
Abstract
Understanding microbial diversity in spacecraft assembly clean rooms is of major interest with respect to planetary protection considerations. A coordinated screening of different clean rooms in Europe and South America by three German institutes [Deutsches Zentrum für Luft- und Raumfahrt (DLR), Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ), and the Institute of Microbiology and Archaea Center, University of Regensburg] took place during the assembly, test, and launch operations of the Herschel spacecraft in 2006–2009. Through this campaign, we retrieved critical information regarding the microbiome within these clean rooms and on the Herschel spacecraft, which served as a model for upcoming ESA mission preparations. This “lessons learned” document summarizes and discusses the data we obtained during this sampling campaign. Additionally, we have taken the opportunity to create a database that includes all 16S rRNA gene sequences ever retrieved from molecular and cultivable diversity studies of spacecraft assembly clean rooms to compare the microbiomes of US, European, and South American facilities. Key Words: Planetary protection—Spacecraft assembly facility—Microbial diversity—Herschel. Astrobiology 13, 1125–1139.
1. Introduction
A
A great deal of time and effort on the part of space agencies is, and has been, necessary to analyze the microbiome of spacecraft and associated clean rooms with regard to planetary protection. These efforts have recently provoked debate over the possibility of overprotecting Mars (Fairén and Schulze-Makuch, 2013). The authors claim that Mars has already been contaminated with Earth life in the past (by either meteorites or spacecraft); thus, current planetary protection efforts could be significantly scaled back for financial and scientific reasons. However, the authors of the aforementioned article do not address the problem of false-positive or false-negative life-detection results that could possibly be obtained by using life-detection instruments contaminated by Earth life signatures. Such a contamination could therefore tremendously affect the ongoing life-detection mission and possibly lead to misinterpretations concerning subsequent science-driven missions or even to mission failure (Conley and Rummel, 2013).
Thus, analyzing and interpreting the microbiome of spacecraft-associated environments is crucial for life-detection mission success.
Microbial contamination in and around spacecraft has been assessed with a variety of cultivation-based (La Duc et al., 2007; Stieglmeier et al., 2009, 2012; Moissl-Eichinger et al., 2012) and cultivation-independent methods (Venkateswaran et al., 2001; La Duc et al., 2009; Cooper et al., 2011; Vaishampayan et al., 2013a). Although the results from microbial analyses are strongly dependent on time, location, method, and other factors (clean room maintenance, mission status, etc.), these studies have revealed a vast diversity of microorganisms in clean rooms. These organisms are mostly human-associated, but hardy (spore-forming) bacteria are frequently detected in close proximity to and on spacecraft. Spacecraft and clean room contamination by these organisms poses a potential threat to planetary protection.
NASA has a long tradition of implementing planetary protection (Puleo et al., 1977), and NASA's endeavors in bioburden assessment have followed established protocols [NASA standard procedures for the microbial examination of space hardware (NASA-HBBK-6022, 2007)] that focus on heat-shock-surviving (spore-forming) microbes. The number of colony-forming units that appear after such a heat shock (e.g., 80°C for 15 min) offers an indication as to the overall cleanliness of a spacecraft and represents the microbial load (“bioburden”). ESA has adopted many of these protocols but has also established other methods to improve detection of possible contaminants (ECSS-Q-ST-70-55 Working Group, 2008; Probst et al., 2010b).
Currently, ESA is preparing the ExoMars mission, which is scheduled for launch in two stages in 2016 and 2018. During the preparation phase of this mission, optimal procedures are being chosen to analyze bioburden and biodiversity in facilities and on spacecraft. Therefore, ESA announced three projects that aimed at the understanding of microbial diversity in close vicinity of a spacecraft: Determination of the Microbial Diversity of Spacecraft Assembly and Testing Facilities (BioDiv, main contractor: DLR, Cologne; subcontractor: DSMZ, Braunschweig), Archaeal and Specific Bacterial Communities in Spacecraft Associated Clean Rooms (main contractor: University of Regensburg), and Characterization, Controlled Storage, and Publication of Planetary Protection Associated Bacterial Isolates (main contractor: DSMZ). Together, these projects will help to elucidate the microbial bioburden and biodiversity in European and South American facilities before the ExoMars mission is implemented. During these surveys, isolation strategies led to the identification of numerous microbial strains that are representative of the clean room diversity. These isolates were collected, archived, and released to the public through the creation of a microbial culture collection, maintained by the Leibniz Institute DSMZ in Braunschweig, Germany (Moissl-Eichinger et al., 2012).
As a model spacecraft for future missions, the Herschel space telescope was followed through different locations in Europe and South America and sampled under assembly, integration, and testing activities in each facility until the launch from Kourou, French Guiana. Although Herschel was not subject to biocleanliness control (it represents planetary protection category 1 according to COSPAR; COSPAR, 2011), the housing clean rooms were operated as ISO 8 and ISO 5 (ISO 146441,
Some data collected through this project (“Herschel mission”) have previously been described (Stieglmeier et al., 2009, 2012), as has the above-mentioned culture collection (Moissl-Eichinger et al., 2012). Another publication with respect to the microbial diversity detected in the spacecraft assembly facilities in Kourou can be found in this issue (Schwendner et al., 2013). The article summarizes and discusses the data that were obtained in order to provide a “lessons learned” document, which could serve as an information basis for the design of future sampling campaigns as well as a source for the comparison with data from other clean room facilities. In addition, we also took this opportunity to create a database that includes all (molecular and isolates') near full-length 16S rRNA gene sequences retrieved and published from any spacecraft assembly facility. This will provide a valuable resource for future surveys that include comparative analyses of previous cultivation and molecular data.
2. Materials and Methods
2.1. Description of the sampling campaign
The microbial analysis of the Herschel space telescope and its housing facilities took place from 2006 to 2009 at different locations in Europe and South America. During this time frame, five sampling events were performed, and the microbial community was analyzed within the framework of the three aforementioned projects. The BioDiv project aimed to follow the European Cooperation for Space Standardization (ECSS) standard of ESA for the cultivation of heat-shock-resistant microbes (bioburden determination; ECSS-Q-ST-70-55 Working Group, 2008) from surfaces, as it is recommended by space agencies for the estimation of contamination by predominantly spore-forming microbes. This included the establishment of the “milliflex-assay” as a rapid method to detect colonies within 7 h. Additionally, the BioDiv project focused on the overall diversity that was cultivable by applying R2A medium (to obtain vegetative organisms, “vegetatives”). Air sampling was also performed to understand the distribution of microbes within the clean room.
