
Abstract
The strong oxidant H2O2 is known to exist in solid form on Europa and is suspected to exist on several other Solar System worlds at temperatures below 200 K. However, little is known of the thermal chemistry that H2O2 might induce under these conditions. Here, we report new laboratory results on the reactivity of solid H2O2 with eight different compounds in H2O-rich ices. Using infrared spectroscopy, we monitored compositional changes in ice mixtures during warming. The compounds CH4 (methane), C3H4 (propyne), CH3OH (methanol), and CH3CN (acetonitrile) were unaltered by the presence of H2O2 in ices, showing that exposure to either solid H2O2 or frozen H2O+H2O2 at cryogenic temperatures will not oxidize these organics, much less convert them to CO2. This contrasts strongly with the much greater reactivity of organics with H2O2 at higher temperatures, and particularly in the liquid and gas phases. Of the four inorganic compounds studied, CO, H2S, NH3, and SO2, only the last two reacted in ices containing H2O2, NH3 making
1. Introduction
T
The persistence and detection of any material within or beneath Europa's ice shell depend strongly on such material's response to the local environment of high-energy radiation, strong oxidants, and low temperatures. In general, one can divide the solid-phase chemistry that is expected to occur on Europa and other icy moons of the Solar System into that which is driven by UV photons (E ∼ 10 eV), by kiloelectronvolt and megaelectronvolt external particle radiation, and by thermal processes at about 30–130 K. The chemistry induced by UV photons is largely confined to surface depths of less than a centimeter, whereas ionizing radiation can alter the chemistry for several meters below the surface (Barnett et al., 2012). Thermal chemistry, which does not rely on an external energy source, will be active at all depths below the surface but may be more important at increasing depths as temperatures rise.
To date, telescopic and spectroscopic observations of Europa have probed its surface composition to depths of at best a few centimeters, with deeper subsurface chemistry remaining hidden. We and others have examined Europa's ice chemistry through extensive photo- and radiation-chemical laboratory experiments on icy materials (Hudson and Moore, 2001; Baratta et al., 2002; Hand and Carlson, 2011; Johnson et al., 2012). However, low-temperature thermal processes relevant to Europa have been studied much less frequently. In a recent paper, we described a thermally driven chemical reaction that can occur at europan temperatures even in the absence of external photons and ionizing radiation (Loeffler and Hudson, 2010). Amorphous ices consisting of H2O and SO2 showed changes in their IR spectra on warming from 50 to 100 K, demonstrating that H2O and SO2 combined to form H3O+ and
That this reaction took place in the absence of far-UV photons and ionizing radiation means that it also can occur beneath Europa's surface. Thus, the results we reported can be regarded as “hidden chemistry” in that they are changes that will occur at depths greater than those which current instrumentation can probe.
Moore et al. (2007b) showed that the irradiation of H2O+SO2 ice mixtures at 86–132 K with 0.8 MeV protons resulted in oxidation of SO2 into
This radiation-driven process is a plausible source of the
Having shown that H2O2 can produce sulfate through Reaction 3 followed by Reaction 4, we now consider whether thermally induced H2O2 reactions also are important in other systems. The ability of frozen H2O2 to oxidize other molecules is of considerable interest to astrobiologists for several reasons. Greenberg (2010) summarized the case for downward transport of surface ices on Europa. Such movement would transfer surface material toward subsurface liquid and higher temperatures, carrying with it Europa's radiolytically generated H2O2 (Carlson et al., 1999a). If the H2O2 reaches the subsurface liquid, then it could significantly alter the chemistry occurring there, as the liquid would become more acidic (Pasek and Greenberg, 2012). Were subsurface oxidation by frozen H2O2 possible, then it could weaken the argument that organics (e.g., CH4), originating from endogenic or exogenic sources (Zolotov and Kargel, 2009), trapped in ices could be available as nutrients for subsurface life. Conversely, any SO2 that is oxidized by H2O2 could provide a potential nutrient for subsurface sulfate-reducing bacteria or their extraterrestrial analogues. There also is a connection to planetary protection in our interest in low-temperature H2O2 reactions. Such reaction chemistry can be used to understand the fate of molecules deposited by accident on icy Solar System surfaces and in subsurface regions that are shielded from direct exposure to UV light or ionizing radiation but which still may contain H2O2.
In the present study, we examined how H2O2 in H2O-ice might promote chemical changes in the absence of ionizing radiation and far-UV photons. Our goal was to determine whether relatively simple molecules, specifically those listed in Table 1, can react in the solid state with H2O2. We point out that for this study we chose a relatively simple reaction system to compare the propensity of hydrogen peroxide to react with a variety of astrobiologically relevant molecules. Including more reactants or adding a known room-temperature catalyst (e.g., metal ions) might have affected our results. Although H2O2 is a potent room-temperature oxidant, to our knowledge the present study and our previous one (Loeffler and Hudson, 2013) are the first to investigate its oxidizing abilities in ices at temperatures relevant to the outer Solar System. Here, we first briefly review our earlier SO2 work and then extend it and move on to other new results obtained by in situ measurements with IR spectroscopy.
