
Abstract
Following the suggestion that nucleoside analogues having their nucleobases joined to ribose via a carbon-carbon bond might easily arise prebiotically, the glycosylation of uracil carrying electron-donating substituents (Me, OH, OCH3, NH2) at its 5 or 6 positions was investigated. Of these, only 6-aminouracil gave glycosylated products in greater than 50% yield under simulated prebiotic conditions. The reaction provided four products, three of which were purified by preparative HPLC. The structure of the isolated compounds was determined by high-resolution mass spectrometry and NMR spectroscopy. The glycosylation products were, as expected, C-nucleosides, with the sugar having either a pyranose or a furanose structure, with the ratio depending on the precise conditions, implying reversible addition. Interestingly, the 6-aminouracil riboside displays two hydrogen bonding patterns, the “acceptor-donor-acceptor” pattern of uridine itself and (upon 180° rotation) the “acceptor-donor-donor” hydrogen bonding pattern. The second, in an artificially expanded genetic information system, is trivially called “V” and pairs with a purine analogue that presents the complementary “donor-acceptor-acceptor” hydrogen bonding pattern, trivially called “J.” Key Words: Prebiotic chemistry—Uracil—Glycosylation—C-glycoside—Artificially expanded genetic information systems. Astrobiology 15, 301–306.
1. Introduction
A
This structural feature of standard nucleobases becomes relevant when we consider how the nucleobase repertoire of nucleic acids might be expanded, for example, to increase the folding, binding, and catalytic potential of RNA. This is one approach needed to manage the relatively poor functional powers of RNA, functional powers that would have been needed in a hypothetical RNA world, where RNA was presumed to be the only genetically encoded biocatalyst. Thus, while both components of the extra nucleobase pair of Hirao (Kimoto et al., 2013) are N-glycosides, and one of Romesberg's pair is (Malyshev et al., 2014), additional pairs that retain Watson-Crick geometry often require that the nucleobase analogue be joined to the ribose by a carbon-carbon bond (C-glycoside) to achieve the desired hydrogen bonding pattern. For example, while the nonstandard isocytosine can present as an N-glycoside the unnatural acceptor-acceptor-donor hydrogen bonding pattern to its complement, the donor-acceptor-donor, donor-donor-acceptor, and acceptor-donor-donor hydrogen bonding patterns on a pyrimidine analogue require a C-glycoside (Fig. 1) (Benner, 2004).
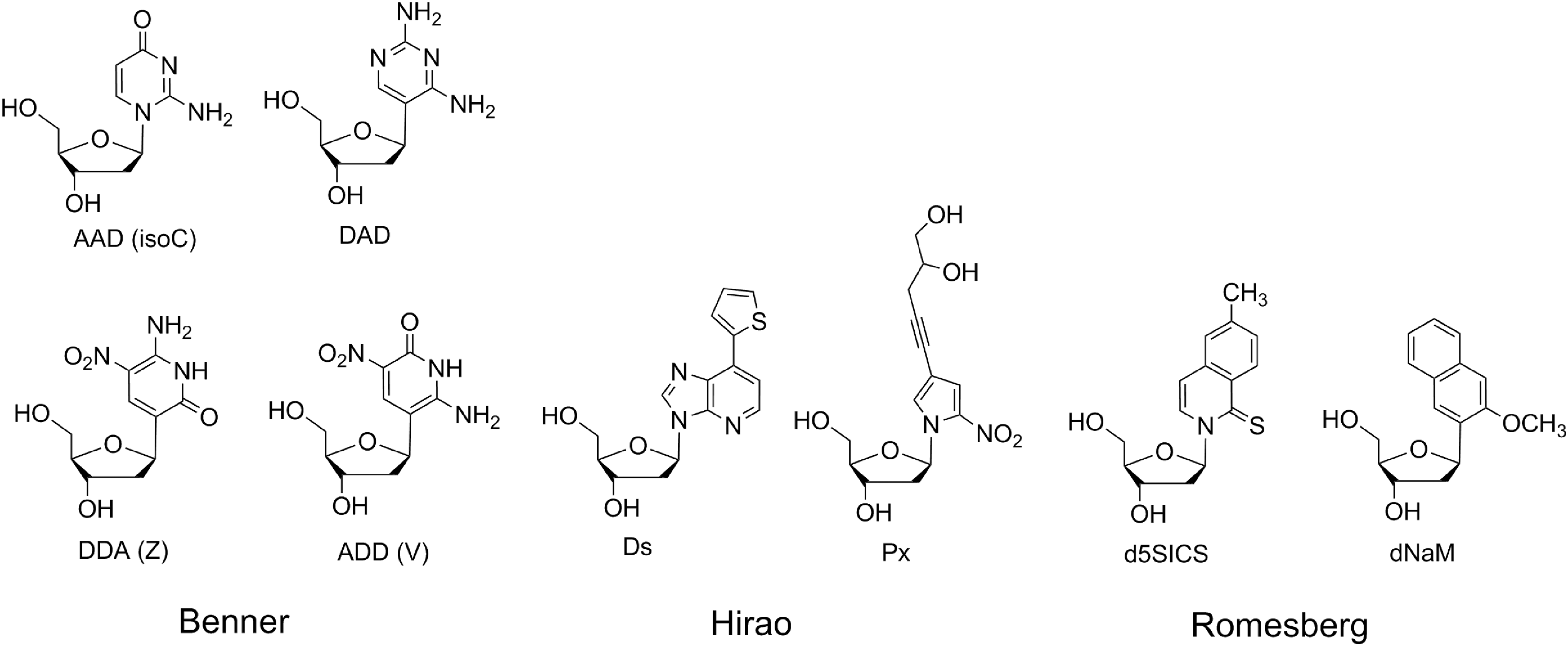
Nucleosides with extra nucleobases from the Benner, Hirao, and Romesberg laboratories. D stands for hydrogen bond donor, and A stands for hydrogen bond acceptor in the first of these expanded genetic systems.
