
Abstract
The major organic component in carbonaceous chondrites is a highly aromatic macromolecular material. Aromatic organic matter and phyllosilicates are colocated in these meteorites, and it is possible that the physical association represents a synthetic chemical relationship. To explore the potential reactions that could take place to produce the aromatic macromolecular material, we heated various simple aromatic units in the presence of montmorillonite with different exchanged cations. The majority of cation-exchanged montmorillonites tested, sodium-, aluminum-, iron-, nickel-, and cobalt-rich montmorillonites, do not produce polymerization products. By contrast, Fe3+ cation-exchanged montmorillonite readily facilitates addition reactions between aromatic hydrocarbons. A feasible mechanism for the process is oxidative coupling, which involves a corresponding reduction of the Fe3+ cation to its Fe2+ counterpart. A similar reduction process for the other metal cations does not take place, highlighting the importance of iron. This simple process is one feasible mechanism for the construction of aromatic macromolecules such as those found in carbonaceous chondrites. The search for a relationship between Fe3+-rich phyllosilicates and aromatic organic structures (particularly dimers, trimers, and more polymerized forms) in carbonaceous chondrites would represent an effective test for constraining the role of clay catalysis in the early Solar System. Key Words: Oxidative coupling—Chondrite—IOM—Montmorillonite. Astrobiology 15, 787–792.
1. Introduction
P
There have been numerous environments and processes proposed for the formation of organic macromolecular material (Kerridge, 1999). In the stellar outflows of carbon stars, acetylene pyrolysis and condensation reactions can take place. In interstellar environments, processes such as ion-molecule reactions and radiation chemistry on grain mantles (e.g., polymerization) can occur. In the nebula equilibration reactions, surface catalysis (e.g., FTT), radiation reactions, and photochemistry may all produce organic materials. Finally, on asteroidal meteorite parent bodies, liquid phase reactions, surface catalysis, and thermal processing may modify or produce organic structures. With the number of possible environments and mechanisms available, it is possible that the organic macromolecular material is not formed from a single source or by one process but has a mixed heritage (Sephton, 2014).
To reveal potential construction mechanisms of the macromolecular material is to constrain the environmental conditions through which its organic units have passed and the relative timing of appearance and interaction of meteorite constituents. One reactant or catalyst that may have played a role in macromolecular material construction is phyllosilicates. The most organic-rich meteorites are also the most phyllosilicate-rich. There is some spatial correlation between organic matter and phyllosilicates that has been visualized using osmium staining techniques (Pearson et al., 2002). Carbonaceous chondrites have undergone varying amounts of aqueous alteration, which results in the formation of secondary phyllosilicate minerals (e.g., Tomeoka And Buseck, 1985; Scott and Krot, 2014). Extensively altered CIs contain saponite (a smectite), whereas the less altered CMs contain rare or no saponite (e.g., Tomeoka and Buseck, 1988). Other secondary hydrated minerals may also be produced which may be able to catalyze some reactions.
Phyllosilicates are well-known catalysts for organic synthesis (Zhou, 2011) and have been implicated in early Solar System organic reactions (Pearson et al., 2002). An unexplored avenue for the possible relationships between the macromolecular material and phyllosilicates is the importance of mineral chemistry, particularly cation content. Establishing a specific chemical control for phyllosilicate mineral–organic matter reactions will provide an additional means to recognize their products in real samples.
Here, we chemically modify montmorillonite to produce a range of cation types. The modified phyllosilicate samples were heated with simple aromatic units before recovering the polymerization products for analysis. Our findings reveal that only specific phyllosilicate chemistries produce organic polymerization products and are, therefore, capable of aiding macromolecular material construction. Furthermore, the organic polymerization mechanisms generate products that are distinct enough to indicate the reactions from which they formed. Although the experiments presented here do not attempt to recreate the conditions found on parent bodies, they do provide an insight and understanding of certain processes that may occur on parent bodies.
