
Abstract
Tetrapyrroles are essential to basic biochemical processes such as electron transfer and photosynthesis. However, it is not known whether these evolutionary old molecules have a prebiotic origin. We have serendipitously obtained pyrroles, which are the corresponding monomers, in laboratory experiments that simulated the interaction of amino acid–containing seawater with molten lava. The thermal pyrrole formation from amino acids, which so far has only been reported for special cases, can be explained by the observation that the amino acids become metal bonded, for example in (CaCl2)3(Hala)2·6H2O (Hala=DL-alanine), when the seawater evaporates. At a few hundred degrees Celsius, sea salt crusts also release hydrochloric acid (HCl). On primordial volcanic islands, the volatile pyrroles and HCl must have condensed at cooler locations, for example, in rock pools. There, pyrrole oligomerization may have occurred. To study this possibility, we added formaldehyde and nitrite, two species for which plausible prebiotic sources are known, to 2,4-diethylpyrrole and HCl. We found that even at high dilution conjugated (oxidized) oligomers, including octaethylporphyrin and other cyclic and open-chain tetrapyrroles, were formed. All experiments were conducted under rigorously oxygen-free conditions. Our results suggest that primitive versions of present-day biological cofactors such as chlorophylls, bilins, and heme were spontaneously abiotically synthesized on primordial volcanic islands and thus may have been available to the first protocells. Key Words: Abiotic organic synthesis—Early Earth—Evaporites—Laboratory simulation experiments—Volcanism. Astrobiology 13, 578–595.
1. Introduction
A
As with any prebiotic synthesis, the simulation of a protocell-independent formation of porphyrins must be consistent with the geochemical and geophysical conditions on primitive Earth, as far as they are known (Strasdeit, 2010). Key steps of prebiotic chemical evolution took place during the first billion years of Earth's history, in the Hadean and early Archean eon (Brack, 2005). In this period, small volcanically active landmasses started to protrude from the ocean (Buick and Dunlop, 1990; Buick et al., 1995; Van Kranendonk et al., 2003, 2008; Westall, 2003; Nisbet and Fowler, 2004; Westall et al., 2006; Taylor and McLennan, 2009; Arndt and Nisbet, 2012). Thus, volcanic eruption clouds were probably a common phenomenon. Lightning in these clouds is thought to have produced organic compounds abiotically (Hill, 1992; Basiuk and Navarro-González, 1996; Navarro-González and Segura, 2004). This idea is supported by laboratory experiments demonstrating that several different amino acids are formed by spark discharges in steam-rich reducing gas mixtures (Miller, 1955; Johnson et al., 2008). Objects from space were additional sources of prebiotic amino acids (Pizzarello et al., 2006; Martins and Sephton, 2009), whereas the contribution of the bulk atmosphere is still unclear (Plankensteiner et al., 2004, 2006; Cleaves et al., 2008; Kuwahara et al., 2012).
It has been proposed that already in prebiotic times the ocean contained substantial concentrations of inorganic salts. In fact, the early seawater could have been even more saline than the modern one (Knauth, 1998, 2005; Izawa et al., 2010). This idea is consistent with the presence of highly saline fluid inclusions in ∼3.75 billion-year-old quartz (Appel et al., 2001); sodium chloride and potassium chloride in chondritic meteorites (Barber, 1981; Zolensky et al., 1999; Bridges et al., 2004); and evaporite minerals in ancient terrains on Mars (Gendrin et al., 2005; Osterloo et al., 2008; Glotch et al., 2010). Present-day observations show that large amounts of seawater evaporate when lava reaches the coast of a volcanic island and flows into the ocean. As a result, solid sea salt is formed and exposed to temperatures up to some hundred degrees Celsius (Edmonds and Gerlach, 2006). Organic molecules present in the seawater are embedded in and heated together with the sea salt crusts. It is reasonable to assume that these processes also occurred at the volcanic coasts of early Earth and that prebiotic amino acids that were dissolved in the ocean were involved.
In the present paper, we first explore the prebiotic chemistry associated with the interaction between seawater and lava. We present experimental results that show that pyrroles can form from amino acid–containing salt crusts. We further demonstrate that pyrroles form oligopyrroles, including porphyrins, under experimental conditions simulating primordial volcanic islands. Based on these results, a prebiotic reaction sequence is proposed whose individual steps proceeded spontaneously in different environments of volcanic islands. This sequence led from volcanic gases to potential photopigments and electron transfer molecules.
2. Materials and Methods
2.1. Chemicals and elemental analyses
Racemic isovaline was prepared as described before (Strasdeit et al., 2001). All other chemicals were purchased commercially and used without further purification. The 13C-substituted DL-alanines were obtained from Sigma-Aldrich (1-13C) and Eurisotop (2-13C and 3-13C), respectively. Biliverdin hydrochloride: 95% (Chemos GmbH); 2,4-diethylpyrrole: 98% (Frontier Scientific); formaldehyde: 16% aqueous solution, methanol free, Ultra Pure (Polysciences); hemin (ferriprotoporphyrin chloride): ≥98% (Roth); octaethylporphyrin: 97% (Aldrich). All metal salts used were of analytical grade. The water used throughout this study was distilled twice in a quartz apparatus.
CHN analyses were obtained from Mikroanalytisches Labor Pascher, Remagen, Germany. Cl analyses were performed by Mikroanalytisches Labor Pascher (compounds
2.2. Gas chromatography–mass spectrometry
An Agilent 6890N/5973 GC/MSD system equipped with a DB-5MS capillary column (30 m length, 0.25 mm inner diameter, 0.25 μm film thickness) was used. Measurement conditions: column temperature 50–200°C, helium carrier gas, inlet pressure 0.874 bar, inlet temperature 280°C, MSD operated in full-scan mode (15–350 amu, 2.36 scans s−1, electron impact ionization at 70 eV, 2.5 min solvent delay). For quantification purposes, a flame ionization detector at 250°C was used instead of the MSD.
2.3. Spectroscopic measurements
The following instruments and measuring conditions were used. UV-visible absorption spectroscopy: Analytik Jena SPECORD 210 spectrometer, resolution 0.5 nm, scan speed 1 nm s−1; the solutions were measured in a gas-tight quartz cuvette. IR spectroscopy: Thermo Nicolet 5700 FT-IR spectrometer, attenuated total reflection mode. Raman microscopy: Horiba Jobin Yvon LabRAM spectrometer, λ excitation=633 nm. Proton nuclear magnetic resonance (1H NMR) spectroscopy: Varian Unity Inova 300 spectrometer, 300 MHz.
2.4. High-resolution/high-accuracy mass spectrometry
The spectra were obtained with a Bruker LTQ Orbitrap XL mass spectrometer (ThermoScientific, Bremen, Germany) equipped with an electrospray ionization (ESI) ion source. An external calibration was performed according to the manufacturer's guidelines (calibration mixture: SDS, caffeine, sodium taurocholate, MRFA, and Ultramark 1621). The mass spectrometer was operated in positive ion mode with an ionization voltage of 4.5 kV. The capillary temperature was 275°C. N2 served as sheath-gas (2 mL min−1). The data were recorded in the 100–2000 Da mass range by the Orbitrap mass analyzer, which was operated with a target mass resolution of 30,000 (defined at m/z 400). Samples were introduced by direct infusion at a flow rate of 10 μL min−1. The data were processed with Xcalibur software (version 2.0, ThermoScientific, Bremen, Germany).
2.5. X-ray crystallography
A suitable crystal of (CaCl2)3(Hala)2·6H2O (
Powder diffraction patterns were measured with CuKα radiation with the use of a Bruker D8 Focus diffractometer equipped with a Sol-X energy dispersive detector.
2.6. Preparation of the salt–amino acid mixtures
NaCl (41.20 g, 705 mmol), KCl (1.12 g, 15 mmol), MgCl2·6H2O (16.26 g, 80 mmol), CaCl2·2H2O (2.21 g, 15 mmol), and the respective α-amino acid (10 mmol) were dissolved in water. The solution was evaporated at room temperature. The resulting salt crust was crushed, and adhering water was removed under vacuum. When amino acid mixtures were studied, between 1 and 6 mmol of each individual amino acid was employed.