The project designated Archaeal and Specific Bacterial Communities in Spacecraft Associated Clean Rooms applied 32 different cultivation conditions in addition to the standard procedure (“alternative” cultivation methods) to investigate a broader spectrum of extremotolerant microbes. Extreme conditions with respect to temperature, pH, salt content, oxygen availability, and nutrient content were used to cultivate tolerant microbes and understand the requirements of a clean room community. The conditions applied were chosen with respect to possible survivability in space, so that media were also used that provided conditions for autotrophic or diazotrophic microbes (primary producers). Additionally, molecular analyses were applied to facilitate understanding of the entire microbial community (uncultivable majority plus cultivable minority), which included quantitative PCR (qPCR) analysis as well as cloning of 16S rRNA gene sequences from Bacteria and Archaea. The resulting isolates were organized and archived at the DSMZ in Braunschweig (Moissl-Eichinger et al., 2012).
2.2. Sampling and sample extraction
Five samplings were performed in three different spacecraft assembly clean room facilities, one in Germany, a second in the Netherlands, and a third in French Guiana. The facilities in Germany (Friedrichshafen) were maintained by EADS Astrium (European Aeronautic Defense and Space Company), while the other clean rooms were located at the ESA European Space Research and Technology Centre (ESTEC) in Noordwijk and at the Centre Spatial Guyanais (CSG) in Kourou, maintained by the French space agency Centre National d'Etudes Spatiales (CNES). More details about the sampling schedule (dates, clean room classes, sampling types, etc.) are provided in Table 1. Table 1 also depicts the abbreviations for each sampling event, which are used as follows: ESTEC samplings: ES1, ES2; Friedrichshafen samplings: FR1, FR2; Kourou sampling: KO. The clean rooms were operated as ISO 8 and ISO 5 (ISO 146441,
Clean room classification according to ISO 14644 (for further details please see Materials and Methods).
Wipes were used for bioburden measurements following the ESA standard; SpongeSicles were used for alternative cultivation analyses.
Samples were taken from clean room surfaces that included mainly floor and ground support equipment (GSE), spacecraft, and clean room air. Throughout the campaign, different sampling tools were used. These included nylon-flocked swabs (552C regular swab, microRheologics, Brescia, Italy), wipes (Spec-Wipe 4, VWR International GmbH, Darmstadt, Germany; 15 cm×15 cm), and SpongeSicles (Biotrace [3M], St. Paul, MN, USA). In the first sampling (Table 1), witness plates (V2A 1.4301, no. 4 finish, 25 mm×5 mm×1 mm) were exposed for 3 days during clean room operations. BiSKits (biological sampling kits, Quicksilver Analytics, Abingdon, MD, USA) were used for the collection of samples for molecular analyses. Air samples were collected with the AirPort MD8 air sampler (Sartorius, Germany), which was supplemented with sterile gelatin membrane filters. Comprehensive descriptions of sampling procedures and extractions are described elsewhere (Stieglmeier et al., 2012) and are summarized together with subsequent cultivation strategies in Table 2.
Processing of witness plates is not included, since this type of sampling was performed only once.
Mainly floor and ground support equipment.
Phosphate-buffered saline solution including 0.02% (v/v) Tween 80.
Gelatin filter was placed on TSA.
n.a., not applicable.
2.3. Sample processing for bioburden measurements according to ECSS standard document ECSS-Q-ST-70-55C
The sampling and sample processing procedure is outlined in document ECSS-Q-ST-70-55C (ECSS-Q-ST-70-55 Working Group, 2008) and was also described in detail previously (Stieglmeier et al., 2012). In brief, premoistened wipes were used for sampling and afterward extracted in a total volume of 40 mL PBST [phosphate-buffered saline solution including 0.02% (v/v) Tween 80] by vortexing and sonication. Aliquots were either heat-shocked (15 min, 80°C; “wipe assay E.1 for mesophilic aerobic spores and heat-tolerant bacteria,” determination of microbial bioburden) or directly processed (“wipe assay E.2 for aerobic mesophilic bacteria,” determination of vegetatives) by pour-plating in trypticase soy agar (TSA) and R2A (Becton Dickinson, Heidelberg, Germany), respectively. All plates were incubated for 72 h at 32°C. Colony numbers were monitored every 24 h; colonies representative in size, color, abundance, and shape were randomly picked for further characterization, purified by two subsequent streak-outs, and subjected to 16S rRNA gene sequencing.
2.4. Sample processing for the cultivation of specialized microorganisms (alternative assays)
SpongeSicle samples were extracted either under anaerobic or aerobic conditions as previously described (Stieglmeier et al., 2009). For the cultivation of specialized microorganisms, more than 30 different media and conditions were applied as follows. Anaerobes and facultative anaerobes: anaerobic thioglycollate liquid medium (TG), anaerobic thioglycollate agar (TGA; this medium was used for colony counts of anaerobes), anaerobic trypticase soy liquid medium (TS), anaerobic trypticase soy agar (TSA; all media described in Stieglmeier et al., 2009). All anaerobes were grown under nitrogen atmosphere or mixtures with hydrogen and carbon dioxide as described previously (Stieglmeier et al., 2009). Diazotrophs were grown in modified Hino and Wilson N-free medium under nitrogen atmosphere or under a nitrogen atmosphere supplemented with 1% oxygen. Autotrophs were isolated on autotrophic homoacetogen liquid medium (AHM) and autotrophic all-rounder liquid medium (AAM; Stieglmeier et al., 2009). For media focusing on the isolation of archaea, refer to Moissl-Eichinger (2011a). Sulfate-reducing microbes were grown on modified DSMZ 63 agar (Moissl-Eichinger et al., 2012). Oligotrophic microorganisms were grown on 1:10 and 1:100 diluted R2A agar (Moissl-Eichinger et al., 2012). Alkaliphiles/alkalitolerants and acidophiles/acidotolerants were isolated on R2A agar with adjusted pH (pH 9, pH 11, and pH 3.5, respectively; Moissl-Eichinger et al., 2012). Halophiles and halotolerants were grown on R2A agar containing 3.5% and 10% NaCl (w/v), whereas psychrophilic/psychrotolerant and thermophilic/thermotolerant microbes were isolated on R2A after incubation at 4°C, 10°C, 50°C, and 60°C, respectively (Moissl-Eichinger et al., 2012). Microaerophilic/microaerotolerant microorganisms were isolated on R2A under a reduced oxygen atmosphere [98% nitrogen, 2% O2 (v/v), Moissl-Eichinger et al., 2012]. All incubations were performed at 32°C unless otherwise stated. Colony counts were obtained from agar plates only. Enriched microbes in liquid media were transferred to agar plates and purified thereon.