2. Materials and Methods
For our studies, montmorillonite was chosen because of the ease at which it exchanges cations, allowing the chemistry of the mineral to be controlled by laboratory procedures. A natural sodium-rich montmorillonite (WSy-2) was supplied by the Clay Minerals Society and was used unmodified. An aluminum-rich K10-montmorillonite (Fluka) was also used with no modification. The interlayers in montmorillonite contain cations that can be exchanged, so additional cation-rich montmorillonites were generated by using ion-exchange reactions. The natural WSy-2 montmorillonite was cation exchanged by taking 5 g of the natural montmorillonite and stirring for 24 h in a 1 M solution (75 mL) of either FeCl3, NiCl2, or CoCl2, and the solution was replenished and stirred for a further 24 h period. The phyllosilicate was subsequently rinsed, stirred, and centrifuged (∼30 times) until no free chloride ions were detected. Hereafter, the sodium-, aluminum-, iron-, nickel-, and cobalt-rich montmorillonites will be referred to as Na+-mmt, Al3+-mmt, Fe3+-mmt, Ni2+-mmt, and Co2+-mmt, respectively. Following the introduction of cations to WSy-2 montmorillonite, some striking colors were produced. Fe3+-mmt is slightly reddish in color, Ni2+-mmt has a green tint, and Co2+-mmt has a pink tint.
A series of aromatic organic compounds were used to explore the phyllosilicate-assisted construction of oligomeric macromolecular material units. These aromatic compounds were used as single compounds (naphthalene, 2-methylnaphthalene, 2,6-dimethylnaphthalene, 2,7-dimethylnaphthalene, biphenyl, phenanthrene, anthracene, pyrene, dibenzothiophene, 2-naphthol) and a simple mixture of naphthalene and phenanthrene. All the hydrocarbons chosen are found in the soluble organic fraction of carbonaceous chondrites (e.g., Sephton, 2014).
Powdered organic standards (4 mg) with phyllosilicate (30 mg) were loaded into clean borosilicate tubes (100 mm × 4 mm i.d.), cooled in liquid nitrogen and flame sealed under vacuum (∼5 Pa), and then heated at 120°C for 24 h in a gas chromatograph oven. After heating, the tubes were cracked open and the contents and tube interior solvent extracted with 4 mL dichloromethane (DCM). An aliquot of the extract (ca. 200 μL) was taken and made up to 1 mL prior to analysis by gas chromatography–flame ionization detection (GC-FID) and gas chromatography–mass spectrometry (GC-MS).
2.1. Gas chromatography–flame ionization detection
Analysis was performed on a PerkinElmer AutoSystem XL system. Injection (1 μL) was on column using the programmed split/splitless injector. Separation was performed using a DB-5HT column (J&W; 15 m × 0.25 mm × 0.1 μm). The gas chromatograph oven was programmed 40°C for 2 min and then ramped at 10°C min−1 to 350°C, where it was held for 10 min.
2.2. Gas chromatography–mass spectrometry
Analysis was performed on an Agilent Technologies 7890-5975 gas chromatograph–mass spectrometer. Injection was splitless, and separation was performed using a DB-5MSui column (J&W; 30 m × 0.25 mm × 0.25 μm). The gas chromatograph oven was programmed 40°C for 2 min and then ramped at 5°C min−1 to 310°C, where it was held for 6 min. Compound identification was achieved using mass spectra and elution orders.
3. Results
3.1. The influence of phyllosilicate cation contents
Of all the different cation-containing montmorillonites, only Fe3+-mmt catalyzes polymerization of naphthalene (Table 1). Following organic reaction, the Fe3+-mmt changes from a slightly reddish color to dark reddish gray. The data reveal that, if the polymerization of aromatic units to form a meteorite macromolecule proceeds by phyllosilicate-catalyzed reactions, then it is only the Fe3+-containing phyllosilicates that take part. A correlation between iron-containing phyllosilicates and characteristic aromatic polymerization products would be a useful line of investigation to test the phyllosilicate catalysis hypothesis.
The presence of the positive sign indicates the appearance of polymerization products following heating at 120°C for 24 h.