2.7. Synthesis of the amino acid complexes
Preparation of (CaCl2)3(Hala)2·6H2O (
The compounds CaCl2(Hala)2·3H2O (
Infrared and Raman band positions of
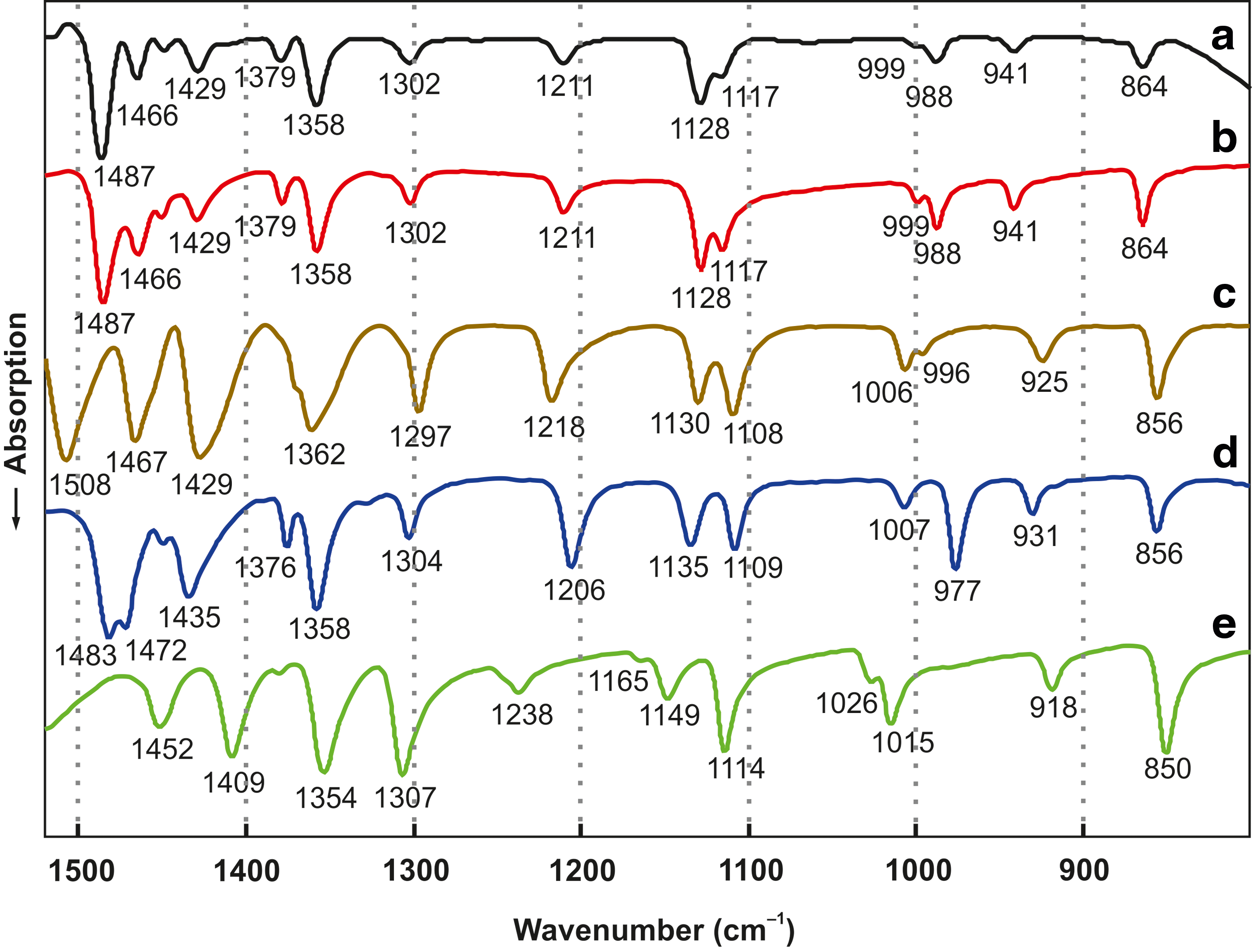
Infrared spectra: (
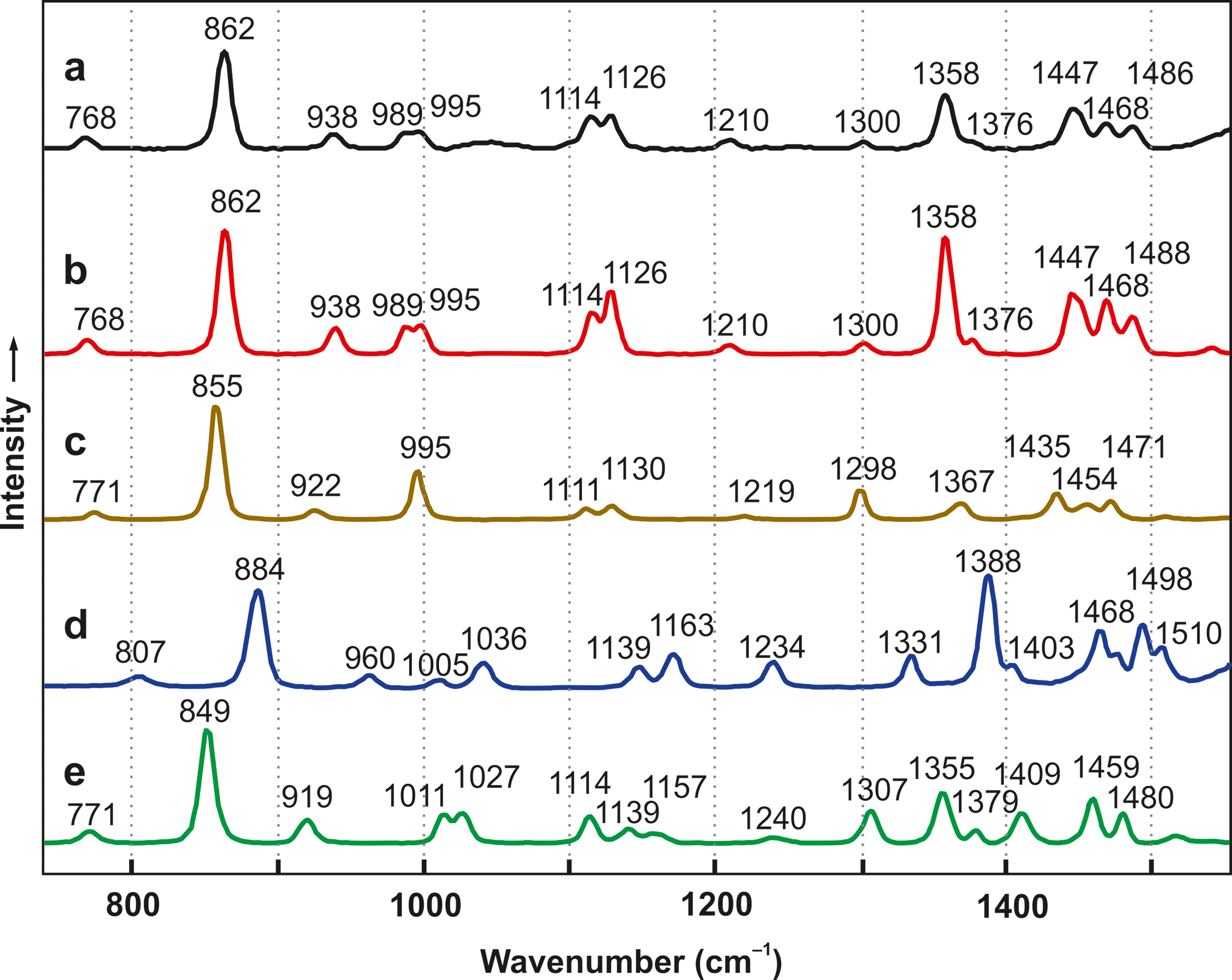
Raman spectra: (
2.8. Thermolyses
The thermolysis apparatus used is shown in Fig. 3. Typically, 55 g of a salt–amino acid mixture, 10 g of a metal complex, or 1 g of 13C-substituted

Two views of the thermolysis apparatus, which consists of a furnace (F), quartz tube (Q), glass rod (G) for placing the sample container (SC) into the heating zone (H), sample storage area (SA), Teflon-sealed connections (C), water-cooled (T1), and liquid nitrogen–cooled traps (T2, T3). Color images available online at
To analyze for ammonia and carbon dioxide, the gas stream was split at the outlet and passed through hydrochloric acid and barium hydroxide solutions where NH4Cl and BaCO3, respectively, formed. The products were identified by IR spectroscopy. In these experiments, the traps T2 and T3 were cooled with ice.