See Tielens (2013). bSee Mumma and Charnley (2011). cSee, for example, Dalton et al. (2010), dLane et al. (1981), eMcCord et al. (1998), fBrown and Calvin (2000), gBauer et al. (2002), hCruikshank et al. (1993), iOwen et al. (1993), jCruikshank et al. (1976), kCruikshank and Silvaggio (1979), lMaguire et al., (1981). mBased on modeling of spectra (Merlin et al., 2012) and nMarten et al. (2002).
2. Experimental Methods
Experiments were performed with a cryostat (T min∼10 K) operating in a stainless steel high-vacuum chamber (P∼1×10−7 torr) interfaced to an IR spectrometer. Ices were prepared by co-deposition of H2O, H2O2, and an organic or inorganic molecule, chosen from those listed in Table 1, onto a pre-cooled (10–50 K) gold-coated aluminum mirror (area≈5 cm2) by using three separate pre-calibrated gas lines. Mixtures that contained CO and CH4 were deposited at 10 K, but all other samples were prepared at 50 K. Pure H2O2 was prepared in a glass manifold as previously described (Loeffler and Baragiola, 2011). During deposition, the increase in the sample's thickness was monitored by interferometry with a diode laser (670 nm), and the deposition was halted when a thickness of about 1.5 μm was reached.
After deposition of a sample, its IR spectrum was recorded before, during, and after warming at 1 K min−1. A closed-cycle helium cryostat and a resistive heater served to maintain ices at the desired temperature within the 10–200 K range studied. Higher temperatures could be reached but were avoided, as they were accompanied by sample loss by sublimation, which complicated the interpretation of our experiments.
Spectra were measured from 7000 to 400 cm−1 with a Bruker Vector 22 Fourier transform infrared spectrometer at 2 cm−1 resolution and with 200-scan accumulations. To obtain a spectrum, the IR reflectance (R) from the ice-coated substrate was divided by the reflectance of the bare metal substrate (R 0), taken before ice formation, and then converted to an absorbance-type scale, -log(R/R 0), for the figures we show in this paper. See the work of Loeffler and Hudson (2010, 2012) for additional experimental details.
Each compound studied was examined in the solid phase mixed with frozen H2O2, both in the presence and absence of H2O-ice. During experiments, ices were held as long as 20 h at the higher temperatures (e.g., 100–170 K) to check for thermal changes. At the end of each experiment, the sample was warmed to room temperature, but no residual material was observed either visually or through IR spectroscopy.
The sensitivity of our spectrometer to detect the products of a thermally induced reaction under the conditions employed was checked with appropriate background and calibration experiments. For example, when working with CO we sometimes observed a weak CO2 feature before warming with H2O2 (or 13CO2 when 13CO was being studied). Blank experiments without H2O2 present traced this initial CO2 in our samples to low-level CO2 contamination, on the order of 0.002%, in our CO source and a small background contamination in our vacuum system. Possible products, such as H2CO, CH3OH, or SO2, from other reactants have weaker IR absorptions than CO2 but were still strong enough to be easily identified if present. We estimate that these possible products were detectable down to ∼1×1015 molecules cm−2 or better, corresponding to a number ratio of ∼2×10−3 (∼0.2%) when compared to the initial reactants. Thus, any nondetection we reported corresponds to an upper limit of these values.
It should be emphasized that the design of our experiments allowed all data to be collected in situ at the temperatures of choice. This avoided the need to raise samples to room temperature for chemical analyses. Also, the co-condensation method used to prepare our ices avoided the uncertainties in freezing room-temperature solutions, which will crystallize to give regions of varying H2O2 concentration in the sample. In short, the value of our results is enhanced both by the way the samples were prepared and by our method of analysis. For recent amino acid work in which similar experimental methods were used, see a study by Gerakines and Hudson (2013).
The reagents and suppliers used were H2O2 (Sigma-Aldrich, 50% by weight), SO2 (Matheson, 99.98%), H2S (Matheson, 99.5%), 12CO (Matheson, 99.998%), 13CO (Sigma-Aldrich, 99% 13C), NH3 (Matheson, 99.9992%), CH4 (Matheson, 99.999%), C3H4 (Sigma-Aldrich, 98%), CH3OH (Sigma-Aldrich, >99.8%), and CH3CN (Sigma-Aldrich, 99.93%). The water used was triply distilled with a resistivity greater than 107 Ω cm.