From a prebiotic perspective, one might think that the carbon-nitrogen bond of N-glycosides might be easier to prepare than C-glycosides under non-enzymatic conditions; replacement of the –OH heteroatomic substituent in the cyclic ribose by a nitrogen heteroatom on a nucleobase appears to be a natural reaction. This need not be the case. For example, 40 years ago, Orgel and his coworkers tried to form the C-N bond in the standard N-glycosidic nucleosides by directly drying and heating ribose with divalent metal ions in the presence of each of the four standard nucleobases: adenine, cytosine, guanine, and uracil (Fuller et al., 1972a, 1972b; Maurel and Convert, 1990). Only adenosine was formed in any detectable amounts, with the noncanonical hypoxanthosine also being observed. This led Orgel and his coworkers to suggest that pyrimidine nucleosides (in particular) might have been prebiotically assembled by constructing the nucleobase around an appendage on a preformed sugar scaffold, much as in the biosynthetic route (Sanchez and Orgel, 1970; Orgel, 2004). This suggestion was recently amplified with a different carbohydrate starting material by Powner et al., in an elegant, although multistep, reaction sequence (Powner et al., 2009, 2011).
However, carbon 1 of ribose in its cyclic form is in equilibrium with an aldehyde, and an aldehyde can be an electrophile in an aldol addition with a sufficiently reactive carbon nucleophile. Miller and coworkers, Hud, and others demonstrated that nucleosides are formed in quite satisfactory yields by reacting electron-rich heterocycles (urazole, 2-pyrimidinone, and 2,4,6-triaminopyrimidine) directly with ribose in an aldol reaction (Kolb et al., 1994; Bean et al., 2007; Chen et al., 2014). This suggests that certain C-glycosides might have been more abundant on prebiotic Earth than certain N-glycosides. As the C-glycosidic bond is thermodynamically more stable with respect to hydrolysis than the N-glycosidic bond (carbon-carbon bonds are typically stronger than carbon-nitrogen bonds), C-glycosides might survive longer in water as well.
Given our longstanding interest in the prebiotic synthesis of RNA (Benner and Ellington, 1990; Kim et al., 2011), and some recent results with laboratory in vitro evolution that suggest that oligonucleotide libraries built from an artificially expanded genetic information system are richer reservoirs of functionality than libraries built from just four standard nucleotides (Sefah et al., 2014), we decided to examine one series of substituted uracils to learn how versatile this condensation reaction is and how the overall electron “richness” of the heterocycle influences the outcome of the reaction. We report these results here.
