6 and then remained the same until the end of induction.
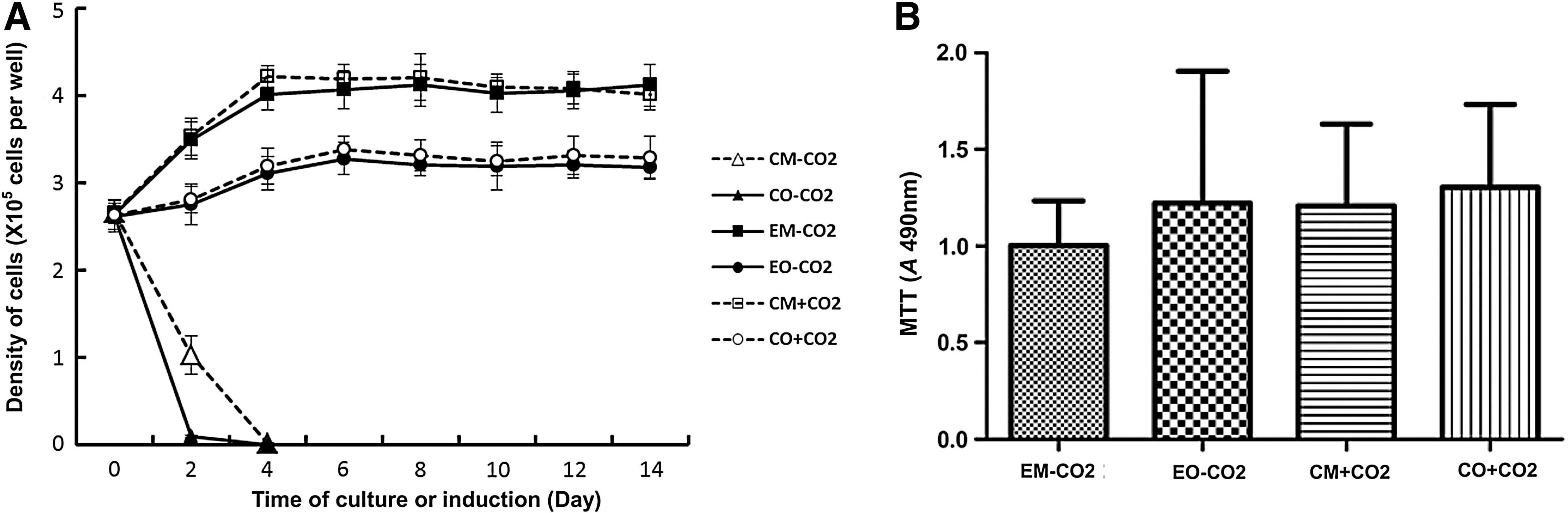
Proliferation and viability of cells from CO2-independent culture and osteogenic induction systems. (
The effect of experimental culture medium or experimental osteogenic medium on cell viability was determined by MTT assay as shown in Fig. 3B. After hMSCs were cultured for 14 days, there was no significant difference in cell viabilities between the experimental culture medium under CO2-independent conditions and the conventional culture medium under CO2-dependent conditions (P>0.05). Also, after hMSCs were induced for 14 days, there was no significant difference in cell viabilities between the experimental osteogenic medium under CO2-independent conditions and the conventional osteogenic medium under CO2-dependent conditions (P>0.05).
3.3. Osteogenic potentials of hMSCs in CO2-independent osteogenic induction systems
According to the pH dynamics of experimental osteogenic medium under CO2-independent conditions described above, we used experimental osteogenic medium to induce the osteogenesis of hMSCs, replacing the medium every 2 days. The osteogenic induction with conventional osteogenic medium under CO2-dependent conditions was used as the positive control, and the cultures with experimental culture medium under CO2-independent conditions and conventional culture medium under CO2-dependent conditions were used as negative controls. All cells induced and cultured for 14 days were histochemically stained to detect the activation of ALP in cells, as shown in Fig. 4A. Cells induced with experimental osteogenic medium under CO2-independent conditions and conventional osteogenic medium under CO2-dependent conditions had higher activation levels of ALP, which indicates that induction systems with experimental osteogenic medium under CO2-independent conditions could induce hMSCs to differentiate into osteoblast-like cells in a similar manner as conventional osteogenic medium under CO2-dependent conditions. Cells cultured with experimental culture medium under CO2-independent conditions and conventional culture medium under CO2-dependent conditions showed a reduction of ALP activation. Cells cultured or induced with each of the different systems for 14 days were harvested and then lysed in lysis buffer for analysis of ALP activity as shown in Fig. 4B. The ALP activity in cells from the induction system with experimental osteogenic medium under CO2-independent conditions was not significantly different from those cells cultured in the induction system with conventional osteogenic medium under CO2-dependent conditions (P>0.05). Those cells cultured with experimental culture medium under CO2-independent conditions showed a lower level of ALP activity similar to the level in cells cultured with conventional culture medium under CO2-dependent conditions.