Finally, we point out that our use of the terms oxidation and reduction refers to the loss and gain of electrons as deduced by both formal charges and oxidation states. The meaning and method for assigning oxidation states continues to be debated and discussed (Gupta et al., 2014; Karen et al., 2014), but for now we still employ the IUPAC-recommended convention (McNaught and Wilkinson, 1997). For a careful review of redox (oxidation-reduction) chemistry in a planetary setting other than Europa, see the work of Nixon et al. (2012).
3. Results
3.1. Inorganic compounds
Table 1 lists the four inorganic compounds we studied. Sulfur dioxide (SO2) was selected since we already have data for it under other conditions (Loeffler and Hudson, 2010, 2013), and hydrogen sulfide (H2S) was chosen as a possible precursor to SO2. Carbon monoxide (CO) and ammonia (NH3) were studied as they are among the simplest of carbon- and nitrogen-containing compounds, respectively. Moreover, each of these molecules is known to be extraterrestrial, with SO2 having been reported for Europa and the other molecules suspected to be present either from cometary or meteoritic delivery or as primordial material.
Figure 1 shows the IR features of a H2O+SO2 ice on warming from 50 K. The most obvious change seen is the appearance of vibrational bands near 1050 cm−1 due to the formation of
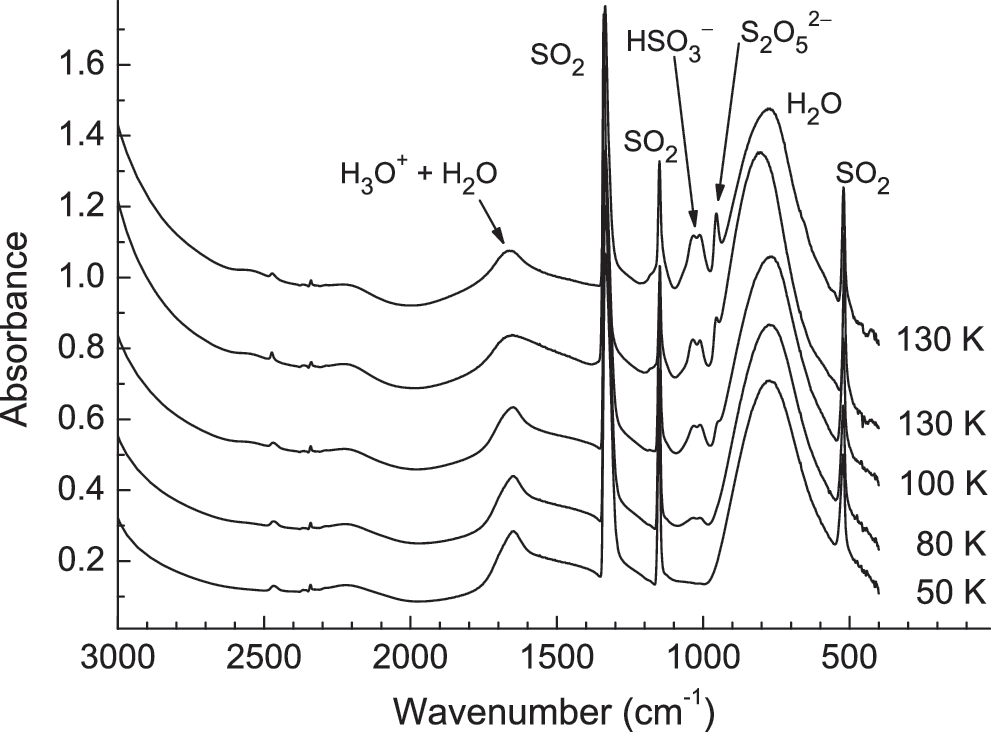
Infrared spectra of an H2O+SO2 (6:1) ice mixture made at 50 K and then warmed at 1 K min−1 to 130 K. The top spectrum was recorded after the sample had been at 130 K for 40 min. Spectra have been offset for clarity.

Infrared spectra of an H2O+SO2+H2O2 (80:14:6) mixture deposited at 50 K and then warmed at 1 K min−1 to 130 K. The top spectrum was recorded after the sample had been at 130 K for 40 min. All spectra have been offset for clarity.

Normalized areas for IR bands of H2O2 (2840 cm−1), SO2 (1150 cm−1), and sulfur oxyanions (940–1170 cm−1) in a H2O+SO2+H2O2 (80:14:6) mixture deposited at 50 K and warmed at 1 K min−1 to 130 K. A temperature of 50 K corresponds to time=0, and the vertical line corresponds to 130 K. The sulfur oxyanions feature has contributions from
Previously, we showed that ion irradiation of H2O+H2S ices near 100 K results in the formation of SO2 at the expense of H2S (Moore et al., 2007a). A straightforward explanation is that reactions such as (5) and (6) convert H2S first into elemental sulfur (S) and then into SO2, with both processes requiring H2O2 and perhaps involving numerous mechanistic steps (e.g., Hoffmann, 1977).