2. Materials and Methods
2.1. Materials
Reagent-grade 5-aminouracil and 6-aminouracil were obtained from TCI America. 5-Methoxyuracil was obtained from Carbosynth; 6-methoxyuracil was obtained from Aces Pharma. All other chemicals were obtained from Sigma-Aldrich and used as received.
2.2. Methods
Glycosylation experiments were conducted in a glass vial (5 mL) containing the uracil derivatives (5 mM), ribose (50 mM), magnesium chloride (12.5 mM), and magnesium sulfate (50 mM) in water (0.3 mL). The vials were first heated at 90°C with lids open until all the water evaporated. The residues were further heated for 1 h and then were resuspended in water and analyzed by reversed-phase HPLC.
The same reactions on a preparative scale were conducted in a beaker (1 L) containing 6-aminouracil (250 mg) and ribose (1500 mg) in water (100 mL). The beaker was heated at 90°C until all the water was evaporated. The residue was then further heated for 14 h, resuspended in water, and resolved into its major components by preparative HPLC.
The stability of the product species under acidic conditions was determined by incubation in a microcentrifuge tube containing each purified fraction (10 μL) and aqueous HCl (0.1 mL, pH 2). The tube was heated at 55°C for 2 h, diluted with aqueous triethylammonium acetate (25 mM, 1 mL), and analyzed by HPLC.
High-performance liquid chromatography analysis was done with a C-18 reversed-phase narrow bore column (3 mm i.d., 150 mm length, 5 μm; SunFire; Waters) on a Waters 2695 separation module equipped with 996 photodiode array detector. The column was eluted with a gradient of (A) aqueous 25 mM triethylammonium acetate and (B) 100% acetonitrile. The elution program created a linear gradient started from 100% (by volume) A to 85% A at 10 min with flow rate of 0.5 mL/min. Peak detection and integration were conducted with the signal at 260 nm; full UV spectra (230∼400 nm) were also obtained.
Preparative HPLC purification was done by using a C-18 reversed-phase column (30 mm i.d., 250 mm length, 5 μm; SunFire; Waters) on a Waters Delta 600 module. The column was eluted with a gradient of (A) aqueous 25 mM triethylammonium acetate and (B) 100% acetonitrile. The elution program created a linear gradient started from 100% A to 3.0 min, 90% A at 28 min, 70% A at 30 min with flow rate of 15 mL/min. Peak detection was conducted with the 260 nm absorbance.
Proton and carbon NMR experiments were conducted with Varian Mercury 300 NMR at room temperature with D2O (0.5 mL) as the solvent. Mass spectra were obtained by the Agilent 6220 ESI-TOF with electrospray ionization.
3. Results
3.1. Glycosylation of 5- and 6-substituted uracils
Uracil derivatives with electron donating groups (CH3, OH, OCH3, NH2) at 5- and 6-position were subjected to glycosylation reaction under Orgel's 1972 reaction conditions. An incubation of substituted uracils and ribose at 90°C for 1 or 14 h in the presence of magnesium chloride and magnesium sulfate did not yield any glycosylated products with the exception of 6-aminouracil (data not shown). The 5-Me and 6-Me uracils were recovered unchanged after the incubation; the methoxy uracils decomposed to give unidentified products. However, incubation of 6-aminouracil and ribose at 90°C drying conditions with magnesium chloride and magnesium sulfate provided four major compounds (with chromatographic retention time of 2.7, 3.5, 4.3, and 6.1 min), as seen by reversed-phase HPLC. These new compounds had the same UV absorption profile as 6-aminouracil, with absorption maxima at 262 nm. This implies that the 6-aminouracil heterocycle did not decompose under these conditions but was incorporated intact into the newly formed compounds.
Similar results were seen with 6-aminouracil without magnesium ions. Only the ratio of products varied when the incubation was done with magnesium sulfate alone (Fig. 2C), or magnesium chloride alone (Fig. 2D), or without any magnesium salt (Fig. 2E). In general, the major products eluted at 2.7 and 6.1 min.