2.5. Sample processing of air samples
Air samples, representing 500 L of filtered air, were processed as described by Moissl-Eichinger et al. (2012).
2.6. Sample processing of witness plates
The witness plates were transferred into sterile tubes containing 50 mL of rinse solution. The tubes were sonicated for 120 s at 35 kHz. R2A plates were inoculated with 5 mL of the suspension by the pour-plate technique for mesophilic vegetative bacteria. Platings were performed in triplicate, and plates were incubated for 72 h at 25°C. The colonies were counted after 24, 48, and 72 h. The remaining suspension from each sample was heat-shocked at 80°C for 15 min, and 5 mL was plated on TSA (in triplicate) for heat-tolerant bacteria and spores. These plates were incubated at 32°C, and the colonies were counted after 24, 48, and 72 h.
2.7. Culture collection and taxonomic analyses
Representative isolates were freeze-dried for long-term storage under ambient conditions. In parallel, cryopreservation of bacterial strains above liquid nitrogen was performed as described (Hippe, 1991). Currently, publicly available bacterial strains as well as their DSM numbers have been published by Moissl-Eichinger et al. (2012). The strains can be ordered from the DSMZ and will be supplied as freeze-dried cultures in glass ampoules or as actively growing culture on request. Detailed information about each strain is accessible at
2.8. Sample processing of samples for molecular analyses
Molecular analysis of samples, including DNA extraction, 16S rRNA gene PCR and cloning, sequencing, phylogenetic analyses, and quantitative 16S rRNA gene PCR, was performed according to the instructions of Stieglmeier et al. (2012). Results have been published by Stieglmeier et al. (2012) and Schwendner et al. (2013) and are revisited in a comparative manner.
2.9. Database construction and statistical analyses
Bacterial 16S rRNA gene sequences that were generated in previous publications by cloning (Moissl et al., 2007; La Duc et al., 2009; Probst et al., 2010a; Vaishampayan et al., 2010; Schwendner et al., 2013; Stieglmeier et al., 2012) and could be found in public databases (NCBI, Greengenes, 5431 sequences, median length 1424 nucleotides; accession numbers: DQ532126–DQ532150; FJ191310–FJ194034; GQ129843–GQ130128; EU704699–EU706281; FJ957429–FJ957855; EU373541–EU888578; HQ434559–HQ434621) were retrieved and used to generate a database that included essential metadata. After SINA-alignment (Pruesse et al., 2012), the multiple sequence alignment was fed into Dnadist (Felsenstein, 1989, 1993) to compute a distance matrix with Jukes-Cantor correction. The distance matrix was used for operational taxonomic unit (OTU) grouping in mothur (Schloss et al., 2009; 2.5% dissimilarity cutoff, furthest neighbor algorithm). In case 16S rRNA gene sequences were non-overlapping, these were grouped in separate OTUs by choosing 5% dissimilarity between them. OTU abundances were either retrieved directly during this method (given that all sequences of a clone library were submitted to a public database) or were manually added from individual publications (e.g., Moissl et al., 2007). The abundance of detected OTUs was normalized to an artificial total sum to ensure their biological/statistical comparability. Hierarchical clustering (based on average-linkage) and non-metric multidimensional scaling (NMDS), both based on Bray-Curtis distance measure, were generated in the R programming environment (R Development Core Team, 2011) with the aid of the Vegan Package. For comparison of the cultivable and molecular diversity, all publicly available 16S rRNA gene sequences from spacecraft-associated clean room facilities were added to the existing database. The entire set of 16S rRNA gene sequences was then classified with the Bayesian method (Wang et al., 2007; Schloss et al., 2009) against a manually updated taxonomy database, whose sequences were grouped at 98% level (available at
3. Results
3.1. Cultivable microbial diversity
As shown in Table 1, a large number of colonies were obtained during the sampling campaign, and almost 900 isolates have been processed (i.e., purified and sequenced). These isolates were obtained via different enrichment conditions (bioburden, vegetative assay, alternative assays). A combination of all these three cultivation approaches resulted in isolates belonging to 130 different bacterial species, whereas the largest number of different taxa was obtained from the Kourou samples. The isolates spanned 53 genera and 4 bacterial phyla [Actinobacteria, Bacteroidetes/Chlorobi, Firmicutes, Proteobacteria (α, β, γ)]., Most of them were deposited at the DSMZ and are available for the public and further research (see Moissl-Eichinger et al., 2012; Schwendner et al., 2013). A summary of all isolated bacterial genera obtained from all assays performed in samplings FR1, FR2, ES2, and KO is given in Figure 1, which also indicates the differences in microbial diversity of each single sampling event.