The experimental setup of Fig. 3, with small variations, was also used to study the thermal stability of biliverdin hydrochloride, hemin, and octaethylporphyrin (OEP). An accurately weighed sample of the tetrapyrrole (1 mg) was held at a fixed temperature in a slow nitrogen stream for up to 24 h. Subsequently, the residue was extracted with methanol, N,N-dimethylformamide, and dichloromethane, respectively. The amount of undecomposed tetrapyrrole in the extract was determined spectrophotometrically. The following extinction coefficients were measured and used in the calculations: biliverdin hydrochloride, ɛ=50,200 L mol−1 cm−1 (376.5 nm, methanol); hemin, ɛ=75,300 L mol−1 cm−1 (399 nm, N,N-dimethylformamide); OEP, ɛ=177,200 L mol−1 cm−1 (398 nm, dichloromethane) [literature value: 176,000 L mol−1 cm−1 at 397 nm (Kinoshita et al., 1992)].
2.9. Synthesis of alkylpyrroles for identification purposes
General procedure: pyrrole or an alkylpyrrole (ca. 0.5 g) was dissolved in anhydrous tetrahydrofuran (10 mL) under an argon atmosphere. The solution was cooled to −20°C, and one equivalent of n-butyllithium was added dropwise. After the solution had warmed up to room temperature, it was again cooled to −20°C, and one equivalent of ethyl or methyl iodide was added. After stirring overnight at room temperature, a saturated solution of ammonium chloride was added. The product (mixture) was extracted twice with diethyl ether (10 mL each) and subjected to column chromatography (silica gel; hexane–ethyl acetate, 9:1, v/v). The absence or low intensity of 1H NMR signals in the 3.1–4.0 ppm range (NCH 3 and NCH 2CH3; CDCl3 as solvent) indicated that N-alkylated products were neglectable.
By using the above procedure, pyrrole and several commercially available alkylpyrroles were C-alkylated. In some cases, only one product was possible, for example in the C-alkylation of 3-ethyl-2,4-dimethylpyrrole. Mostly, however, mixtures were obtained that often contained positional isomers. They were nevertheless useful in identifying the products of the thermolysis experiments. Positional isomers of alkylpyrroles exhibit virtually identical mass spectra. However, the GC retention times can be used to distinguish them. A straightforward example is shown in Fig. 4. When 2,4-dimethylpyrrole was monomethylated, both trimethyl isomers were formed. In contrast, the monomethylation of 2,5-dimethylpyrrole can only yield 2,3,5-trimethylpyrrole (t=9.8 min). The peak at t=10.5 min, which shows the same mass spectrum, must therefore belong to the 2,3,4-isomer. Thus, it was possible to unambiguously assign the corresponding peaks in the GC of the thermolysis products. In addition, the complete C-methylation of both 2,4-dimethylpyrrole and 2,5-dimethylpyrrole yielded 2,3,4,5-tetramethylpyrrole (t=13.3 min).
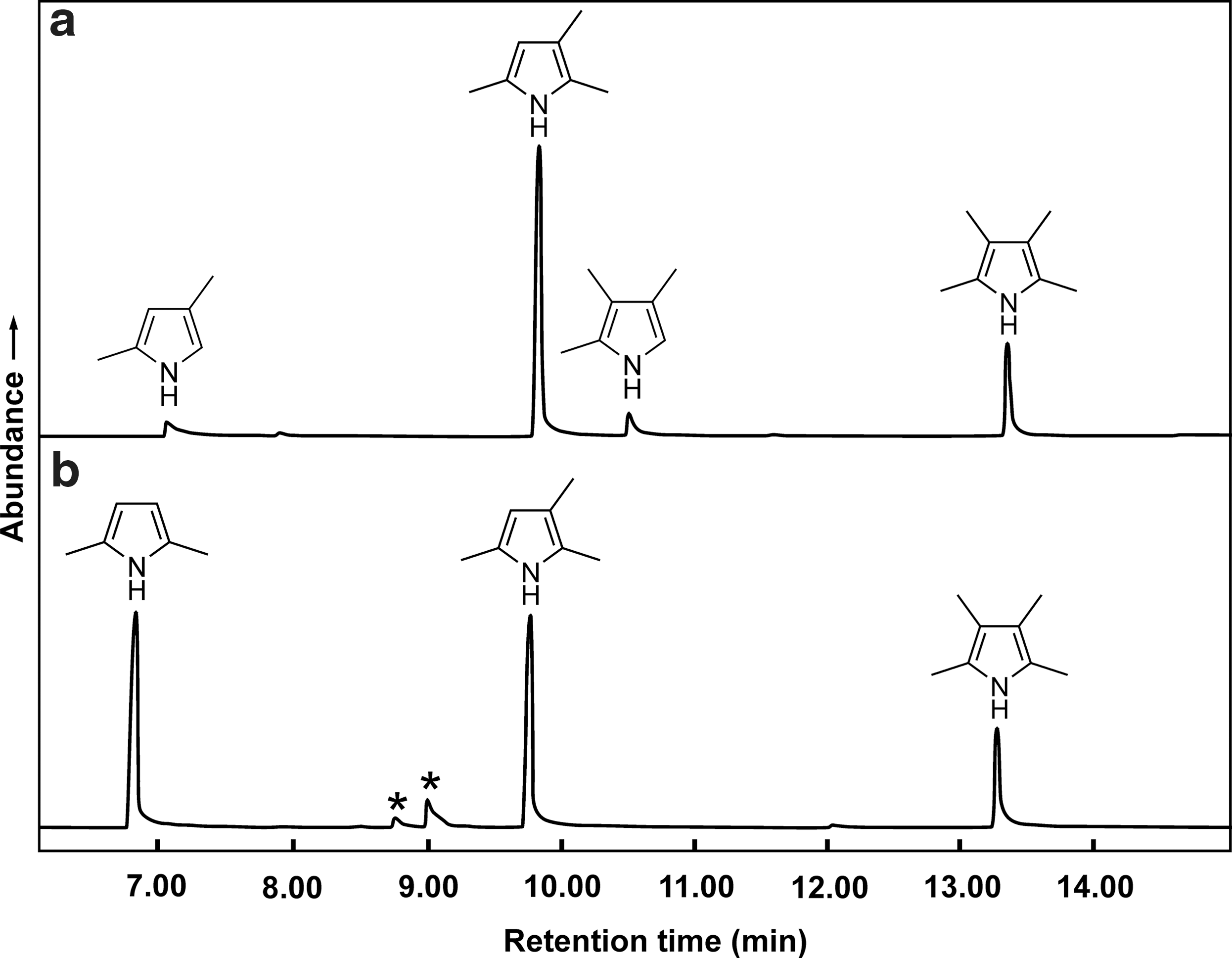
Gas chromatograms (total ion detection) of the products obtained by methylation of (
2.10. Estimation of the alkylpyrrole yield
A thermolysis experiment with 10.0 g of (CaCl2)3(Hala)2·6H2O (
2.11. Oligopyrrole formation
A typical experiment was performed as follows. A solution of NaCl (705 mM), MgCl2 (80 mM), CaCl2 (15 mM), KCl (15 mM), and HCl (10 mM) was prepared. NaNO2 (8.3 mg, 0.12 mmol) was dissolved in a 240 mL portion of this stock solution. The resulting solution was thoroughly degassed and transferred into a glove box filled with pure argon gas. Then we added 3,4-diethylpyrrole (16.5 μL, 0.12 mmol) and a 16% aqueous solution of formaldehyde (21.6 μL, 0.12 mmol HCHO). The reaction mixture was briefly shaken. After standing undisturbed for 5 days at 30°C, the mixture was analyzed by UV-visible spectroscopy.