To examine the ability of H2O2 to effect the H2S→SO2 conversion in unirradiated ices, we prepared frozen H2O2+H2S and H2O+H2O2+H2S mixtures at 50 K and slowly warmed them to sublimation. We saw no evidence for H2S decomposition and SO2 formation, and the H2O2 bands remained relatively constant as the temperature increased. We conclude that no thermally induced redox reactions occurred between H2O2 and H2S at even the highest temperature studied (∼200 K), or at least there was no formation of detectable products.
Turning to ammonia (NH3), we can envision two types of reactions with H2O2. Our previous work (Moore et al., 2007a) showed that no reactions are seen on warming H2O+NH3 ices, which we verified in the present study (see Fig. 4). For the present experiments, an oxidation sequence for NH3 such as
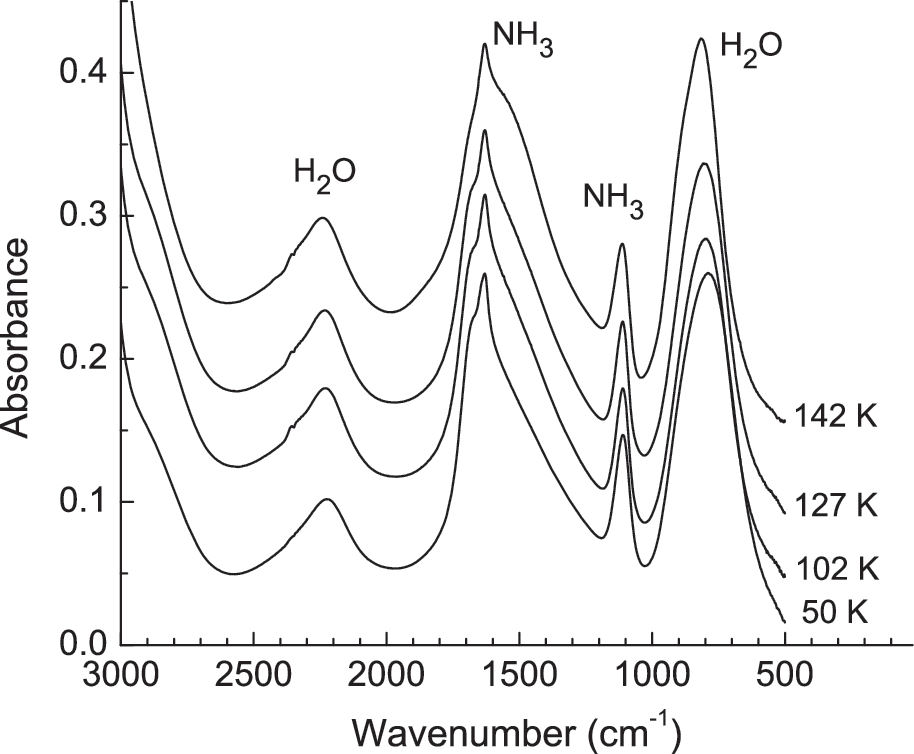
Infrared spectra of an H2O+NH3 (10:1) mixture deposited at 50 K and then warmed at 1 K min−1. Spectra have been offset for clarity.
can be envisioned, giving both
might be possible. To test these ideas, we conducted warming experiments with and without H2O-ice present. Figure 5 shows the IR spectrum of an anhydrous H2O2+NH3 mixture deposited at 50 K and then warmed to sublimation. The deposited sample possessed absorptions that do not belong to either H2O2 or NH3, indicating that a reaction occurred. The strong, sharp IR feature of NH2OH near 1190 cm−1 (Nightingale and Wagner, 1954) is not seen, arguing for this molecule's absence. However, there are multiple absorptions (see Discussion) that can be assigned to
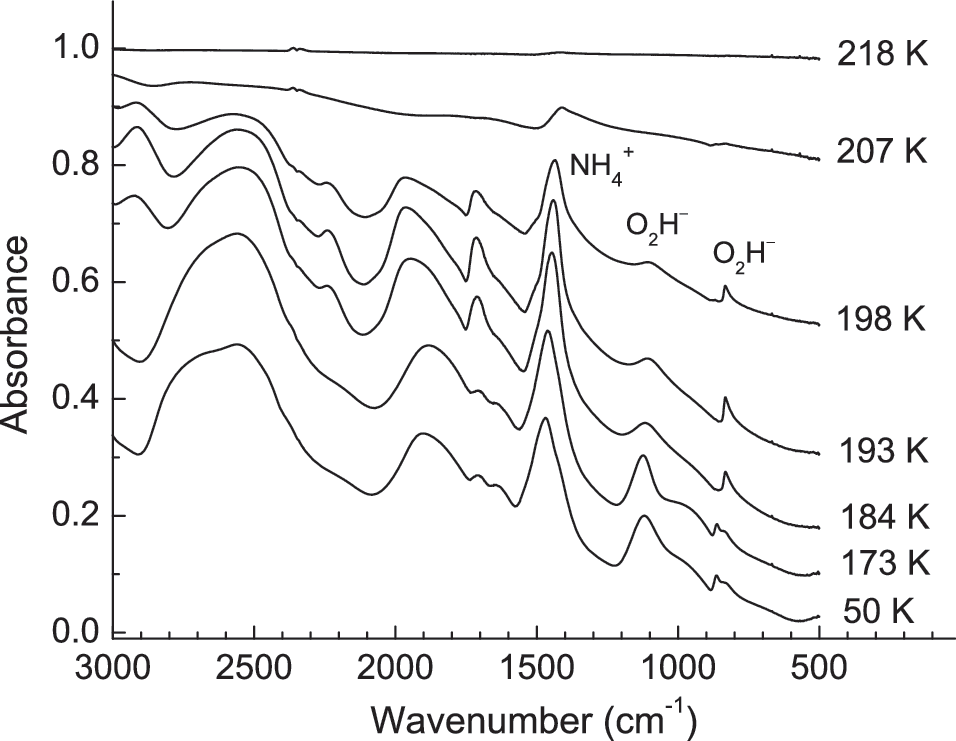
Infrared spectra of an H2O2+NH3 (1:1) mixture deposited at 50 K and then warmed at 1 K min−1. Spectra have been offset for clarity.