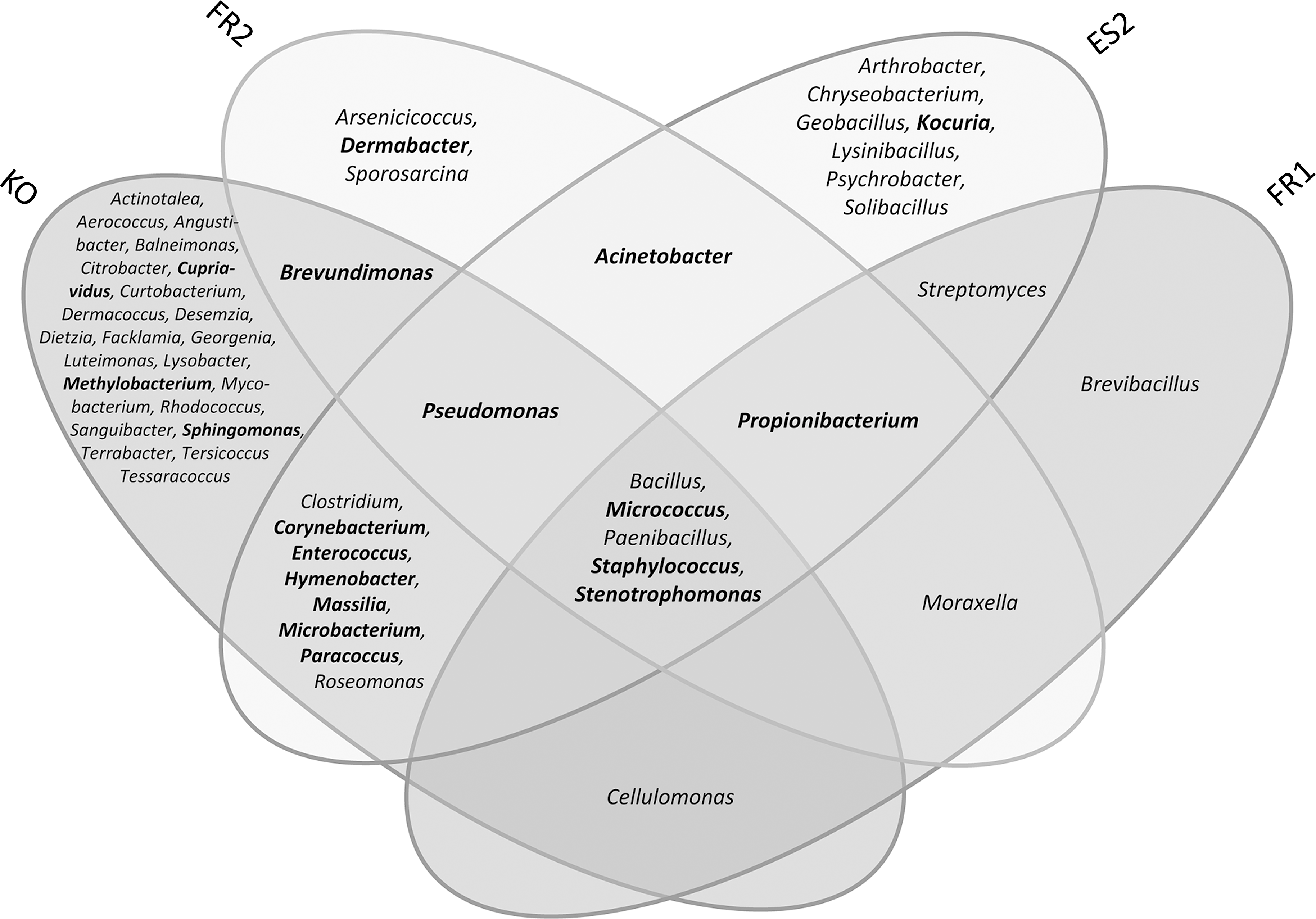
Venn diagram of the microbial diversity (genera) detected by cultivation. Microbial genera that were detected by cultivation and molecular detection methods are highlighted in bold. Results are shown for FR1, FR2, ES2, and KO only, since ES1 sampling and analyses were performed without alternative cultivation methods.
Bacteria that were isolated from each of the facilities included the spore-forming microbes Bacillus and Paenibacillus but also non-spore-forming Gram-positives (Micrococcus, Staphylococcus) and Gram-negative Stenotrophomonas. The success of the alternative enrichment strategies is demonstrated by the isolation of, for example, strictly anaerobic propionibacteria, retrieved from three samplings (FR1, FR2, ES2).
Further details with respect to the overall cultivable microbial diversity have been given in other publications (Stieglmeier et al., 2009, 2012; Schwendner et al., 2013).
3.2. Cultivable, heat-shock-resistant microbial diversity (“bioburden”)
The heat-shock-resistant microbial diversity obtained following the ECSS standard for bioburden determination spanned three phyla (Table 3). The overwhelming majority of isolates were assigned to the phylum Firmicutes, with genus Bacillus dominating. Bacillus pumilus was found in each of the locations, as were representatives of the B. subtilis group.
Heat-shock-resistant isolates from FR1 samplings were not characterized phylogenetically and are therefore not included in this table.
Isolates capable of forming spores.
B. subtilis refers to representatives of the B. subtilis group due to an insufficient resolution of the 16S rRNA gene sequence analysis; no further characterization was performed.
Besides Gram-positive, spore-forming bacteria, a broader variety of non-spore-formers and Gram-negative microorganisms were obtained. In particular, Staphylococcus strains appeared frequently on agar plates, apparently resisting the application of heat shock (80°C for 15 min; Table 3).
3.3. Microbial diversity with respect to collection and cultivation strategy
The cultivation success of microorganisms is strongly dependent on the enrichment medium used and cultivation strategy applied. To visualize these differences, the four different sampling and enrichment methods were displayed according to their success in covering the bacterial diversity (Fig. 2).
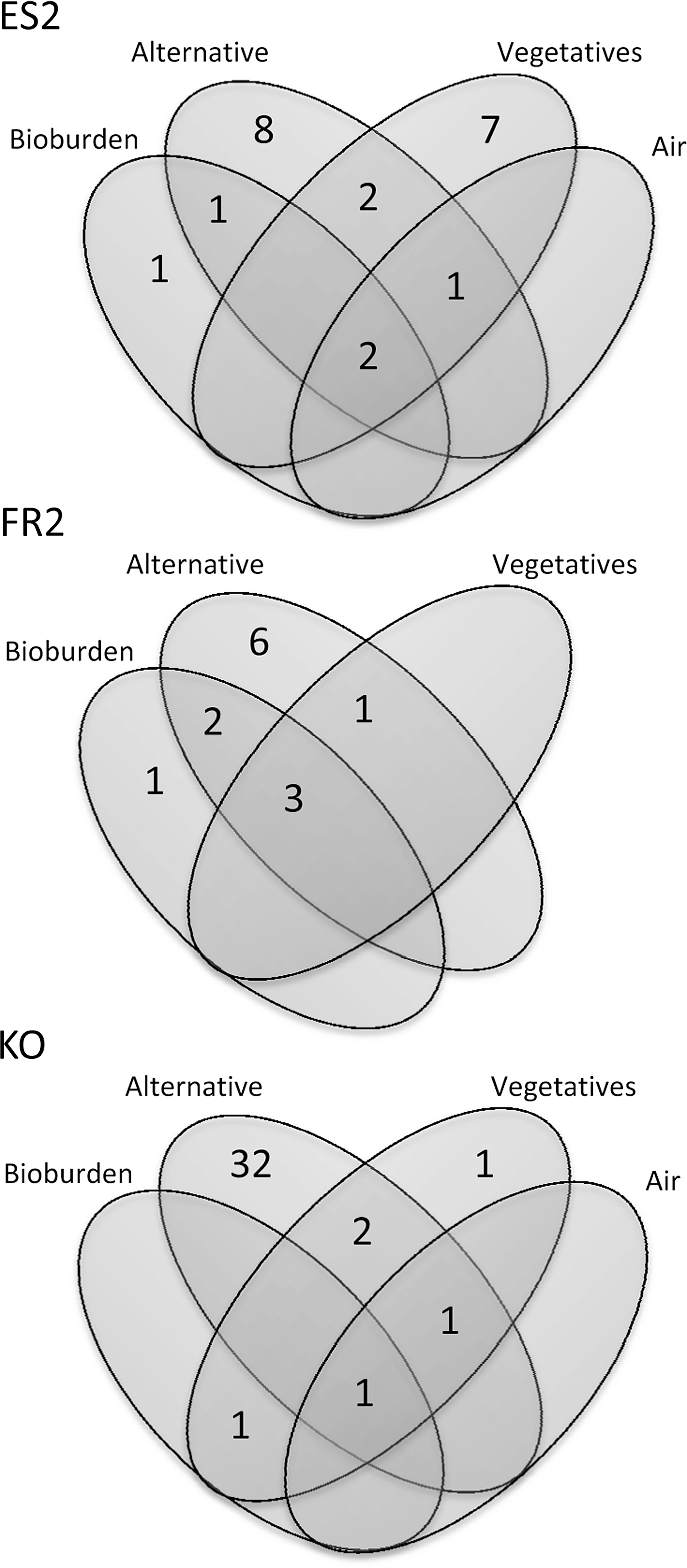
Number of bacterial genera obtained by different cultivation strategies (isolates obtained after heat-shock treatment and enrichment on TSA (“Bioburden”), isolates grown on R2A without prior heat shock (“Vegetatives”), and isolates obtained under alternative cultivation conditions (“Alternative”). ES1 and FR1 are not shown (alternative assay not performed/low number of analyzed isolates). Air sampling was not performed at the FR2 sampling event.