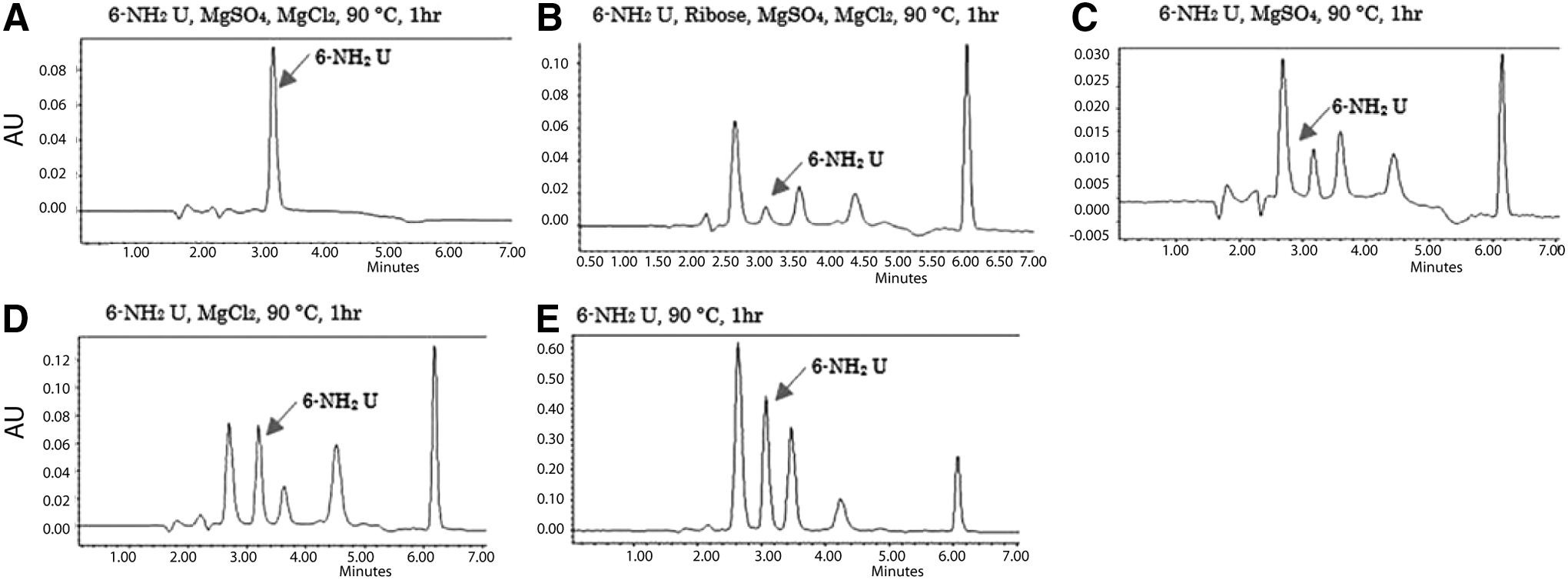
HPLC analysis of the products from the glycosylation reaction between ribose and 6-aminouracil.
3.2. Structure determination of glycosylated products
To determine the structure of the glycosylation products, the reaction was carried out in preparative scale in the absence of magnesium. 6-Aminouracil (200 mg) and ribose (1.5 g) were dissolved in water and placed in an oven at 90°C until the water had evaporated. Further heating for 12 h yielded three major glycosylated products (with chromatographic retention times of 2.7, 3.5, and 6.1 min in the analytical HPLC analysis), in addition to unreacted 6-aminouracil. The other glycosylation product (4.3 min in the analytical HPLC analysis) was produced in very small amounts and was not recovered from the preparative HPLC. The three major products were isolated by preparative HPLC, and their chemical structures were determined by high-resolution mass spectrometry and NMR spectroscopy.
The molecular weights of all products, as determined by high-resolution electrospray ionization time of flight, was MW=259 (detected as sodiated ions, MW=282). This corresponds to a condensation product of 6-aminouracil and ribose. However, mass spectrometry was unable to distinguish between the various possible isomers of the product of 6-aminouracil and ribose (Fig. 3).