Na2+-mmt is the natural WSy-2 from the Clay Minerals Society, Al3+-mmt is K10 from Fluka, the remainder were generated by ion-exchange reactions.
3.2. The influence of organic structures
In this experiment with Fe3+-exchanged natural montmorillonite, there is a large conversion (ca. 40%) of 2-naphthol to its dimer 1-(2-hydroxy-1-naphthyl)naphthalen-2-ol (see Fig. 1). Oxidative coupling of 2-naphthol has previously been reported for iron-exchanged K10-montmorillonite (Bhor et al., 2008). However, in the Bhor et al. (2008) study, samples were sonicated unheated, and an oxidant was also used to reform Fe3+. Our results are similar to the previously published work but with a lower conversion yield, which can be explained by Bhor et al. (2008) using a modified montmorillonite and an oxidant to reform Fe3+. It is not unexpected that a natural montmorillonite as used in our study displays reduced reactivity when compared to a K10-montmorillonite.

GC-MS total ion chromatograms of the products of reactions between Fe3+-rich montmorillonite and some simple aromatic compounds heated at 120°C for 24 h. The unreacted starting material elutes at low retention times, while the polymerization products appear at higher retention times. (
The main products produced from naphthalene are its dimers and trimers, binaphthalenes (1,1′- > 1,2′- > 2,2′-), and minor amounts of ternaphthalenes (Fig. 1). As mentioned previously, no polymerization products were observed from the montmorillonites containing no Fe3+ cations. Following this observation, all subsequent experiments used only the Fe3+-mmt.
Biphenyl experiments produced minor amounts of its dimers, the two possible quaterphenyls that can be seen in Fig. 1. Phenanthrene experiments produced two major peaks and a minor peak, which all appear to be its dimers, the biphenanthrenes. For the anthracene experiments, its dimers the bianthracenes were present in the products. An additional notable product was anthraquinone (4a,9a-dihydroanthracene-9,10-dione), which is an oxidation product of anthracene. The presence of the anthracene oxidation product reflects the ability of heating in the presence of Fe3+ phyllosilicates to cause the oxidation of organic compounds; anthracene is particularly sensitive to oxidation reactions at the 9 and 10 positions. Dibenzothiophene produced small amounts of a dimer that were just above the detection limits of the instrument (not shown).
Gas chromatography–mass spectrometry conditions were not suitable for the detection of any dimers for pyrene; however, peaks probably reflecting the presence of pyrene dimers were detected by GC-FID when using a high-temperature column perfectly suited to the analysis of relatively high-molecular-weight hydrocarbons (not shown).
2-Methylnaphthalene experiments produced a series of dimethylbinaphthalenes and minor amounts of trimethylternaphthalenes (Fig. 2). Hydrogenation of the produced dimethylbinaphthalenes also occurred to generate units with one ring saturated and a small number of products with both rings saturated. No hydrogenation of the 2-methylnaphthalene starting material occurred.