Bacillus, Micrococcus, and Staphylococcus were detected by at least three different isolation strategies, whereas a broad diversity of microbes (in, e.g., Kourou samples) was obtained only by applying a greater number of alternative cultivation conditions.
3.4. Diversity of cultivable, strictly and facultatively anaerobic bacteria
The procedures and the successful isolation of strictly and facultatively anaerobic bacteria from clean room samples have been previously described (Stieglmeier et al., 2009; see also Probst et al., 2010a). During the Herschel campaign, most of the isolates recovered under anaerobic cultivation conditions were facultative anaerobes. Also, some strictly anaerobic microbes were retrieved from each location, adding up to 12% of all isolates obtained from alternative assays (Table 4). Different strains of Propionibacterium were isolated from FR1, FR2, and ES2. Spore-forming clostridia were isolated from the ESTEC (ES2) and South American facilities.
3.5. Diversity of extremotolerant bacteria
The use of alternative media for the enrichment of microorganisms from spacecraft assembly facilities increased the overall isolate diversity by a factor of 2.6 (18 and 47 genera for standard and alternative cultivation, respectively). In total, 29 microbial genera were exclusively obtained by providing alternative cultivation conditions. The cultivable clean room microorganisms showed a clear preference for alkaline media, as well as media with reduced organic compounds (Table 5).
Only colony counts from solid media are shown. For comparison, CFU obtained from bioburden measurements as well as from the vegetative assay are given in the last two rows. ES1 is not shown since no enrichment on alternative media was performed.
According to ECSS standard
n.a., not applicable due to overgrowth of the plates (swarming colonies); n.d., not done.
Two Bacillus strains, one Paenibacillus strain, and Micrococcus flavus were exclusively isolated on R2A medium, pH 11. Dermacoccus sp. was enriched on plates with pH 5, as was Mycobacterium chubuense, Angustibacter sp., Terrabacter sp., and Hymenobacter rigui, the only representative of the Bacteroidetes phylum.
A broad diversity of oligotrophic and oligotolerant bacteria was obtained on 1:100 diluted R2A: Acinetobacter, Balneimonas, Brevundimonas, Citrobacter, Kocuria, Microbacterium, Micrococcus, Moraxella, Paenibacillus, Sanguibacter, Staphylococcus, Stenotrophomonas, Streptomyces (Moissl-Eichinger, 2011b). Colony counts obtained from media for oligotrophs were often comparable to or exceeded counts from classical R2A medium (Table 5).
Sporosarcina globispora (FR2 sampling) was only isolated under low-temperature conditions (10°C) and was the sole isolate that was unable to grow at 32°C in subsequent cultivations. All other microbes isolated at 4°C and 10°C were capable of growing at the standard incubation temperature, and most of them were also detected under other growth conditions provided. Growth at lower temperatures was delayed compared to elevated temperatures. The only thermotolerant microorganism isolated was Geobacillus sp. (ES2 sampling).
Some isolates revealed multitolerance with regard to different cultivation approaches. For instance, Micrococcus [isolation conditions: R2A 1:100, pH 11, and salt concentrations of 10% (w/v)] and Paenibacillus (isolation conditions: R2A 1:100, pH 11, anoxic, and 50°C) were also grown on medium for CO2- and N2-fixing microbes. CO2-fixing activity was found for Staphylococcus, but isolates of this genus also showed the ability to grow under reduced organic concentrations, alkaline pH, elevated salt concentration, and anaerobic conditions. Moreover, isolates classified as Bacillus turned out to be physiologically versatile, being enriched even at pH 11, without oxygen, 50°C or elevated salt concentrations.
3.6. Molecular microbial diversity based on 16S rRNA gene cloning
Details with respect to molecular analyses based on bacterial 16S rRNA gene pool cloning and sequencing from locations FR1, FR2, ES2, and KO have been published earlier (Stieglmeier et al., 2012; Schwendner et al., 2013). Overall, the molecular analyses revealed the presence of signatures of up to 10 bacterial phyla within the spacecraft assembly clean rooms [Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Deinococcus/Thermus, Firmicutes, Gemmatimonadetes, Planctomycetes, and Proteobacteria (α, β, γ)], whereas the highest diversity was obtained from KO samples and the lowest diversity from FR1.
Propionibacterium and Staphylococcus were the only microbial genera that were detected in all facilities (Fig. 3). Interestingly, the molecular analyses revealed a relatively good overlap with cultivation results, and 18 microbial genera were detected by both methods. Staphylococcus, for instance, was identified by cultivation and cultivation-independent methods in all sampled facilities. Propionibacterial molecular signatures were found in each clean room, and isolates were cultivated from three samplings (FR1, FR2, ES2).

Venn diagram of the microbial diversity (genera) in the four sampling events FR1, FR2, ES2, and KO, detected by molecular methods based on 16S rRNA gene analyses of the gene pool. Microbial genera that were detected by cultivation and molecular detection methods are highlighted in bold. Sequences that could not be phylogenetically classified on genus level are not shown.
Signatures of putative spore-forming microbes were not detected with the molecular approach, verifying our previous assumption that spore formers seem to be present mainly as spores, which reduces the chance of being recognized by DNA extraction methods without bead-beating.