For the ESI-Orbitrap MS measurements, it was necessary to conduct the above-described experiment without metal chlorides. The absence of these salts only slightly changed the electronic absorption spectrum, with the differences being mainly in the relative intensities of the absorption maxima. Immediately before measuring the mass spectra, we added acetonitrile to the reaction mixtures. The electronic spectra of the solutions with and without acetonitrile were virtually identical except for some minor shifts in band positions.
The total yields of the oligomers of 3,4-diethylpyrrole and 3-ethyl-2,4-dimethylpyrrole were estimated at 1–2% from the visible region of the absorption spectra. The relatively high molar extinction coefficients (in dichloromethane) of OEP (Kinoshita et al., 1992) and protonated 4,4'-diethyl-3,3',5,5'-tetramethyl-2,2'-dipyrromethene (Semeikin et al., 2003), respectively, were assumed for all products. This method inevitably underestimated the concentrations of the less-oxidized products, which have lower extinction coefficients or no absorption at all in the visible region. Consequently, the actual yields were probably higher.
2.12. Reaction between a volcanic rock and hydrochloric acid
We used unweathered picrobasalt sand that was rich in olivine [(Mg,Fe)2SiO4]. It had been collected near the coast at the Piton de la Fournaise volcano on the island of La Réunion. Five grams of the picrobasalt was added to 50.0 mL of hydrochloric acid of pH 2.0. The mixture was stirred in a closed measuring cell. The pH was monitored with a pH electrode. After 6 days, dissolved Mg2+ and Fe2+ ions were identified in the supernatant by precipitation of characteristic MgNH4PO4·6H2O (struvite) crystals and Turnbull's blue, respectively.
3. Results
3.1. Amino acids in artificial sea salt crusts
To simulate the formation of primordial amino acid–containing sea salt crusts, we prepared aqueous solutions of NaCl, MgCl2, KCl, CaCl2, and an α-amino acid. At least ∼5 mmol of the amino acid was necessary for an unambiguous interpretation of the vibrational spectra and X-ray diffractograms. In most experiments, including the thermolyses, 10 mmol was employed, resulting in an amino acid content in the 1–2% range. Glycine, DL-alanine, DL-valine, DL-glutamic acid, α-aminoisobutyric acid, and DL-isovaline (2-amino-2-methylbutanoic acid) were used. In principle, the solids that remained after evaporation to dryness could have contained amino acid crystals; or, alternatively, the amino acids could have reacted with sea salt components. Raman and IR spectroscopy and X-ray powder diffraction clearly showed that amino acid crystals were absent. For alanine, for example, this can be easily seen by comparing the spectra a and e in Figs. 1 and 2. In fact, new compounds that contained magnesium chloride or calcium chloride had been formed. Glycine (Hgly), alanine (Hala), and α-aminoisobutyric acid (Haib), for example, were present as CaCl2(Hgly)·H2O (Yusenko et al., 2008), (CaCl2)3(Hala)2·6H2O (
(CaCl2)3(Hala)2·6H2O (
From the composition and spectral properties of

Crystal structure of (CaCl2)3(Hala)2·6H2O (
3.2. Formation of pyrroles from salt-embedded amino acids
The amino acid–containing salt crusts described above were subjected to thermolyses at 350°C. A slow stream of nitrogen gas was used to simulate the non-oxidizing atmosphere and local atmospheric movements on a primordial volcanic coast. The volatile product fraction that condensed near room temperature was analyzed by GC-MS. It was found that the amino acids had formed several methylated and ethylated 1H-pyrroles. The only exceptions were glycine (no pyrroles) and glutamic acid (only 2-methylpyrrole detected). Salt-embedded amino acid mixtures [e.g., glycine (3 mmol), DL-alanine (2 mmol), DL-valine, DL-glutamic acid, α-aminoisobutyric acid, and DL-isovaline (1 mmol each)] similarly generated various alkylpyrroles. An example [glycine (6 mmol) and DL-alanine (4 mmol)] is shown in Fig. 6. The mass spectra reveal that the pyrroles were exclusively C-alkylated. In addition to pyrroles, a few alkylpyridines and alkylindoles were detected, but in much lower concentrations.
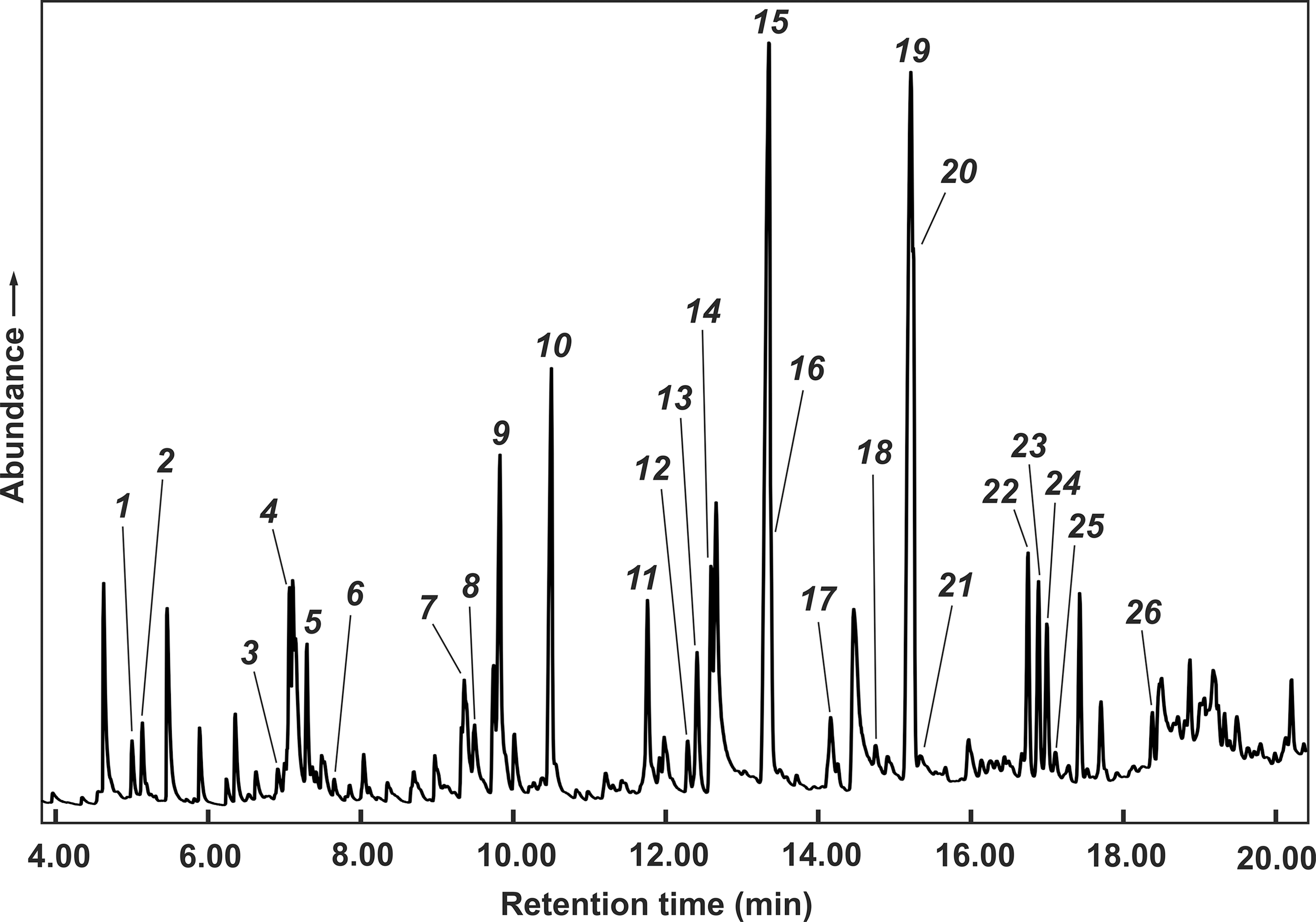
Gas chromatogram (total ion detection) of volatile products obtained by thermolysis of a glycine–DL-alanine mixture embedded in artificial sea salt. Composition of the salt crust (mmol): NaCl 705, MgCl2 80, KCl 15, CaCl2 15, glycine 6, DL-alanine 4. Thermolysis conditions: 350°C, N2 atmosphere. In the following, abbreviations are used for the names of the pyrroles, e.g., 3-Et-2,4-Me2 stands for 3-ethyl-2,4-dimethylpyrrole. Peak
In our study of alkylpyrrole formation, we focused on alanine, which is thought to be one of the prebiotically most relevant α-amino acids (Zaia et al., 2008). Under standard thermolysis conditions (350°C, N2), neat alanine did not produce pyrroles. Instead, the amino acid mostly sublimed and partly formed the cyclic dipeptide (3,6-dimethylpiperazine-2,5-dione). In a mixture with NaCl (705 mmol) and KCl (15 mmol), obtained by evaporation, alanine (10 mmol) behaved in the same way. In agreement with this, vibrational spectra reveal that the mixture simply contained crystals of the pure amino acid. The same observations were made with α-aminoisobutyric acid, except that here the sublimation occurred without decomposition. These results showed that the alkylpyrrole formation was not caused by a physical effect of the salt crystals but by chemical interaction between the amino acids and MgCl2 or CaCl2 (see above).