Possible products arising from the glycosylation reaction between ribose and 6-aminouracil.
However, it was possible to assign the products by 1H NMR. The 1H NMR of all three showed an NH2 peak (exchangeable 2H peak) at ∼6 ppm. However, no aromatic C-H resonance was seen. This indicated glycosylation had not occurred on the exocyclic amino group of 6-aminouracil, implying that the glycosylation products are C-nucleosides.
Further structural information was obtained from 13C NMR. The C-glycosylation nucleosides can have 4 stereoisomers—2 furanoses and 2 pyranoses. The furanose has a 5-membered tetrahydrofuran ring and one exocyclic hydroxymethyl group, with a chemical shift of 61–62 ppm. However, in the pyranose 6-membered ring, all the carbon atoms are in the ring, and all have chemical shifts of 65–80 ppm. The 13C NMR of compounds of 2.7 and 6.1 min showed ribose carbon signals at 66–76 ppm and none at 61–62 ppm. In contrast, the compound that eluted at 3.5 min had a resonance at 62 as well as several at 72–83 ppm. Therefore, compounds 1 (emerging from the HPLC after 2.7 min) and 4 (emerging from the HPLC after 6.1 min) were assigned to be pyranose epimers, while compounds 2 (emerging from the HPLC after 3.5 min) and 3 (emerging from the HPLC after 4.3 min) were assigned to be furanose epimers.
3.3. Acid-catalyzed epimerization of the glycosylation products
Further evidence that the glycosylation products are C-nucleosides came from their acid-catalyzed epimerization. It has been known for decades that many C-nucleosides epimerize in the presence of acid (Cohn, 1960; Chambers et al., 1963). If our glycosylation products are C-nucleosides, they should also epimerize in acid. Usually, pyranoses are thermodynamically more stable, and furanose is kinetically favored product (Hutter and Benner, 2003). To measure the rates of epimerization in acidic solution, each HPLC-purified fraction was incubated in pH 2 solution and analyzed by HPLC (Fig. 4). Each fraction (initially a single peak in HPLC) was transformed into a mixture of three compounds, with the mixture comprising mostly pyranose (2.7 and 6.1 min in HPLC) and a small fraction of furanose (3.5 min in HPLC). Interestingly, the other furanose (4.3 min in HPLC) was not detected after the epimerization was complete. These results confirm that the glycosylation products are C-nucleosides.