3.7. Molecular microbial diversity comparison
A database was generated with all molecular and cultivable 16S rRNA gene sequences from clean room environments that were publicly available at NCBI (National Center for Biotechnology Information, March 2012). Using classification of each sequence with the Bayesian algorithm against a manually curated and updated Greengenes database (see Materials and Methods), we identified 230 different microbial genera present in clean room environments. One hundred forty-nine of these 230 microbial taxa were identified via cloning only, and 53 were detected via molecular and cultivation tools. Twenty-eight genera were retrieved in cultivation only and completely escaped the molecular assay. The amount of cultivable bacteria in clean room environments was estimated to be 35% based on genus level, which is extraordinarily high and can mainly be attributed to the application of alternate cultivation assays (see above).
With regard to the molecular diversity only, a statistical comparison of all data generated with 16S rRNA gene cloning from American and European clean room facilities was performed. Non-metric multidimensional scaling (Fig. 4) showed an outgroup of anaerobic enrichment cultures published by Probst et al. (2010a) and therefore confirmed our statistical approach. Microbial communities detected by Moissl et al. (2007) spanned the major part of the ordination analysis, which suggests that the study covered a great proportion of the microbial diversity of clean room facilities. Samples from ESA spacecraft assembly facilities were not as closely related to samples from NASA clean rooms as expected, which indicates that the geographic relatedness of a clean room may have influence on the microbial community structure.

Non-metric multidimensional scaling (NMDS) of normalized clean room diversity (generated via 16S rRNA gene cloning). Pink lines represent a curve-fitting model, which is based on the number of OTUs observed in the samples. Names are constructed as follows: the first letter or letters gives the last name of the first author of the publication in which the library was released, the following number gives the year, and the code after the underscore is the sample ID in the study. Studies: Moissl et al., 2007; La Duc et al., 2009; Probst et al., 2010b; Vaishampayan et al., 2010; Stieglmeier et al., 2012; Schwendner et al., 2013. For instance, M07_JPL1A is from Moissl et al., 2007, sample JPL1A. Displayed names are only those of Moissl et al., 2007, as this study spanned the greatest diversity, ESA samples (further investigated in the current study) and samples from Probst et al., 2010b. The latter study used an enrichment procedure before DNA extraction and thus examined a different microbial community. These samples form an outgroup separated along NMDS1 axis from other samples and therefore validate the statistical approach performed herein. Circles with cross=European clean rooms; black dots=US American clean rooms; circles=enrichment from US American clean room; gray=potential outlier. Color images available online at
3.8. Microbial abundance based on cultivation, bioburden measurement, and molecular analysis
Spore-forming isolates were cultured from each of the facilities. The heat-shock application as well as various enrichment conditions influenced the percentage of spore-forming organisms obtained compared to the total number of isolates (Fig. 5). The bioburden protocol clearly supports the selective enrichment of spore formers and yields percentages from 29% (FR2) up to 90% (ES2) with 62% on average. Lower percentages were obtained by following the approach for the enrichment of vegetative microbes as given in the standard document (14–47%) or by performance of alternative cultivation experiments (0.5–24%). In general, the lowest percentage of spore-forming microbes was observed in samples from FR2 (all three enrichment methods resulted in 23% on average).

Cultivated number of spore formers proportional to all isolates (in %), dependent on the cultivation approach used: isolates obtained after heat-shock treatment and enrichment on TSA (“Bioburden”), isolates grown on R2A without prior heat shock (“Vegetatives”), and isolates obtained under alternative cultivation conditions (“Alternative”). ★: The alternative approach was not performed for ES1. “Bioburden” and “Vegetatives” counts from FR1 were not considered since only a very low number of isolates were phylogenetically analyzed.
The colony-forming unit counts for alkaliphilic or alkalitolerant bacteria ranged from 1.3×103 up to >2.0×104 per square meter (pH 9) and from 4.6×102 up to >2.0×104 per square meter (pH 11). However, acidic media (pH 5 and pH 3) were accepted only marginally for growth (Table 5).
Although the distribution of microbes in the clean rooms turned out to be heterogeneous (for details see Stieglmeier et al., 2012), the average values from different locations were quite consistent. Interestingly, the bioburden measurements revealed, independently from the clean room class, a contamination in the range of 102 heat-shock-resistant cultivables per square meter of clean room floor and GSE, whereas the cultivation approach without heat shock (vegetatives) resulted in one order of magnitude higher colony counts (Table 6). Nevertheless, compared to the data derived from wipe sampling, the swab samples delivered inconsistent data, most likely due to the small area sampled, which resulted in a higher variation (25 cm2, compared to up to 1 m2 sampling area for wipes; Stieglmeier et al., 2012).
The range of colony counts or cell counts detected is given in parenthesis.
n.a., not applicable due to overgrowth of the plates (swarming colonies); n.d., not done.
Spacecraft samples were taken from Ariane 5 fairing (Schwendner et al., 2013).
Number of bacteria was estimated based on 16S rRNA gene copies, corrected by factor 4.1 [each bacterial cell contains several copies of 16S rRNA genes; please refer to Lee et al. (2009) for further details].
3.9. Archaea
Archaea were detected in samples from FR2, ES2, and KO (other sampling campaigns were not used for the detection of archaea). ES2 samples even revealed archaeal fluorescence in situ hybridization (FISH) signals of rod-shaped cells. The average, estimated cell number of archaea per square meter of clean room floor did not exceed 1.2×104 (Moissl-Eichinger, 2011a; based on qPCR results), although their constant presence suggests a larger role within the clean room environment (further details, see Moissl-Eichinger, 2011a). The detected archaeal diversity spans two different archaeal phyla: Thaumarchaeota and Euryarchaeota (methanogens and halophiles).
3.10. Fluorescence in situ hybridization (FISH) analyses
Fluorescence in situ hybridization was used to visualize microbes in original samples and to assign a phylogenetic affiliation to certain morphologies. Although FISH is not feasible for use in low-biomass samples, we were able to visualize microbes in each sample. Different morphologies were detected (cocci, rods, diplococci), revealing all good signals with bacteria-targeted probes. Examples for positive FISH were given by Schwendner et al. (2013) and Moissl-Eichinger (2011a). Paenibacilli were visualized with specifically designed probes in some of the Kourou samples (Schwendner et al., 2013). Strong fluorescence signals suggest a high content of ribosomal RNA and are an indicator for microbial activity. Interestingly, many microbes were found to form aggregates with particles, which made their detection quite difficult. Domain-specific FISH revealed also the presence of archaea (Moissl-Eichinger, 2011a).