In the artificial sea salt mixture, (CaCl2)3(Hala)2·6H2O (
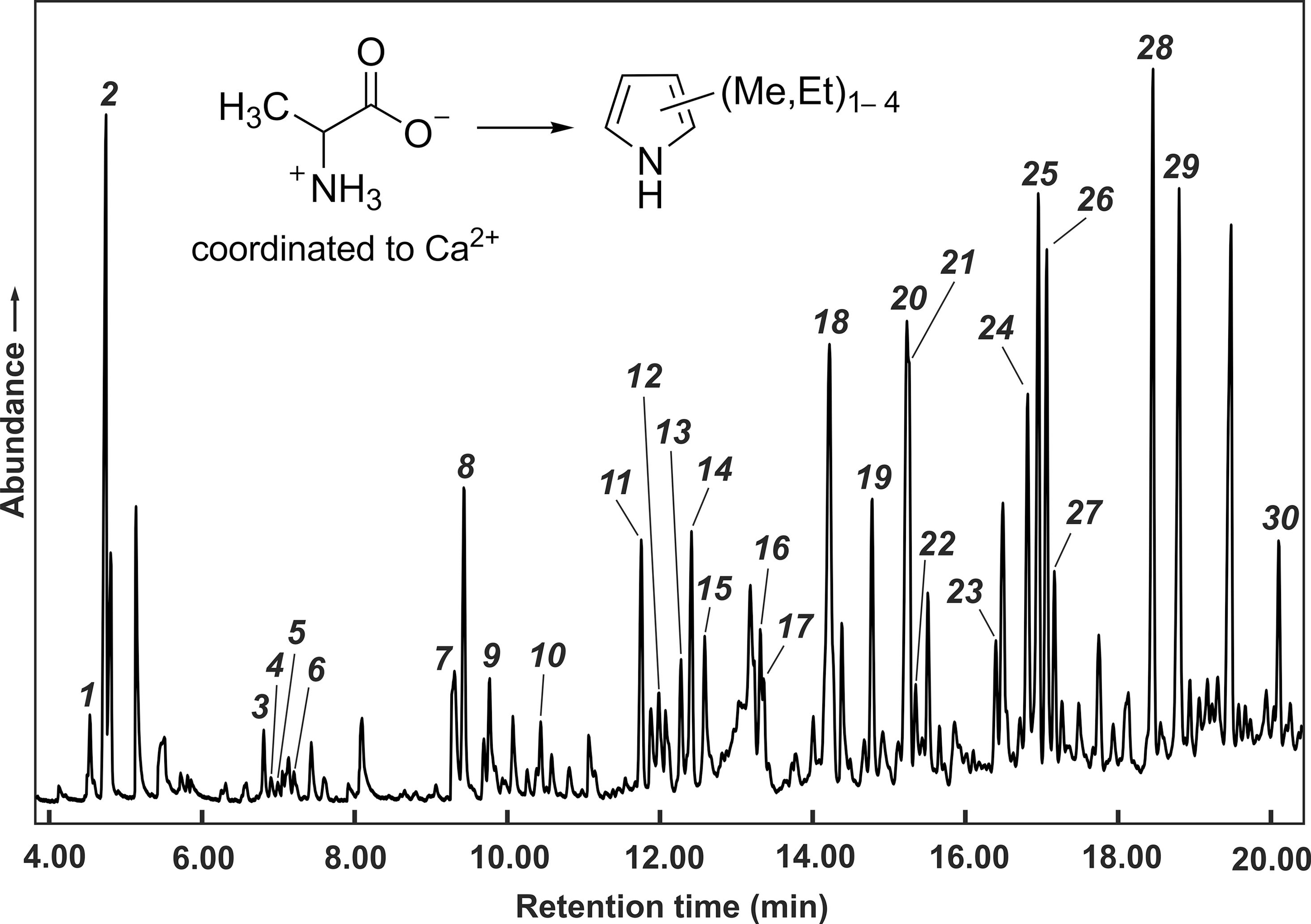
Gas chromatogram (total ion detection) of volatile products obtained by thermolysis of (CaCl2)3(Hala)2·6H2O (
Among the volatile products, a few mono-, di-, and trimethylpyridines were present in low concentrations as well as carbon dioxide and ammonia. Elemental analyses (Table 1) and 1H NMR spectroscopy showed that the nonvolatile residue consisted almost entirely of calcium chloride and the cyclic alanine dipeptide. Under the reasonable assumption that calcium chloride remained completely in the residue, one can easily calculate from the values in Table 1 that at least 45% of the alanine molecules had been transformed into the cyclic dipeptide.
The values in column 3 were calculated for a mixture of 83% calcium chloride, 16% cyclic alanine dipeptide, and 1% water.
To elucidate the mechanism of pyrrole formation, we thermolized three isotopomers of compound

Distribution of the alanine carbon atoms in the 3-ethyl-2,4-dimethylpyrrole molecule. Color images available online at
m/z
First row: molecular ion; second row: molecular ion minus one methyl group; third row: molecular ion minus two methyl groups.