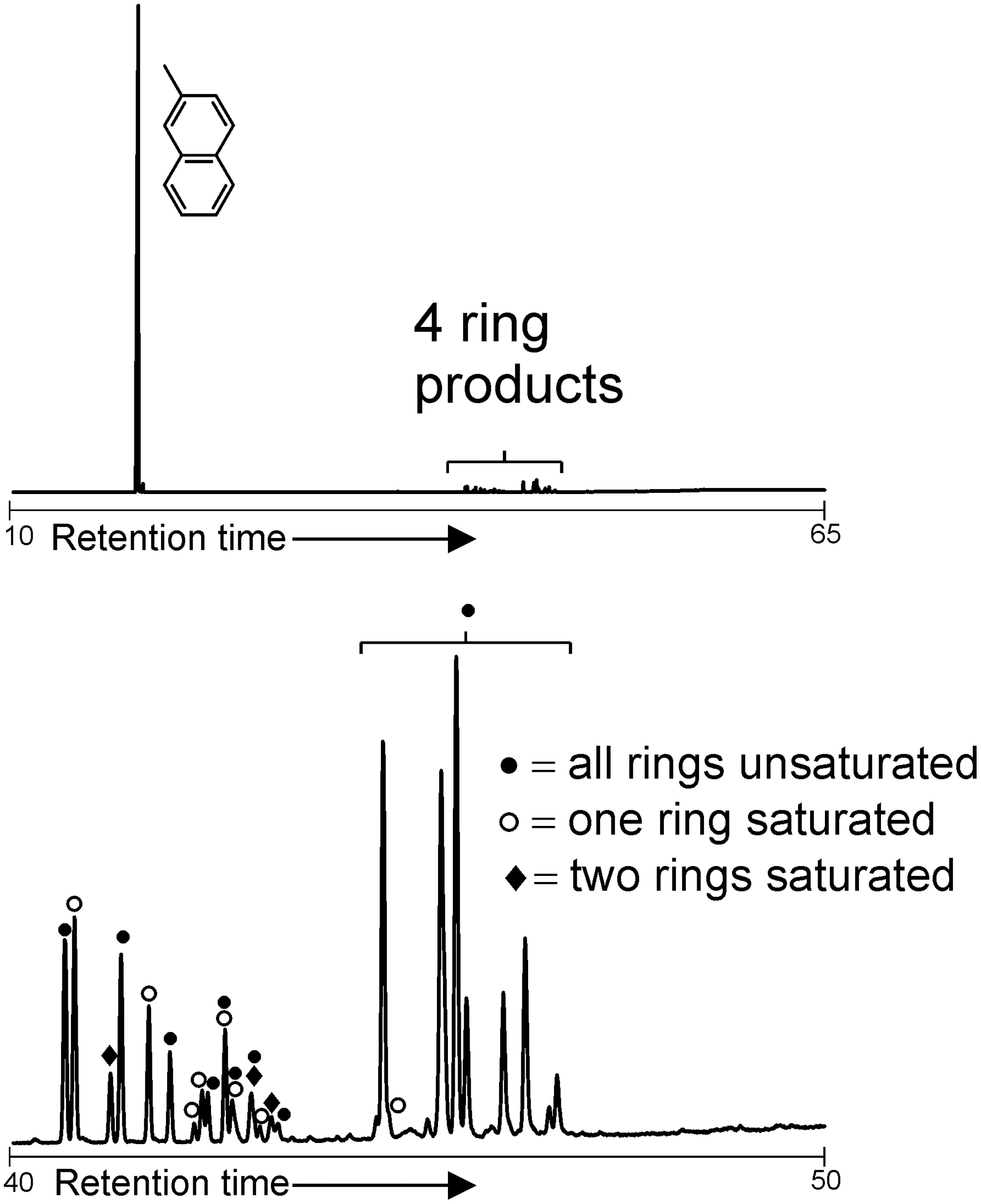
GC-MS total ion chromatograms of the products of reactions between Fe3+-rich montmorillonite and 2-methyl naphthalene heated at 120°C for 24 h. The unreacted starting material elutes at low retention times, while the polymerization products appear at higher retention times. Detection of some highest homologues may be limited by the analytical window of the instrumentation.
Both 2,6-dimethylnaphthalene and 2,7-dimethylnaphthalene produced a series of tetramethylbinaphthalenes and hexamethylternaphthalenes. No hydrogenated products were observed. As displayed in Fig. 3, the position of the methyl substituents on the dimethylnaphthalene starting materials had a significant effect on their polymerization products; the distributions of products from the two starting products were very different.