4. Discussion and Lessons Learned
In preparation for the upcoming ExoMars mission, ESA has increased its efforts to analyze and characterize the microbial diversity and abundance on and around spacecraft during assembly, test, and launch operations. Although not subject to planetary protection requirements, the Herschel spacecraft was used as a model for establishing and practicing microbiological methods to analyze bioburden and biodiversity. The microbiome of built environments and in particular of confined clean rooms is indeed special. Due to a restricted exchange with the environment, microbes are transferred via items or humans only and subsequently suppressed by harsh environmental conditions within the clean room. The confinement and applied cleaning procedures in classified clean rooms (ISO 146441) reduce the microbial load significantly and select hardy microorganisms, such as spore formers. The Herschel project covered different methodologies to analyze spacecraft-associated microorganisms. We used a cultivation approach with more than 30 different media to isolate bacteria and to feed the novel ESA culture collection (Moissl-Eichinger et al., 2012), and used molecular methods that allowed for insight into the uncultivable part of the microbiome. In the following section, we summarize the major findings of the project and make recommendations for changes in procedures or to improve protocols.
Throughout this campaign, surface sampling was performed with different sampling tools, such as wipes, swabs, witness plates, BiSKits, and SpongeSicles. Each of these tools was functional and returned useful data. However, future ESA studies will mainly use wipes and swabs for a number of reasons. First, different sampling tools have different recovery efficiencies (Probst et al., 2011) and can, therefore, considerably affect comparability of results across multiple studies, which can only be overcome by standardizing protocols for sampling and for sample processing. Although the sponge-based sampling tools (BiSKits, SpongeSicles) delivered good results, the possibility of sponge particle loss or buffer residues on a spacecraft or in its close vicinity was a concern and resulted in their exclusion (G. Kminek, personal communication).
In addition, analyses reported severe problems with the BiSKit tool with respect to DNA contamination of the included buffer (Kwan et al., 2011). Recent efforts have focused on selection of wipes and swabs for sampling of clean room and spacecraft surfaces, which can be used for both cultivation and molecular assays. The nylon-flocked swab has been evaluated previously for the efficient recovery of spores from various surfaces (Probst et al., 2010b), but a lower efficiency compared to cotton swabs in recovering free DNA has been found (Kwan et al., 2011). However, cotton swabs are made of natural fibers, which are, as mentioned for sponges, a problem with respect to particle control. In addition, interference with subsequent DNA-based detection methods could become a problem. A similar problem occurs with wipes, which cannot currently be delivered (certified) DNA-free. This has necessitated the inclusion of a step to remove any potential DNA contamination (dry-heat treatment; 170°C, 24 h; A. Auerbach, personal communication). Based on these arguments and the current status of knowledge, polyester wipes (for surfaces up to 1 m2) and nylon-flocked swabs (for surfaces up to 25 cm2) remain the sampling tools of choice. Future studies should be based on the two selected sampling tools for spacecraft and clean room surfaces. Such an approach for bioburden and biodiversity studies will minimize bias potentially introduced by differing sampling techniques and subsequent extraction protocols.
Since only 1% of all microorganisms can be cultivated under defined laboratory conditions (Amann et al., 1995), molecular techniques are generally useful to detect microorganisms that cannot be found by cultivation. Molecular analyses performed in this study were based on DNA extraction without bead-beating, qPCR analyses, and classical 16S rRNA gene cloning (Stieglmeier et al., 2012; Schwendner et al., 2013). Molecular technologies that have been used to characterize clean room microbial diversity include 16S rRNA gene cloning (Venkateswaran et al., 2001; La Duc et al., 2004; Moissl et al., 2007, 2008; Probst et al., 2010a; Moissl-Eichinger, 2011a; Stieglmeier et al., 2012; Schwendner et al., 2013), PhyloChip DNA microarray of the generation 2 and 3 (La Duc et al., 2009; Probst et al., 2010a; Cooper et al., 2011; Vaishampayan et al., 2013a), and 16S rRNA gene pyrosequencing (La Duc et al., 2012; Vaishampayan et al., 2013a). PhyloChip and pyrosequencing were reported to detect a greater level of diversity than classical technologies but have only been conducted on a restricted number of samples. Although these two state-of-the-art technologies allow greater insight into the biodiversity present, the majority of clean room samples have only been analyzed with classical 16S rRNA gene cloning.
Our comparative analysis demonstrated the Herschel clean room microbial diversity to somewhat differ from that reported in NASA clean rooms. Furthermore, the Herschel clean room contained organisms not previously reported in NASA clean room samples. The reasons for this observation are currently unclear but could be assigned to the geographical location of the clean rooms (see also Moissl et al., 2007) or to method biases (DNA extraction technique, selected primers, PCR conditions, etc.; Von Wintzingerode et al., 1997).
Nevertheless, this analysis demonstrated that synchronizing the protocols would also allow more comprehensive comparisons, since this would reduce methodological biases. In addition, it is necessary to further characterize individual clean rooms to create a more complete picture of the microbial signatures that might vary temporarily and spatially. Stieglmeier et al. (2012) clearly showed microbial contamination within a clean room to be extremely variable and highly heterogeneous. As a consequence, frequent sampling and sampling of multiple locations within a clean room is imperative in establishing an accurate picture of the clean room microbiome.
Another suggested change to processing methods is the inclusion of a bead-beating step in the DNA extraction protocol; although appearing frequently in cultivation assays, spore-forming bacteria were not at all detectable via 16S rRNA gene cloning in the Herschel campaign. This suggests that the DNA extraction method employed by these studies was unable to open spores but nevertheless showed the presence of those bacteria as spores and not as vegetative cells.
Answering the question of whether microorganisms are alive or dead in clean room facilities can be a crucial factor when assessing the potential risk of a forward contamination. RNA-targeted studies may provide better evidence for microbial viability than DNA-based studies as RNA degrades faster; it is, however, also more difficult to extract (Vaishampayan et al., 2013a). As an alternative, studies have recently used propidium monoazide (PMA) as a viability marker (Probst et al., 2012; Vaishampayan et al., 2013a). PMA has proven useful for the analysis of intact cells in various microbial communities (Nocker et al., 2007). Masking DNA of disrupted cells for PCR downstream processing revealed the biodiversity, which was alive or at least undisrupted at the time point of sampling. The application of this technology to US American clean rooms demonstrated a lower microbial diversity and abundance than previously assumed by standard qPCR, pyrosequencing, and PhyloChip G3 (Vaishampayan et al., 2013a). It has also been shown that mission-critical clean rooms exhibited a lower microbial richness and abundance than non-mission clean rooms with respect to the living microbial proportion (Vaishampayan et al., 2013a). This indicates that cleaning protocols were highly efficient. However, conservative planetary protection considerations must assess on an individual basis the risk of each possible biological contaminant—alive or dead. PMA-combined technologies may prove useful in future clean room analyses. These molecular methods will not, however, replace time-consuming cultivation efforts. Only cultivation can conclusively demonstrate the viability of organisms recovered from clean rooms. More important, this approach allows microbial isolates to be collected, which can serve as test objects for cleaning and sterilization efforts. The analysis of resistances and adaptations of those microbial isolates by the scientific community can add critical information to planetary protection considerations (Venkateswaran et al., 2003; Link et al., 2004).