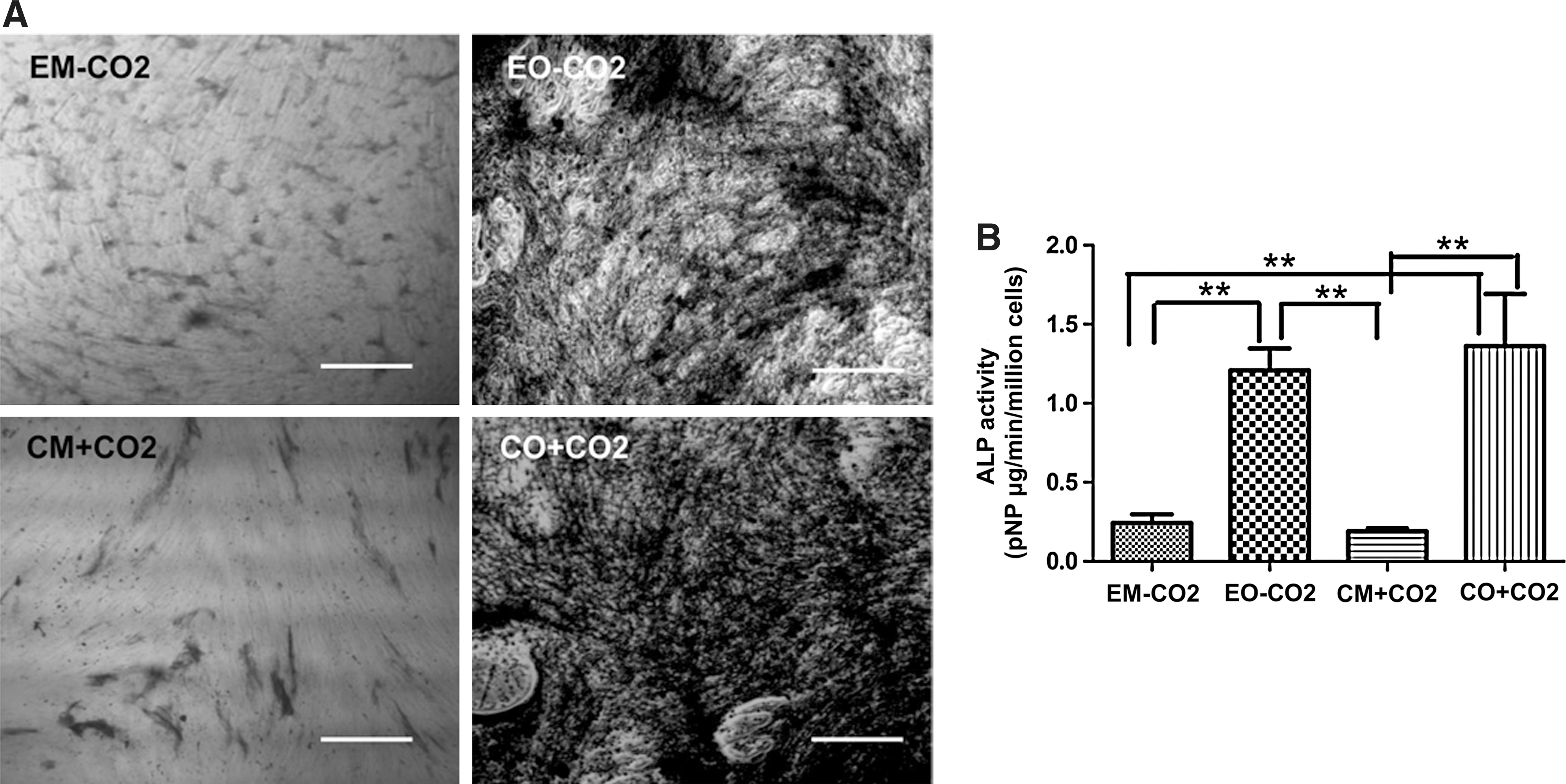
Activity of ALP in cells from CO2-independent osteogenic induction systems. (
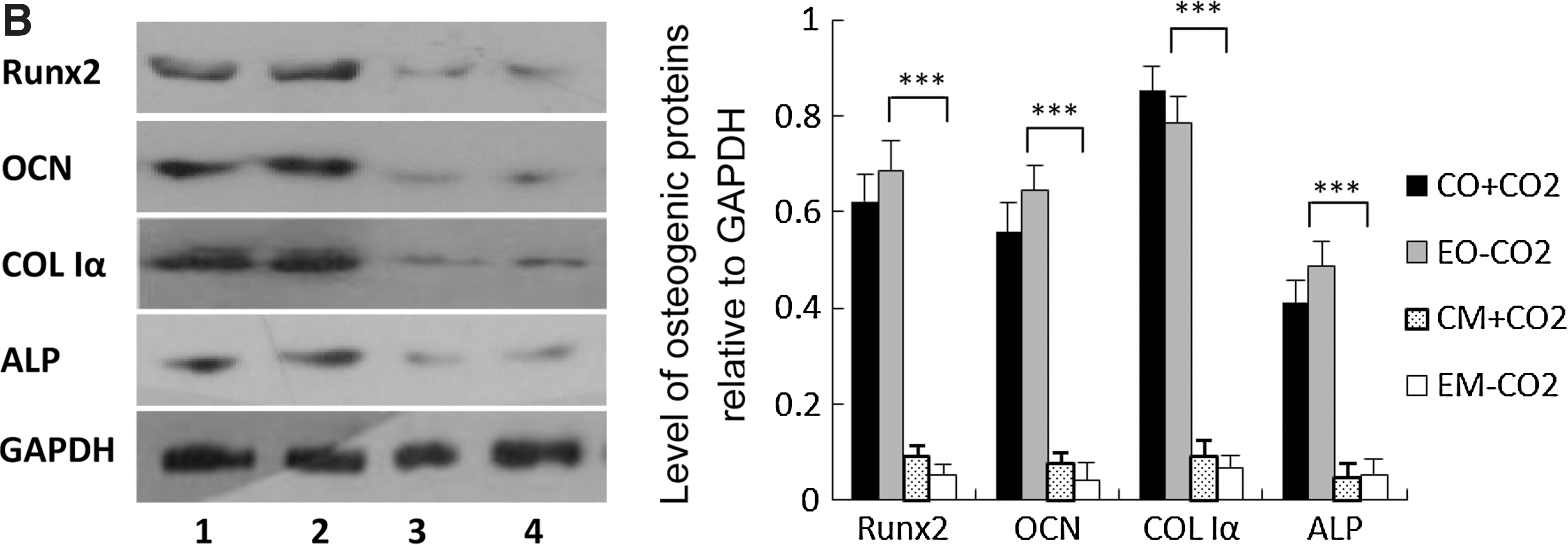
High expression of ALP, OCN, and COL I genes is characteristic of osteogenesis. After culture or induction for 3, 6, 9, 12, and 14 days, the expression level of ALP, COL Iα, and OCN was examined by semiquantitative RT-PCR. The mRNA levels of these three osteoblast-specific genes in cells from the induction system with experimental osteogenic medium under CO2-independent conditions were significantly higher than those in cells from culture systems with experimental culture medium under CO2-independent conditions and conventional culture medium under CO2-dependent conditions (P<0.001) and were not significantly different from those in cells from the induction system with conventional osteogenic medium under CO2-dependent conditions (P>0.001) (Fig. 5A). Runx2 is an important osteogenic transcription factor that can promote the expression of osteogenic genes such as ALP, OCN, and COL I. We also examined the expression level of this gene in cells from different systems. The experimental osteogenic medium under CO2-independent conditions could promote the expression of Runx2 in hMSCs, and the expression level of Runx2 was not significantly different from that in cells induced by conventional osteogenic medium under CO2-dependent conditions. The effect of experimental osteogenic medium on the expression of osteogenic genes in hMSCs was also further confirmed at the protein level (Fig. 5B). The high expression of osteogenic genes showed that experimental osteogenic medium could promote hMSCs to differentiate into osteoblast-like cells under CO2-independent conditions.