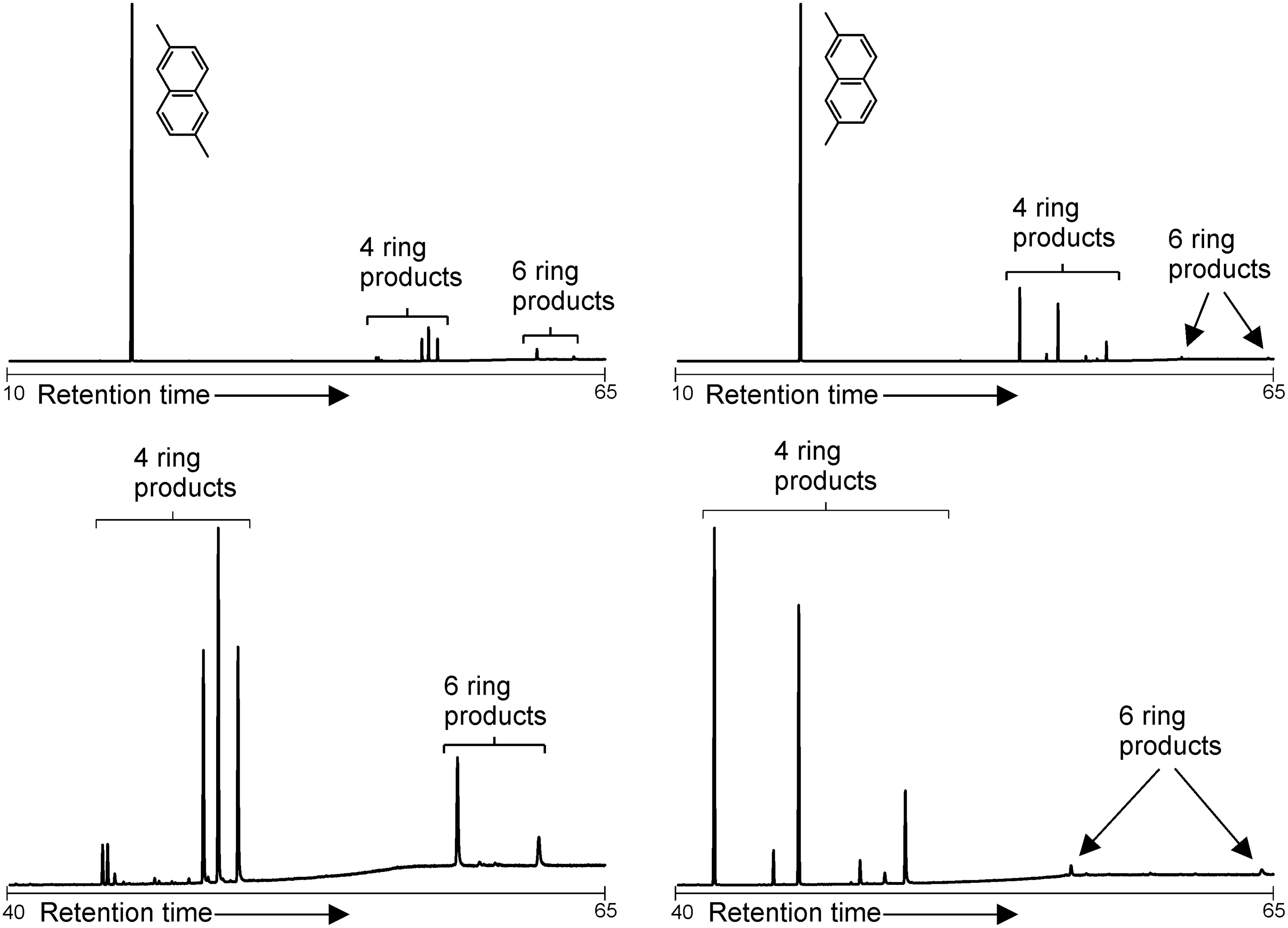
GC-MS total ion chromatograms of the products of reactions between Fe3+-rich montmorillonite and dimethylnaphthalenes. The unreacted starting material elutes at low retention times, while the polymerization products appear at higher retention times. Detection of some highest homologues may be limited by the analytical window of the instrumentation.
The mixture of naphthalene and phenanthrene with Fe3+-mmt produced binaphthyls and biphenanthrenes, which could be observed alongside the individual starting compounds (Fig. 4). In addition, naphthylphenanthrenes were also produced.