All these observations can be interpreted to mean that Reaction 8 took place. Interestingly, a subsequent warming of this same sample had little effect on its spectrum until ∼180 K, where the sample appeared to crystallize. In addition, lowering the deposition temperature to ∼10 K did little to alter the initial spectrum, suggesting that the energy released on deposition was enough to cause Reaction 8 to occur. No IR features were found for the oxidized nitrogen-containing species of (7).
From this anhydrous binary system, we then examined a more complex, three-component sample. Figure 6 shows IR spectra of a three-component H2O+H2O2+NH3 ice (10:1:1) after deposition at 50 K and warming to sublimation. Initially, the only IR absorptions seen were for those of the three reactants. However, on heating the sample, the H2O2 bands began to decrease at ∼100 K and reached the noise level by 130 K. Accompanying this decrease, we observed an increase in the
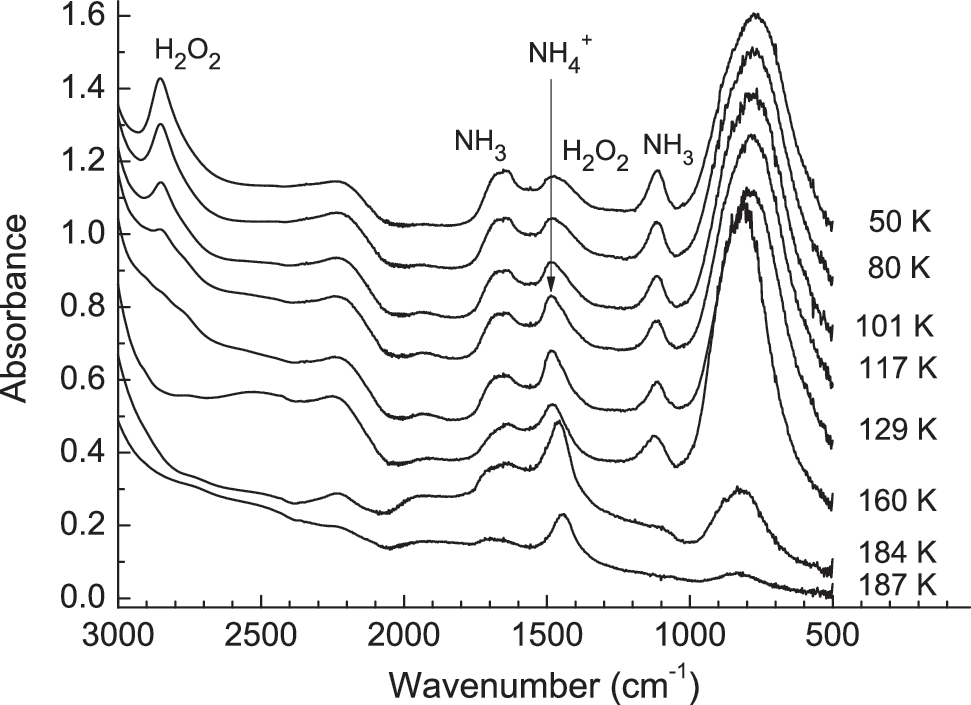
Infrared spectra of an H2O+H2O2+NH3 (10:1:1) mixture deposited at 50 K and warmed at 1 K min−1. Spectra have been offset for clarity.