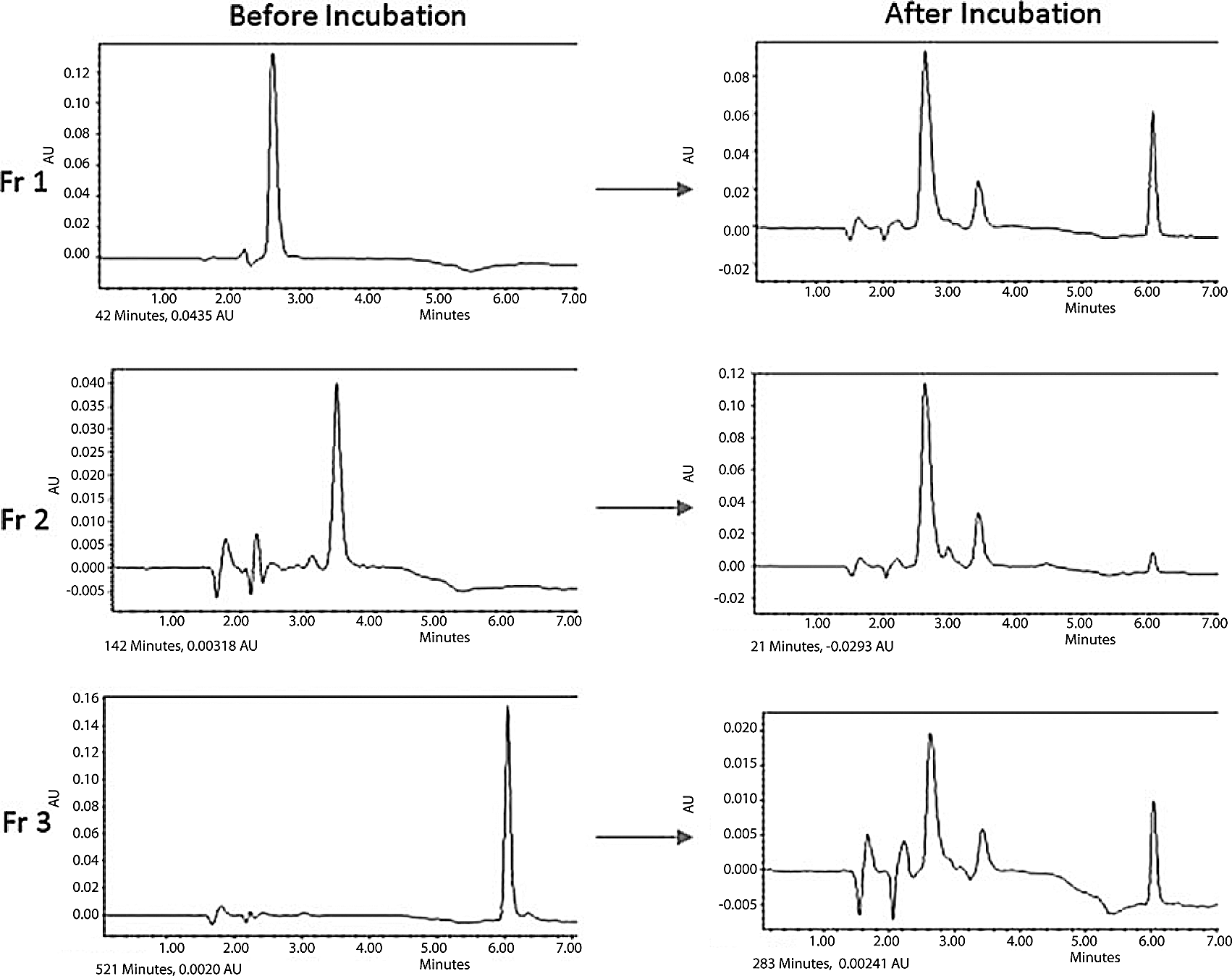
Acid-catalyzed epimerization of purified fractions (Fr 1, 2.7 min; Fr 2, 3.5 min; Fr 3, 6.1 min in analytical HPLC) generated by the reaction between 6-aminouracil and ribose.
4. Discussion
These results expand on recent reports covering the glycosylation of modified pyrimidine nucleosides (Chen et al., 2014). Two aspects of these results are especially interesting.
First, all the uracils tested had an electron-donating substituent. However, only the amino-substituted uracils gave the desired products; the methoxy substituent did not give glycosylated products, even though the methoxy substituent is also quite electron-donating. One possible explanation arises because the methoxyuracil was seen to decompose during the reaction, while the aminouracil does not.
These results can be compared to the glycosylation reaction reported by Hud and his coworkers using 2,4,6-triaminopyrimidine, an extremely electron-rich pyrimidine (Chen et al., 2014). They also reported yields of approximately 50%. The results reported here extend the glycosylation reaction to a considerably less nucleophilic heterocycle. Also noteworthy is the observation that the reaction proceeds without any magnesium based on HPLC peak integration.
The 6-aminouracil gave only C-glycosylated product, despite the availability of three nitrogen nucleophiles. This may, of course, reflect a thermodynamic outcome, as the formation of the N-glycoside under these conditions is likely reversible. Evidence for this arises from the observation of epimerization. The major glycosylation products were pyranose nucleosides in all cases examined, and the pyranose:furanose ratio was ∼3:1. These are all consistent with a reaction pathway similar to that for 2,4,6-triaminopyrimidine glycosylation (Fig. 5). This mechanism explains the emergence of four glycosylation products (two furanoses and two pyranoses) from the ring closure with the 4- or 5-position hydroxyl group (by ribose nomenclature) on the proposed α,β-unsaturated iminium ion intermediate.