Samples from the South American clean room were found by this study to have the broadest diversity of cultivable bacterial genera. The external environment has been proposed to exhibit a major influence on the clean room microbiome (Moissl et al., 2007), and this seems to be true in this case, as the humid and warm climate supports biodiversity (Fierer et al., 2012; Schwendner et al., 2013). Cultivable biodiversity is also influenced by the maintenance procedures and is significantly reduced under higher particulate control, as observed for the FR1 sampling event. However, microorganisms that were cultivated in samples from all four clean rooms were the typical clean room contaminants: Bacillus, Micrococcus, Paenibacillus, and Staphylococcus. Interestingly, Stenotrophomonas, a contaminant not previously considered a typical clean room inhabitant, was cultivated from all locations on standard media. Stenotrophomonas species were found in air filters in two shopping malls in Singapore (Tringe et al., 2008) and have attracted interest with respect to multi-drug-resistance and causing nosocomial infections in immunocompromised people (Denton et al., 1998). In the literature, representatives of these microbes were reported to be resistant to a number of toxic metals, including cobalt, silver, mercury, and cadmium. This may be important for planetary protection considerations (Pages et al., 2008).
Although heat shock was applied, a relatively low percentage of spore formers were recovered on culture media. Micrococcus, Staphylococcus, Rothia, Acinetobacter, Stenotrophomonas, Paracoccus were among the heat-shock-surviving, non-spore-forming bacteria found in this study. Micrococcus and Staphylococcus are typical clean room contaminants and could therefore significantly influence colony counts retrieved in bioburden measurements. For a detailed discussion please refer to Stieglmeier et al. (2012).
A large number of isolates have not been assigned to a species due to larger (>2.5%) phylogenetic distance of the 16S rRNA gene compared to closest related type species, or an unclear phylogenetic situation. One isolate (Tersicoccus phoenicis gen. nov, sp. nov.) was found to represent a novel genus and was recently described (KO_PS43; Vaishampayan et al., 2013b). Interestingly, this species was isolated in parallel in a US clean room (Kennedy Space Center) independent of this study. Another novel isolate was described 2 years earlier (Behrendt et al., 2010). A spore-forming organism, Paenibacillus purispatii sp. nov., was isolated from a sample taken at the ISO 8 clean room in ESTEC (ES2 sampling). The enrichment of both bacteria was achieved in alternative media under anaerobic conditions (Hino and Wilson NH3-free medium and TSA, respectively).
The application of alternative media has increased the cultivable microbial diversity immensely. For instance, the appearance of Propionibacterium is worth mentioning. This microorganism was isolated from three European clean rooms, and its cultivation was only possible by the use of alternative cultivation media (anaerobic conditions). In accordance with previous studies (e.g., La Duc et al., 2007), many isolates were found to tolerate “extreme” cultivation conditions. Media at pH 9 or 11 and low-nutrient plates, such as R2A, revealed similar or higher colony counts. This could indicate an adaptation of the microorganisms to (alkaline) cleaning detergents and to low nutrient availability in clean room environments.
The application of these two conditions and the cultivation under strictly anaerobic conditions has added crucial insights with respect to the cultivable microbiome from clean rooms. As a consequence, ESA has proposed to add these three cultivation strategies as an optional procedure to the standard document in order to analyze not only the heat-shock-resistant bioburden but also possibly adapted microorganisms.
Archaea were detectable in low, but constant, abundance in all clean rooms analyzed to date; furthermore, in one case these organisms were even shown to be intact and physiologically active (Moissl et al., 2008; Moissl-Eichinger, 2011a). The role of Eury- and Thaumarchaeota in such built environments is currently unclear. Similarly, it is also unclear whether these organisms pose a risk to planetary protection efforts. Recent experiments have proven the association of Thaumarchaeota with human skin (Probst et al., 2013), which is in accordance with the observation that most microbial contaminants in clean rooms are human-associated.
Fluorescence in situ hybridization has proven useful for the visualization of (active) archaea and bacteria in clean room samples. Although the overall number of cells was rather low, detection of coccoid and rod-shaped microbes was possible, which were mainly attached to particles. This observation supported the decision to sonicate the wipes during extraction steps in order to loosen the attachment of microbial cells to particle or wipe surfaces.
Although a presumably large portion of the microbial community present on and around spacecraft can be detected, analyzed, and visualized, the microbiome in clean rooms is still mysterious in many ways. However, each analysis performed will contribute important data for future sampling campaigns as did the Herschel study. The space agencies' efforts are substantial to reduce microbial contamination and to confine the risk for extraterrestrial environments and life-detection missions as much as possible. This is an effort that will require constant adaptation and improvement of protocols in order to deliver a better understanding of the microbial diversity and biocontamination in space travel.
Footnotes
Acknowledgments
Part of the research described in this paper was carried out by DLR under contract with ESA, ESTEC contract no. 20234/06/NL/EK. The work performed in Regensburg was funded by ESA, ESTEC contract no. 20508/07/NL/EK. We thank the Herschel Project Team for support during sampling. We thank Gerhard Kminek for critical discussion. Alexander J. Probst was supported by the National German Academic Foundation (Studienstiftung des deutschen Volkes). We are grateful to Anna Auerbach and Melanie Duckstein for providing information, lab management, and technical assistance. We thank Gabriel Milinovich for critically reading the manuscript.
Abbreviations
DLR, Deutsches Zentrum für Luft- und Raumfahrt; DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH; ECSS, European Cooperation for Space Standardization; ESTEC, European Space Research and Technology Centre; FISH, fluorescence in situ hybridization; GSE, ground support equipment; OTU, operational taxonomic unit; PMA, propidium monoazide; qPCR, quantitative PCR; TG, thioglycollate liquid medium; TGA, thioglycollate agar; TS, trypticase soy liquid medium; TSA, trypticase soy agar.