Expression of osteogenic genes in cells from CO2-independent osteogenic induction systems. (
4. Discussion
The vital activities of cells are maintained by biological energy that is obtained from glucose metabolism in cells. Unlike in vivo cellular metabolism, cells cultured in vitro consume glucose in the glycolysis pathway and produce abundant amounts of lactic acid. In vitro, relatively small amounts of glucose can be exhaustively oxidized through the tricarboxylic acid cycle. Fitzpatrick et al. (1993) found that 96% glucose was catabolized through the glycolysis pathway, and only 0.21% and 3.3% glucose were catabolized through the tricarboxylic acid cycle and pentose phosphate pathway, respectively, when hybridoma cells were cultured in the medium with 10 mM glucose. Therefore, the production of lactic acid in vitro may decrease the pH level of culture environment. To antagonize the decrease of pH in culture environment, the conventional medium is usually supplemented with a high concentration of sodium bicarbonate that composes a buffering system with CO2 in the culture environment and plays a synergistic role in balancing the pH level in medium. As shown in the results above, the pH level in conventional culture medium was increased after cells were cultured for a given time under CO2-independent conditions. This effect was attributed to hydrolysis of HCO3 − into CO3 2+, which resulted in the increase of pH in the medium and led to cell death. Thus, a culture system composed of a given concentration of conventional medium, serum, O2, CO2, and 37°C constant temperature has been used for in vitro culture of mammalian cells. However, this culture system has limited application in some specific environments, such as those encountered during spaceflight. Therefore, it is necessary to develop an experimental CO2-independent culture system.
In previous studies, some modified culture systems have been used to culture mammalian cells, such as the hydroxyethyl-piperazine-ethanesulfonic acid (HEPES) buffer system. HEPES is a zwitterionic organic chemical buffer agent with a pKa of 7.3 at 37°C. In the physiological context, HEPES has an excellent buffer capacity in a pH range of 7.2–7.6. Media supplemented with HEPES exhibit more effective buffering in the physiological pH range with most cell cultures than media in which the normal bicarbonate buffering system alone is used. Since the buffering capacity of HEPES is independent of the CO2 concentration, the HEPES system has been widely used in cell culture. Some medium formulations incorporate HEPES in addition to CO2/sodium bicarbonate. These media have the advantage of maintaining optimal pH in an open system when the culture vessel is removed from the enriched CO2 atmosphere of the incubator. However, this compound can be toxic, especially for some differentiated cell types. HEPES has been shown to greatly increase the sensitivity of media to the phototoxic effects induced by exposure to fluorescent light (Zigler et al., 1985; Lepe-Zuniga et al., 1987). In addition, the components of medium have also been modified for CO2-independent cell culture. Gao et al. (2010) developed a CO2-independent culture system by adding various components to DMEM and used this culture system to culture bladder cancer cells T-24. They found that the experimental, CO2-independent medium under CO2-independent conditions could promote the proliferation of T-24 cells in a similar manner as CO2-dependent DMEM under CO2-dependent conditions. However, T-24 cells also had proliferation potential in the culture system with CO2-dependent DMEM under CO2-independent conditions, although their proliferation potential decreased compared to the culture systems with experimental, CO2-independent medium under CO2-independent conditions and CO2-dependent DMEM under CO2-dependent conditions.