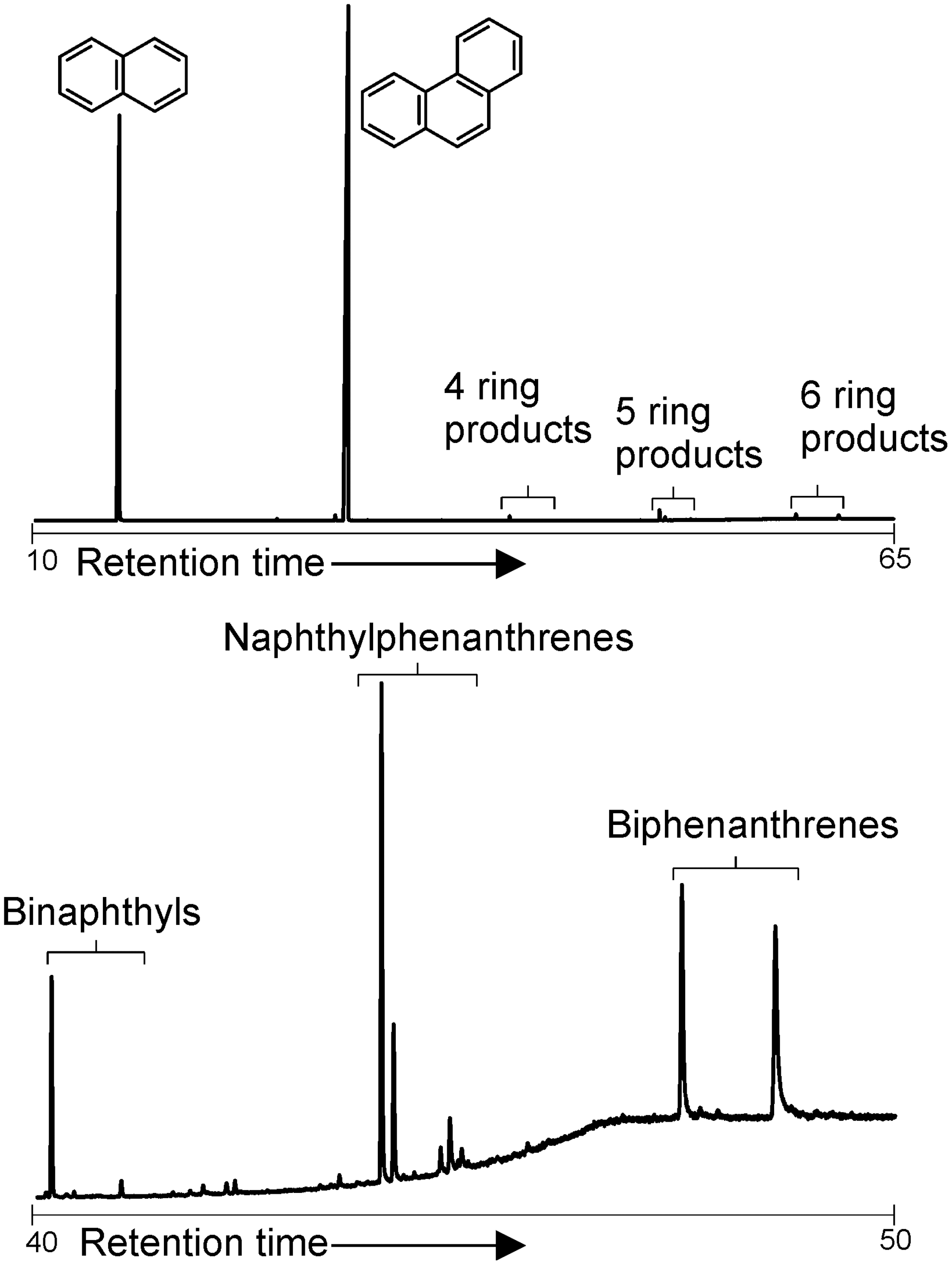
GC-MS total ion chromatograms of the products of reactions between Fe3+-rich montmorillonite, naphthalene, and phenanthrene. The unreacted starting material elutes at low retention times, while the polymerization products appear at higher retention times. Detection of some highest homologues may be limited by the analytical window of the instrumentation.