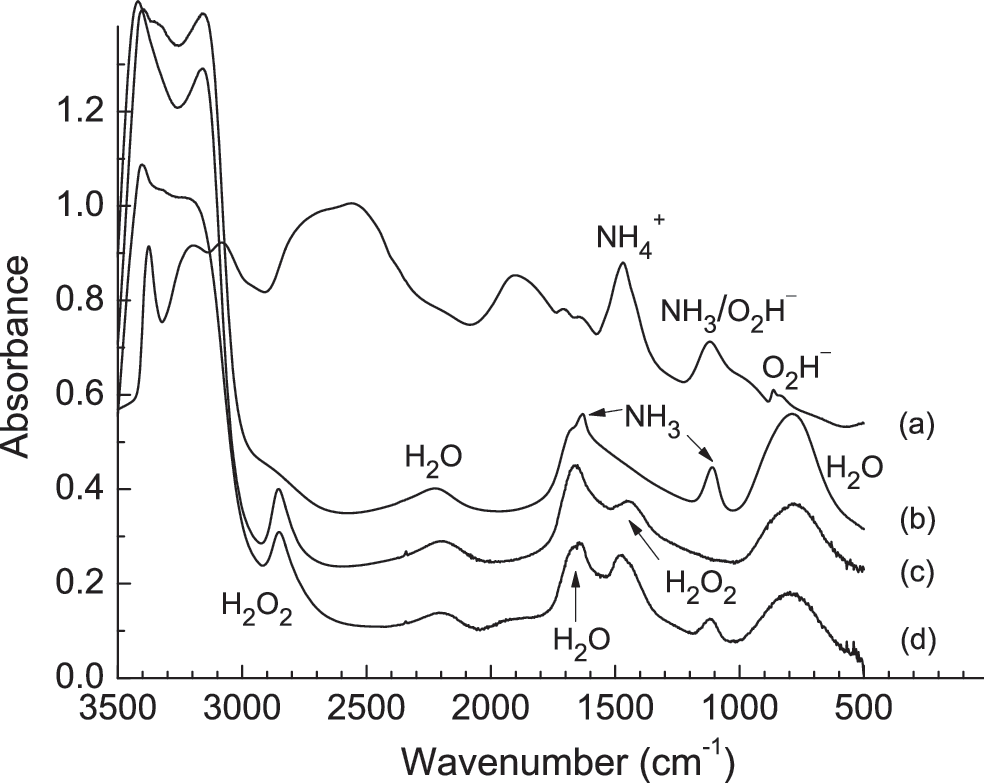
Infrared spectra of four ice mixtures each at 50 K: (
The fourth inorganic molecule we examined was CO. Ices made of H2O2+CO and H2O+H2O2+CO were warmed from 10 to sublimation at 1 K min−1 with an eye toward CO2 formation. However, Fig. 8 shows that no CO2 formation was observed above the estimated background level in our experiments, with the only major spectral changes corresponding to H2O2 crystallization near 160 K. We observed that much of the CO initially present sublimed from the ice as the temperature increased. We suspect that any residual CO present above ∼100 K was likely too small to produce a detectable amount of CO2, with the large intrinsic strength of the latter's IR features aiding their detection.
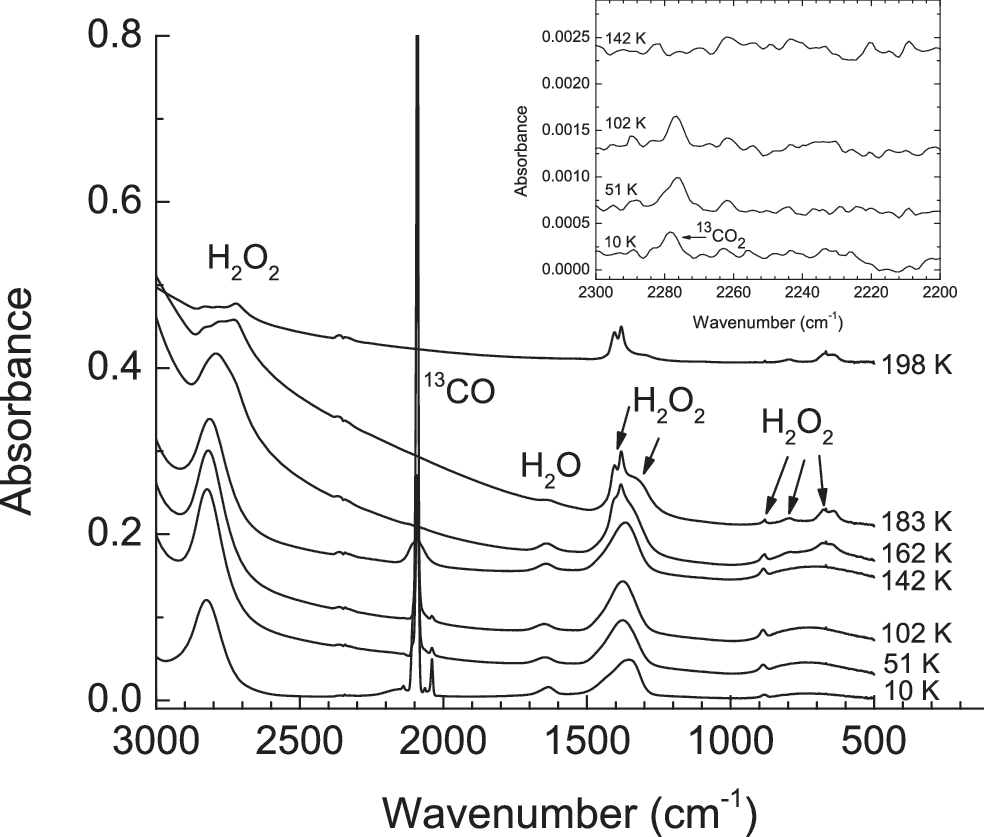
Infrared spectra of a H2O2+13CO (1:1) mixture deposited at 10 K and warmed at 1 K min−1. Spectra have been offset for clarity. The inset uses an expanded scale to illustrate the detection limits in our measurements. The 13CO2 band shown at 10 K is an impurity on the level of 0.002% in our 13CO source.