In the present study, we developed an experimental culture medium through modification of inorganic salt components based on L-DMEM, that is, changed the concentration of sodium bicarbonate, disodium hydrogen phosphate dodecahydrate, and potassium dihydrogen phosphate. This modification of the salt buffer system was designed to improve the buffering capacity of the medium pH under CO2-independent conditions. Our results show that culture of hMSCs with conventional culture medium under CO2-independent conditions would result in a quick increase of medium pH in a short time. Culturing with this system for 4 days resulted in a sharp decrease of cells. Therefore, culture of hMSCs requires a narrower range of pH than culture of cancer cells. The increase of pH in the medium may be attributed to a high concentration of sodium bicarbonate in the conventional culture medium that may be hydrolyzed into a high concentration of CO3 2+ (NaHCO3→Na++HCO3 −, HCO3 −+H2O=H2CO3+OH−). This results in an increase of pH in the medium in conditions that lack additional CO2 and thereby leads to a sharp decrease in the number of hMSCs. However, whether the sharp decrease of hMSCs should be attributed to necrosis and/or apoptosis of hMSCs resulting from an increase of pH in the medium needs further examination. For the experimental culture medium, the concentration of sodium bicarbonate was decreased from 3700 mg/L to 410 mg/L, which should reduce significantly the level of CO3 2+ produced in the medium. The decrease of cation concentration in the medium had been compensated by adding sodium dihydrogen phosphate dodecahydrate and potassium dihydrogen phosphate to the medium. The experimental culture medium showed a slight decrease of pH when hMSCs were cultured for a given time. It is inferred that lactic acid produced by cells through the glycolysis pathway may result in this slight decrease of pH level due to the lower concentration of sodium bicarbonate in the medium. However, the experimental culture medium could promote the proliferation of hMSCs under CO2-independent conditions if the medium was replaced every 2 days despite a slight pH decrease in the experimental culture medium. After culture of hMSCs, the pH level of experimental culture medium was decreased from 7.2 to 7.02, and the reduced magnitude of pH was about 0.2. This suggests that this buffer system should be conducive to growth and proliferation of hMSCs.
Based on the experimental culture medium above, an experimental osteogenic medium was prepared by adding a given concentration of dexamethasone, ascorbic acid, and sodium β-glycerophosphate into experimental culture medium. The conventional osteogenic medium was prepared by adding a given concentration of these three osteogenic factors to the conventional culture medium. This conventional osteogenic medium has been used for osteogenic induction experiments of hMSCs (Huang et al., 2008; Yang et al., 2010). Here, the experimental osteogenic medium should have the same concentration of cation as the experimental culture medium. Therefore, we tried to use the experimental osteogenic medium with these three factors to examine the effect of this buffer system on the osteogenesis of hMSCs under CO2-independent conditions. The results showed that the experimental osteogenic medium could promote the osteogenesis of hMSCs under CO2-independent conditions. The expression of osteogenic genes, such as ALP, OCN, and COL Iα, was increased in the osteogenic system with experimental osteogenic medium under CO2-independent conditions in a similar manner as the osteogenic system with conventional osteogenic medium under CO2-dependent conditions. The expression of Runx2, an important osteogenic transcription factor that can bind to osteoblast-specific-acting element in the promoter region of osteogenic genes to initiate the expression of target genes (Ducy et al., 1997; Franceschi and Xiao, 2003), was also upregulated in cells induced by experimental osteogenic medium under CO2-independent conditions. These results demonstrate that the addition of osteogenic factors in the experimental culture medium should not change the buffer system and is suitable for osteogenesis of hMSCs.
5. Conclusion
The experimental culture medium developed by our lab can promote the proliferation of hMSCs under CO2-independent conditions. Also, the experimental osteogenic medium prepared on the basis of the experimental culture medium can induce the osteogenesis of hMSCs under CO2-independent conditions. These experimental systems should be considered in some specific and CO2-independent research, such as culture or differentiation induction of cells in a spaceflight environment. However, further improvement of experimental medium, specifically the pH-buffering potential, remains for future study.
Footnotes
Acknowledgments
The authors thank the healthy donors of the First People's Hospital of Zhejiang for kindly providing bone marrow and thank Dr. Matthew Lynes at Harvard University for critically reading this manuscript. This work was supported in part by grants from National Basic Research Program of China (2012CB967902), Strategically Guiding Scientific Special Project from Chinese Academy of Sciences (XDA04020202-23), Opening Foundation of the State Key Laboratory of Space Medicine Fundamentals and Application (SMFA12K02), and Special Project of Strategic Precursor Science and Technology in CAS (XDA04020202-23).
Abbreviations
ALP, alkaline phosphatase; COL Iα, collagen Iα; FBS, fetal bovine serum; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HEPES, hydroxyethyl-piperazine-ethanesulfonic acid; hMSCs, human mesenchymal stem cells; MEM, minimum essential medium; OCN, osteocalcin; Runx2, runt-related transcription factor 2.