4. Discussion
These data demonstrate for the first time that cation-exchanged natural phyllosilicates are able to promote simple polymerization reactions under moderate conditions. The results suggest a feasible mechanism, which was previously unstudied, to form aromatic macromolecules under certain natural conditions relevant to the chemical evolution of carbonaceous chondrite meteorite parent bodies. Using gas chromatography–based instrumentation, we were limited by quite a narrow analytical window; therefore, it is feasible that larger molecules were also being formed and that the ability of the recognized mechanism to produce aromatic polymers is even more efficient than documented.
The envisaged mechanism by which dimers and polymerization products can form is when a free radical is produced by hydrogen-subtraction, which can then react with another molecule (see Grzybowski et al., 2013). The dimer can undergo further hydrogen-subtraction to generate higher-molecular-weight products. Different positions on the starting molecule have different reactivities that will be reflected in the products formed. It is important to note that this distinct starting material control on reaction products (e.g., position of methyl substitution) may provide the potential to recognize the isomeric form of the starting materials from the nature of the polymerized products. The overall mechanism is called “oxidative coupling” because both hydrogen and electrons are removed from the starting molecule, and the Fe3+ facilitates the reaction by being the electron accepter and is thus reduced to Fe2+.
A number of partially hydrogenated products were produced in the 2-methylnaphthalene experiment. Hydrogen for the hydrogenation reactions is a by-product of the polymerization reactions. If oxidative coupling was an important process in the early Solar System, it is reasonable to assume that hydrogenated products should be found in the meteoritic organic macromolecule. Hydropyrolysis of the Murchison meteorite does liberate a significant amount of partially hydrogenated aromatic products (unpublished data), which is consistent with this hypothesis.
Meteoritic macromolecules are extremely complex in their nature, which presents a number of analytical challenges in studying this material (Anders and Kerridge, 1988). If polymerization reactions between simple molecules occur, it is reasonable to assume that coupling reactions could occur between the organic macromolecules and free components. From isotopic studies, there appears to be a genetic link between free aromatic units in meteorites and their macromolecular counterparts (Sephton et al., 2000).
The phyllosilicate content of carbonaceous chondrites changes with type, and serpentine and clay minerals vary in chemistry and relative abundance (Brearley, 2006). It follows therefore that a testable hypothesis for the role of Fe3+-rich phyllosilicates in the polymerization of aromatic units to form macromolecular material would be a maximum relative abundance of dimers and trimers within those meteorites that have or had Fe3+-rich phyllosilicates. The oxidative polymerization reaction could occur prior, during, or after parent body accretion, and the correlation of dimers and trimers with mineralogy could also indicate the timing of any Fe3+-assisted polymerization reactions. During aqueous alteration, although phyllosilicates become more enriched in Mg2+, there is also an increase in the overall Fe3+/Fe2+ ratio (Browning et al., 1996; Beck et al., 2012). A future combined organic geochemical and mineralogical investigation of carbonaceous chondrites would be an effective way to convert our plausible laboratory-investigated mechanism for macromolecule formation to one that can be recognized as having occurred on the asteroidal meteorite parent bodies in the early Solar System.
5. Conclusions
Cation contents of phyllosilicates exert a strong control on their ability to promote organic chemical reactions. A study of montmorillonites reveals that only the Fe3+ cation version reacts with aromatic hydrocarbon starting materials to produce polymerization products. The reduction of Fe3+ to Fe2+ is accompanied by the oxidative coupling of aromatic units. Dimers and trimers are characteristic products of the phyllosilicate-catalyzed process. Concentrations and types of organic matter and phyllosilicates vary between carbonaceous chondrites. Searching across meteorite types for a correlation between potential oxidative coupling products such as dimers and trimers and Fe3+-rich phyllosilicates would test the hypothesis that mineral-catalyzed reactions aided the construction of macromolecular materials in the early Solar System.
Footnotes
Acknowledgments
This work was supported by The Leverhulme Trust (Grant # RPG-213). We thank three anonymous reviewers for constructive reviews that improved the manuscript.
Author Disclosure Statement
No competing financial interests exist.