3.2. Organic compounds
Each of the four inorganic molecules we combined with H2O2 presented a particular appeal, but the present work initially was motivated by the possibility of observing thermal reactions of organics below 200 K. Table 1 lists the four organic compounds we studied. Methane (CH4) was selected for its simplicity, its presence in a variety of astronomical environments, and its potential presence in many astrobiological settings, both as a possible metabolic product and as a nutrient. Propyne (C3H4) was chosen as a typical unsaturated molecule and as one that is potentially more reactive than CH4. Methanol (CH3OH) and acetonitrile (CH3CN) were taken as simple representatives of the alcohols and nitriles, respectively. Oxidation products can be written easily for each of these organic molecules, with those of C3H4 perhaps being the most varied. Suffice it to say, however, that no oxidation products were seen within the detection limits in any of our experiments with H2O+H2O2+X ices (X=CH4, C3H4, CH3OH, and CH3CN) or in the simpler H2O2+X ices. This suggests that these four molecules are not easily destroyed by thermal reactions with H2O2 in either H2O-rich ices at 50–170 K or the simpler two-component anhydrous mixtures with frozen H2O2.
4. Discussion
4.1. H2O2 oxidation of SO2 in ices
Thermal reactions between SO2 and H2O2 have been investigated extensively within the atmospheric science community due to their importance in removing SO2 from Earth's atmosphere (e.g., Clegg and Abbatt, 2001). Based on those studies, (3) and (4) are believed to be the primary reactions between H2O2 and SO2 in the presence of H2O. Although Earth's clouds are generally much warmer than surfaces of icy satellites, the SO4
2- we have seen in our experiments is likely from similar processes. Critical tests of our analysis of the chemistry are shown in Fig. 3. As required by our interpretation, the decrease in the IR features of the limiting reagents SO2 and H2O2, near 60 K in our experiments, is accompanied by a simultaneous increase in the IR absorbance of sulfur oxyanions, primarily
The subsurface depth on Europa to which these H2O2 observations, and others in this paper, apply will depend on the thickness and other properties of that satellite's ice shell. Numerous studies have addressed the ice shell's thickness and have yielded values between about 1 and 40 km (Billings and Kattenhorn, 2005). Taking 100 K as the temperature of surface ices and 270 K for the bottom of the ice shell, as is typically done (e.g., Bray et al., 2014), and making a simple assumption of linearity, we estimate a temperature gradient of 17 K km−1 for an intermediate shell thickness of 10 km. Thus, the highest temperature reported in this paper (∼190 K) corresponds to a depth of about 5 km below Europa's surface.
4.2. H2O2 reactions with NH3
Besides SO2, the only other molecule in Table 1 that reacted with H2O2 was NH3. However, unlike SO2, where we observed that H2O-ice was needed to initiate the reaction, NH3 combined with H2O2 in the absence of H2O. The first study to show that NH3 and H2O2 react to form a stable compound was published nearly 100 years ago by Maass and Hatcher (1922). The white crystalline powder that formed in their experiments melted near 25°C, leading Knop and Giguere (1959) to propose that the bonding in the reaction product might be ionic and not the weaker bonding found in hydrates of NH3 and H2O2. Knop and Giguere's spectroscopic work, along with a previous study (Simon and Kriegsmann, 1955), suggested that the new ionic species was ammonium hydroperoxide (NH4O2H), consisting of
A comparison of Figs. 5 and 6 shows that the addition of excess H2O-ice to the H2O2+NH3 system significantly changed the IR spectra and the associated chemistry. Warming the 50 K sample produced
4.3. H2O2 reactions and astrobiology
Redox reactions with liquid- and gas-phase H2O2 readily occur at room temperature and above but not necessarily at temperatures expected in the outer Solar System. In the present work, we were particularly interested in determining how the eight molecules we examined (Table 1), which included several with biological connections, might respond to frozen H2O2 at low temperatures. The concentrations of H2O2 used in our experiments are much higher than what has been detected (Carlson et al., 1999a) and are expected in extraterrestrial environments, so negative results in our work (i.e., no reaction seen) suggest no reaction under less-harsh, more-realistic conditions.