3.3. Oligopyrrole formation under simulated volcanic island conditions
We investigated the possibility that pyrroles oligomerized at cooler locations on primordial volcanic islands, for example, in rock pools. Our simulated rock pool contents consisted of acidic artificial seawater (10 mM HCl≙pH 2) to which an alkylpyrrole, formaldehyde (HCHO), and an oxidant had been added. Three alkylpyrroles displaying different degrees of substitution were selected for the experiments, namely, 3-methylpyrrole, 3,4-diethylpyrrole, and 3-ethyl-2,4-dimethylpyrrole. Nitrite (
Each of the three model pyrroles gave deeply colored products with HCHO and either or both of the oxidants
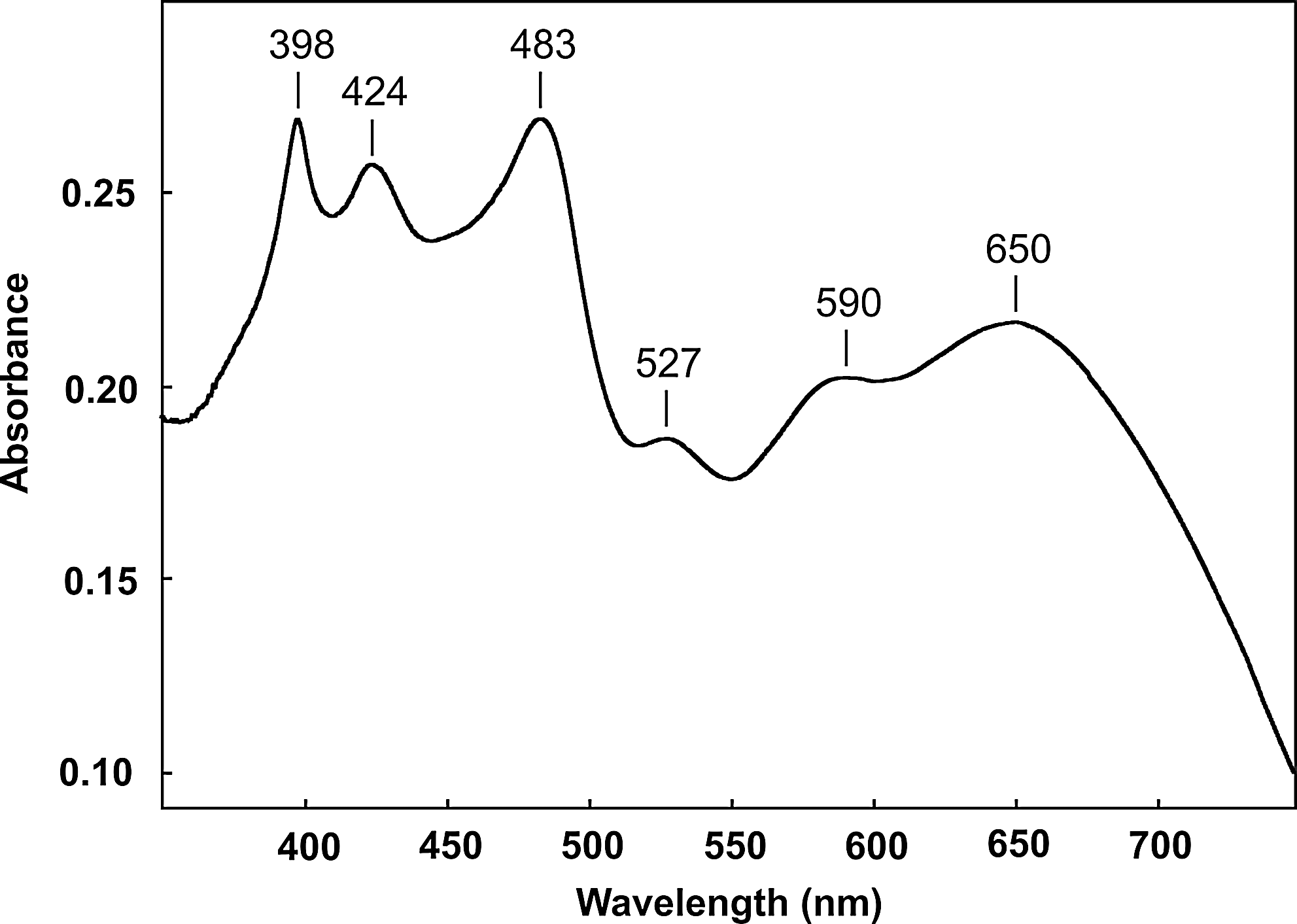
Visible absorption spectrum of the products of the reaction system 3,4-diethylpyrrole/HCHO/NaNO2 (dichloromethane extract of a metal chloride–free reaction mixture).
The electronic absorption spectrum of our test reaction system indicates that several products had been formed, among them the porphyrin (Soret band at 398 nm, Fig. 9). Further examination by ESI-Orbitrap MS revealed the presence of 38 oligopyrroles ranging from dimers to hexamers (Figs. 10 and 11). The elemental compositions of these molecules were calculated from the accurate masses (Table 3). The observed product diversity can, in part, be attributed to different degrees of double bond conjugation (i.e., oxidation) for a given oligomer size. This is exemplified for two series of tetramers in Fig. 12. One series consists of cyclic molecules, including OEP and octaethylporphyrinogen, the other of open-chain ones. Adjacent members of each series differ by two hydrogen atoms. We found that the tetrameric molecules showed the highest signal intensities, with OEP dominating the mass spectra. The intensity sequence was as follows: OEP>other cyclic tetramers≈open-chain tetramers>trimers≈pentamers>dimers≈hexamers. For 3,4-diethylpyrrole and 3-ethyl-2,4-dimethylpyrrole, the oligopyrrole yield was estimated at 1–2% from electronic absorption spectra. Given the experimental procedure, this value must be regarded as the lower limit of the yield (see Materials and Methods).

Structural formulas of the molecules with entry numbers 1 to 20 in Table 3. The configurations at the exocyclic double bonds of the open-chain structures are unknown and have been arbitrarily assigned. If tautomeric forms are possible, only one is shown here.

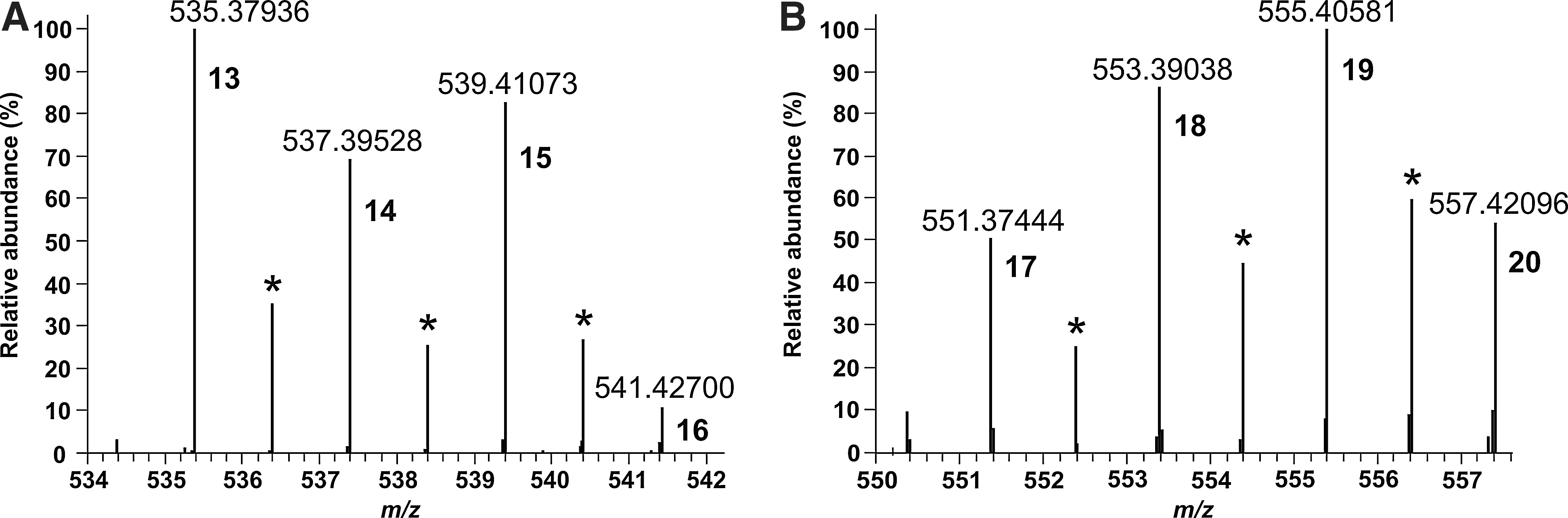
High-resolution/high-accuracy mass spectra of (
To gain an impression of the thermal stability of OEP, we performed experiments where this compound was exposed to different temperatures in a nitrogen atmosphere. The results showed that OEP was stable for at least 24 h at 200°C and for 30 min at 240°C. Even after 30 min at 350°C, ∼13% of the OEP was still intact. It had survived mostly in the thermolysis residue but also by sublimation. For comparison, we also studied the biomolecule derivatives hemin (ferriprotoporphyrin chloride) and biliverdin hydrochloride. They proved to be less thermally stable than OEP. In 24 h experiments, hemin began to decompose at ∼150°C and biliverdin hydrochloride already at ∼100°C.
3.4. Role of hydrochloric acid
During the thermolysis of sea salt, hydrogen chloride is formed by decomposition of magnesium chloride hydrates. This reaction has been observed at present-day volcanic coasts (Edmonds and Gerlach, 2006) as well as in our pyrrole formation experiments where the aqueous product fractions were strongly acidic (pH≤1). Thus, if pyrroles were produced from salt crusts at primordial volcanic coasts, they were necessarily accompanied by hydrogen chloride. Therefore, we included hydrochloric acid in our oligomerization experiments with pyrroles. In these experiments, hydrochloric acid in fact turned out to be essential for oligopyrrole formation in the model system 3,4-diethylpyrrole/HCHO/nitrite. The reaction, which was usually conducted with 10 mM HCl, still proceeded when the HCl concentration was reduced to 1 mM but no longer when the acid was omitted.