For our experiments with solid H2O2, we selected as reactants the four organic and four inorganic compounds of Table 1. Of the inorganics, no thermally induced reactions were observed between H2O2 and either CO or H2S. Reaction of the former could have made CO2, while the latter could have produced SO2, as given in (5) and (6). No such thermal conversion was seen. Extraterrestrial thermal oxidation of H2S at low temperatures appears to require a more complex set of conditions than those we employed, perhaps even biological conditions akin to those relevant to sulfur-oxidizing bacteria. In contrast to the reluctance of solid H2S to react with H2O2, here and elsewhere we have documented the relative ease of the SO2→
Ammonia (NH3) was the fourth inorganic compound we examined. Our initial thoughts were of a possible thermal nitrification of NH3 in H2O2-containing ices to make the oxyanions
Turning to the organic compounds we examined, solid methane (CH4) and solid methanol (CH3OH) conceivably could have undergone multiple thermal oxidations in the presence of H2O2, such as through the sequence
to make CO2, but we observed no CO2 on warming ice mixtures containing either CH4 and H2O2 or CH3OH and H2O2. Each product molecule in (9) has at least one fairly strong IR spectral feature that would easily have been detected, but none were seen in the 10–150 K region examined. At room temperature and above, propyne (C3H4) can undergo oxidative cleavage to make CO2 and CH3COOH (acetic acid), but no evidence for either was found in our low-temperature experiments, and both CO2 and CH3COOH would have been relatively easy to detect with our IR methods. Finally, from earlier work we already knew that low-temperature radiation-induced oxidation of acetonitrile (CH3CN) first makes OCN- and then CO2 (Hudson and Moore, 2004), but neither of these products was seen in our warmed, unirradiated CH3CN-containing ices.
This leads to perhaps the most significant astrobiological implication of our experiments with organics, namely, the stability of these four typical, relatively simple organic molecules in the presence of solid H2O2. One can envision a situation in which, for example, vertical transport of material on Europa brings surface oxidants, such as H2O2, down to subsurface regions and into contact with organics trapped in ice (Greenberg, 2010). Our experiments showed that such encounters do not necessarily destroy those same organic compounds. In the particular case of CH4, whether it is primordial, endogenous, or exogenous, it would remain available for methanotrophs.
Finally, this study emphasizes that understanding and predicting Europa's low-temperature solid-phase chemistry, and its contribution to astrobiology, will require substantial laboratory efforts. For example, with standard tables of E θ values (reduction potentials) for half-cell reactions, one can predict that all of the eight compounds we combined with H2O2 should be oxidized by that molecule, which does not agree with our observations. However, such reference data are nearly always for a standard set of conditions near 25°C, 1 bar, and 1 molar concentration, conditions markedly different from those of Europa ices. Furthermore, extrapolating from calculations on systems with liquid phases to those, like ours, that are entirely formed from solids is not straightforward. Since the influence and interplay of both thermodynamic and kinetic contributions to ice chemistry cannot yet be predicted a priori, we anticipate a significant role for experimental work for the foreseeable future.
5. Summary and Conclusions
Laboratory results have been presented here on the reactivity of H2O2 with selected organic and inorganic molecules in ice mixtures at temperatures of the outer Solar System. Using IR spectroscopy, we observed that two of the inorganic compounds studied, SO2 and NH3, were consumed in H2O2-containing ices by thermally induced reactions, yielding
In contrast, at the timescales of our experiments we found that none of the four organic compounds examined underwent low-temperature thermally induced reactions with solid H2O2 in either the presence or absence of H2O-ice. We believe that these observations of a nonreaction speak positively for future attempts to find organics on Europa. On a frozen world where H2O2 could move downward to meet trapped subsurface organics, it could descend without modification by them, leaving those organics unaltered, so we can anticipate their discovery through future explorations. Put another way, the formation of surface radiolytically generated H2O2 on Europa, and that molecule's downward movement with H2O-ice, does not necessarily mean that all organics encountered in icy subsurface regions will be destroyed by H2O2 oxidation.
Footnotes
Acknowledgments
The support of NASA's Planetary Geology and Geophysics and Outer Planets Research programs is gratefully acknowledged. The authors particularly acknowledge support from the NASA Astrobiology Institute through a grant to the Goddard Center for Astrobiology. Perry Gerakines is thanked for assistance in day-to-day operations of the equipment in our laboratory.
Author Disclosure Statement
No competing financial interests exist.