Present-day biological oligopyrroles must often form metal complexes to be able to perform their functions. Prominent examples are chlorophylls (Mg2+ complexes) and hemes (Fe2+/3+ complexes). Experiments on the possible prebiotic formation of metal–oligopyrrole complexes were not part of this study. However, we investigated whether dissolved metal ions, which are necessary for complex formation, could have been available in a volcanic island environment. To this end, we exposed a basaltic rock rich in olivine [(Mg,Fe)2SiO4] to 10 mM hydrochloric acid (pH 2.0). The solution pH increased to 3.9 within 24 h. A practically constant value of pH 4.8 was reached after 6 days. The pH increase from 2.0 to 4.8 was equivalent to the neutralization of 99.8% of the initial amount of acid. Under the experimental conditions, chloride did not form insoluble salts. Therefore, the loss of protons from the solution must have been compensated for by the release of an equivalent amount of metal ions in order to maintain electroneutrality. Accordingly, Mg2+ and Fe2+ were identified in the solution. The natural pH of water standing over the basaltic rock was 9.0.
4. Discussion
In the early Archean, volcanic islands provided various environments with different geochemical and geophysical conditions. The close mutual proximity of these environments facilitated the chemical exchange between them. Thus, prebiotic organic molecules could have been transferred from certain environmental conditions to other very different ones. In the chemical laboratory, such changes of reaction conditions are usually prerequisite for multistep syntheses of complex organic molecules. Alternatively, three or more components can react simultaneously to form a single product with no need for different conditions. These so-called “multicomponent reactions,” however, have relatively limited potential, in contrast to stepwise syntheses. These experiences from synthetic organic chemistry probably also applied to abiotic chemical syntheses on early Earth. Two volcanic island environments formed the basis for our simulation experiments: (i) the coasts where the interaction between seawater and molten lava produced and heated sea salt crusts and (ii) rock pools not in immediate vicinity of lava flows.
We started by preparing artificial amino acid–containing seawater. The six α-amino acids employed were chosen because of their high relative abundances in carbonaceous chondrites (Zaia et al., 2008; Martins and Sephton, 2009) and in product mixtures obtained in electric discharge experiments (Johnson et al., 2008). Lightning has been estimated to have produced a steady-state amino acid concentration of 0.3 mM in the primitive ocean (Miller, 1987). This number might be too high because the bulk atmosphere was probably not sufficiently reducing. However, besides extraterrestrial delivery, lightning in volcanic ash-gas clouds must also be considered (Hill, 1992; Basiuk and Navarro-González, 1996; Navarro-González and Segura, 2004). This source may have efficiently supplied amino acids to the surface ocean water near active volcanic islands. The exact composition of the early Archean seawater is unknown. Today the six ions Na+, Mg2+, K+, Ca2+, Cl−, and
The salt crusts that remained after evaporation of the amino acid–containing artificial sea salt solutions did not contain free amino acids. Instead, metal–amino acid compounds had been formed, for example (CaCl2)3(Hala)2·6H2O (
Heating of amino acid–containing artificial sea salt crusts produced pyrroles when the amino acids were DL-alanine, DL-valine, α-aminoisobutyric acid, or DL-isovaline (either alone or in amino acid mixtures). The model compound
Mass spectrometric results obtained by using 13C isotopomers of alanine (see above) enabled us to suggest a possible mechanism of pyrrole formation (Fig. 13). In this mechanism, first a ketone is formed from two calcium-coordinated alanine molecules. This step is analogous to the long-known thermal decomposition of simple calcium carboxylates such as the acetate (Krönig, 1924; Lee and Spinks, 1953). Next, the ketone becomes α,β-unsaturated and undergoes intramolecular ring closure. Finally, aromatization and alkylation follow. Details of the alkylation step are unknown. Consistent with this mechanism, carbon dioxide and ammonia appeared as thermolysis products. It is noteworthy that the formation of pyrroles from serine proceeds via a completely different pathway (Yaylayan and Keyhani, 2001). From Figs. 8 and 3 it can be deduced that each alkylpyrrole molecule required between two and six alanine molecules for its formation, depending on the degree of substitution.
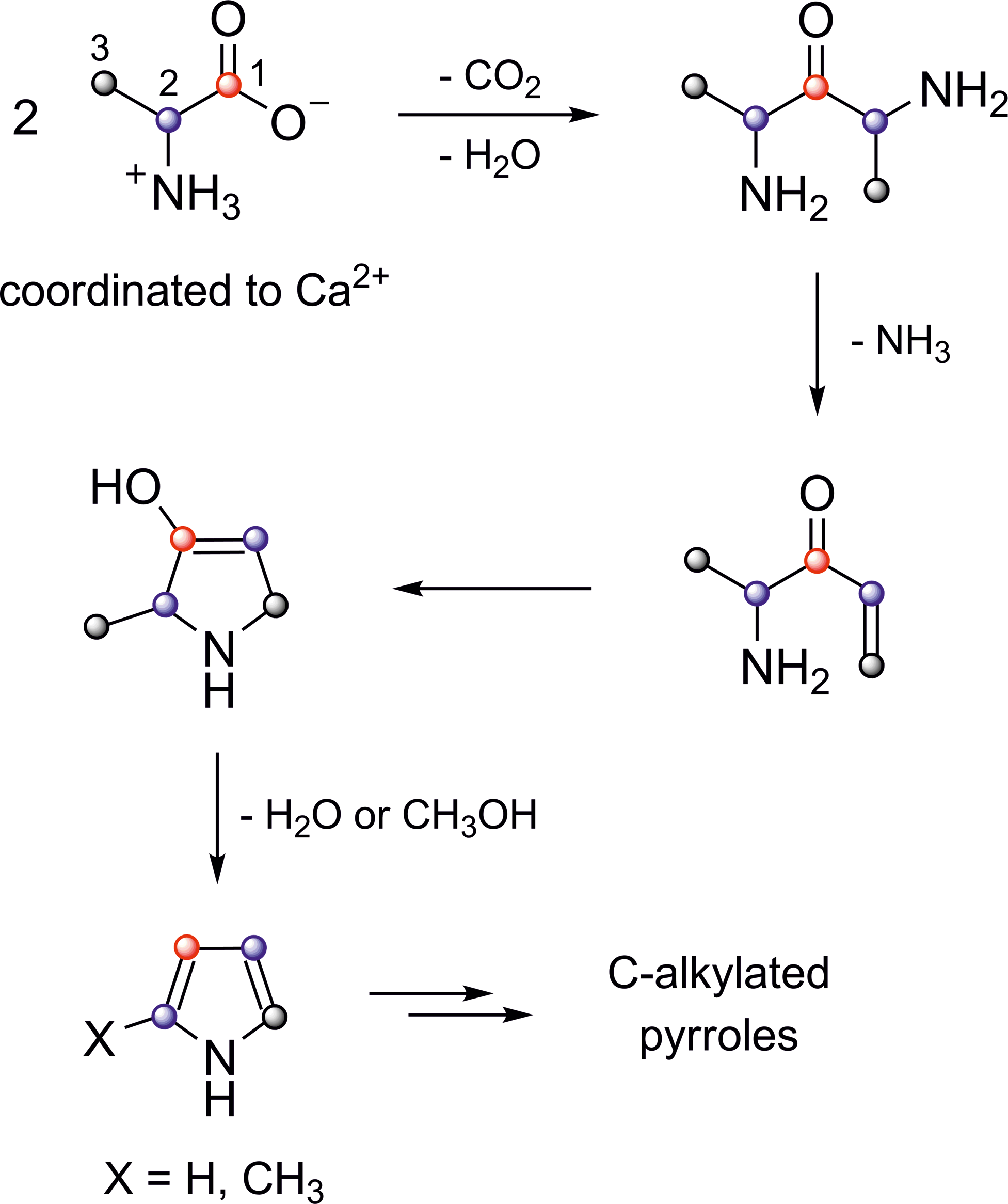
Possible mechanism of formation of pyrroles from salt-embedded alanine. Color images available online at
Knowing the origin of every carbon atom allowed us to estimate the yield of pyrroles at ∼2%. For comparison, the yield of the main organic product, the cyclic alanine dipeptide, is at least 45%. The relatively low percentage yield of pyrroles, however, translates into large masses in a volcanic island scenario. During a larger eruption of an island volcano, more than 108 m3 of lava can enter the sea and evaporate the same volume of seawater (Edmonds and Gerlach, 2006). If the amino acid concentration in the early ocean was 0.3 mM (see above), 3·107 mol of amino acids would have been affected. We conservatively assume that only 10 mol % of these amino acids could, in principle, have produced pyrroles. Taking into account our experimental result that 32.3 mol of alanine yielded 0.126 mol of alkylpyrroles, ∼104 mol (≈103 kg) of alkylpyrroles would have been formed. Even if the oceanic amino acid concentration was 2 orders of magnitude lower, that is, in the micromolar range, still several kilograms of pyrroles could have been formed in a single volcanic eruption. Thus, substantial amounts could have accumulated over longer periods of time. Another interesting aspect is that the high local temperatures must have caused a spatial separation between the salt residue and the volatile products. The latter included not only the pyrroles but also hydrogen chloride from salt decomposition and huge amounts of water. Today, the steam clouds that originate from lava–seawater interaction predominantly move inland due to thermal convection. Its has been reported that such clouds, together with eruption plumes, caused acidic rain of pH<2 (Staudacher et al., 2009).
Prebiotic volatiles almost certainly condensed in cooler regions and accumulated in depressions (rock pools), analogous to the processes on modern volcanic islands. Therefore, we have experimentally addressed the question of how pyrroles could have reacted in rock pools. Formaldehyde (HCHO), which is generally regarded as a prebiotic molecule (Navarro-González, 1992; Cleaves, 2008), was employed as an additional organic component. The prebiotic bulk oceanic concentration of HCHO probably did not exceed ∼1 mM (Cleaves, 2008). Consequently, we have limited the HCHO concentration to 0.5 mM and made no attempts to optimize the product yields by increasing the concentrations of the starting materials. It is remarkable that even at this high dilution the condensation of alkylpyrroles and HCHO proceeded, albeit to a rather limited extent. The initial products, for example the porphyrinogen 16 (Fig. 10), are unstable in the sense that oligopyrrole formation is easily reversible unless an oxidation step follows. We therefore added the potential oxidants nitrite (
The presence of hydrochloric acid proved essential for the formation of conjugated oligopyrroles. In fact, HCl has been reported to catalyze the condensation of unsubstituted pyrrole with HCHO in aqueous solution (Sobral et al., 2003). There are good reasons for assuming that hydrochloric acid was catalytically or stoichiometrically involved in the oxidation step, too. For example, it is well known that acidic conditions enhance the oxidizing power of nitrite (Wiberg, 2001). However, due to the low concentrations, we were unable to identify the reduction product(s) of nitrite and thus cannot propose an equation for the overall reaction. In contrast to HCl, the metal chlorides of the artificial seawater were not essential for oligopyrrole formation. This observation is prebiotically interesting because rock pools that were not situated in or near the intertidal zone could have been provided with sea salt only by aerosols. Thus, water in these rock pools may have contained little or no sea salt. In this context, we note the suggestion by Monnard et al. (2002) that life may have originated in a freshwater environment (see also Deamer, 2004).
Our results indicate that the prebiotic synthesis of oligopyrroles from amino acids gave relatively low yields. However, fully conjugated oligopyrroles, especially the aromatically stabilized porphyrins, are remarkably resistant compared to other presumably prebiotic oligomers such as peptides and oligonucleotides. For example, alkylporphyrins of biological origin are found in sediments hundreds of millions of years old (Callot and Ocampo, 2000). Our experiments showed that OEP withstood 200°C for several hours. It partially survived much higher temperatures for some minutes, which is in part due to its ability to sublime. Consistent with our findings, it has been reported that OEP vaporized without decomposition at 610°C under flash pyrolysis conditions (Gelin et al., 1996). Hence, it can be expected that, at least locally, substantial steady-state concentrations of abiotically formed porphyrins built up. Moreover, early cell ancestors may have benefited even from small, “catalytic” amounts of oligopyrroles.
Porphyrins are not the only oligopyrroles that can act as strong ligands toward metal ions. For example, the dipyrrins, which are “half-porphyrins,” also have a rich coordination chemistry (Wood and Thompson, 2007). The dipyrrin structural motif occurred in several reaction products of our 3,4-diethylpyrrole/HCHO/NaNO2 model system, with compound 1 (Fig. 10) being the simplest representative. Metal complexes of porphyrins and dipyrrin-like open-chain oligopyrroles could have performed similar functions in early cell ancestors, for example, as electron transfer molecules. However, the availability of suitable metal ions was a prerequisite for complex formation. As part of the present study, we have experimentally demonstrated that Fe2+ and Mg2+ ions were released when hydrochloric acid reacted with picrobasalt. Picrobasalt, also referred to as oceanite, is an igneous rock found on ocean volcanic islands. On primordial volcanic islands, where hydrochloric acid must have been abundant, this reaction may have provided dissolved metal ions for the formation of Fe2+- and Mg2+-oligopyrroles. Our results suggest that in primordial volcanic environments hydrochloric acid was involved in (i) oligopyrrole synthesis (see above), (ii) solubilization of metal ions, and (iii) pH modification/regulation in rock pools. Thus, it seems that HCl is a more important prebiotic molecule than previously recognized.
5. Summary and Conclusions
Taken together, our results support a prebiotic two-step scenario in which oligopyrroles were spontaneously formed from α-amino acids: (i) near coastal lava flows, amino acids were trapped in solid sea salt and subsequently thermolyzed to pyrroles; (ii) the pyrroles and hydrochloric acid, which also appeared as a thermolysis product, condensed in rock pools where they reacted with formaldehyde and nitrite to give conjugated oligopyrroles, including porphyrins and open-chain tetrapyrroles. Plausible prebiotic sources are known for all starting materials. Furthermore, the scenario refers to a specific geological setting that can still be observed today on volcanic islands. It is interesting to note that contemporary biosynthetic pathways to tetrapyrroles also start from α-amino acids (Beale, 1999; Voet and Voet, 2011).
There is no reason to assume that the substitution patterns of modern biological tetrapyrroles existed from the beginning. In fact, basic concepts of evolution suggest that these patterns evolved from simpler ones (Strasdeit and Fox, 2013). Consistent with this idea, the methyl–ethyl substitution in some of our alkylpyrroles is already reminiscent of the methyl–vinyl and methyl–2-carboxyethyl substituent pairs in modern heme b. It is conceivable that in protometabolic reaction networks, oligopyrroles were able to perform rudimentary functions even without possessing well-defined, uniform substitution patterns. Initially, early cell ancestors may have utilized spontaneously formed oligopyrroles in redox reactions and for UV protection. Later on, they developed their own cyclic-tetrapyrrole syntheses, perhaps by starting from α-aminonitriles (Ksander et al., 1987) or aminoketones and diketones (Lindsey et al., 2009, 2011; Soares et al., 2012; Taniguchi et al., 2012). It has been proposed that UV protection by porphyrins and related pigments preceded light harvesting (e.g., Larkum, 2006). Protection against short-wavelength UV radiation was certainly necessary if early cell ancestors evolved in the subaerial environment of volcanic islands. Prebiotic porphyrins with only simple alkyl substituents were less vulnerable to thermal damage than modern biological ones so that they could accumulate despite occasionally hot conditions.
Finally, we note that key environmental prerequisites for our prebiotic volcanic island scenario probably also existed on Noachian Mars, namely, volcanoes (e.g., Xiao et al., 2012), standing bodies of water (e.g., Head et al., 1999), and soluble metal chlorides (Osterloo et al., 2008; Glotch et al., 2010). As a consequence, the usefulness of porphyrins as biomarkers in future Mars missions should be reevaluated (Suo et al., 2007).
Footnotes
Acknowledgments
We thank P. Guni, B. Hillers, I. Klaiber, F. Leißing, S. Ringer, M. Wälz, and M. Wagner for technical assistance; D. Haase and W. Saak for the X-ray diffraction data of compound
Author Disclosure Statement
The authors state that no competing financial interests exist.
Abbreviations
1H NMR, proton nuclear magnetic resonance; ESI, electrospray ionization; GC, gas chromatogram, gas chromatography; Haib, α-aminoisobutyric acid; Hala, DL-alanine; Hgly, glycine; MS, mass spectrometry; OEP, octaethylporphyrin.