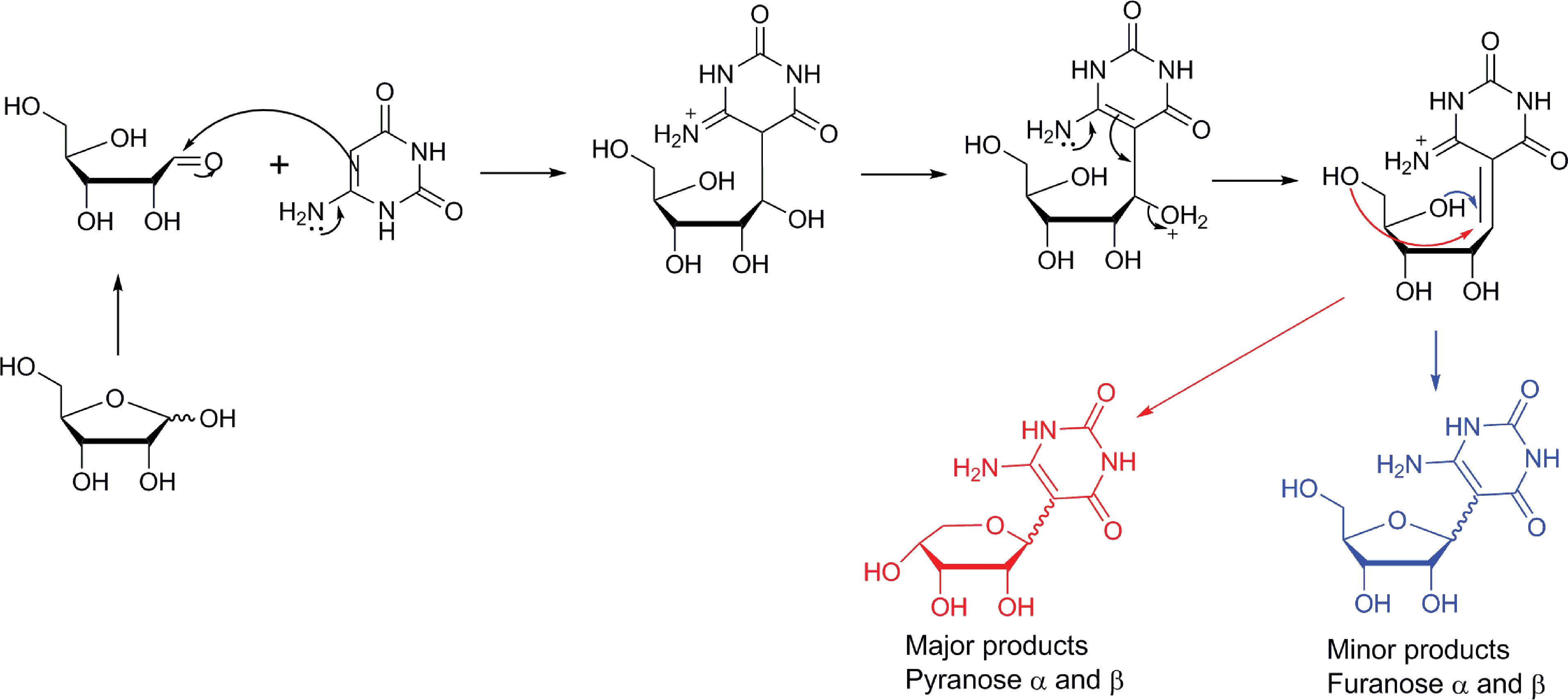
Proposed reaction pathway of the reaction between 6-aminouracil and ribose to form a C-glycoside nucleoside analogue. (Color graphics available at
Second, the C-glycosylation nucleoside arising from the reaction between ribose and 6-aminouracil has two hydrogen bonding patterns. On one side, the heterocycle presents (in a Watson-Crick geometry proceeding from the major to the minor groove) the acceptor-donor-acceptor pattern, the same as uracil. On the other side, however, the pattern is acceptor-donor-donor, which is the nonstandard pattern seen in the AEGIS base V (Fig. 1). Because it has two different hydrogen bonding patterns in one base, this nucleoside product, formed under “prebiotic” conditions, could be a good candidate for a “biversal” nucleobase to complement the purine nucleobases presenting the donor-acceptor-donor and donor-acceptor-acceptor hydrogen bonding patterns (Liang et al., 2013).
Footnotes
Acknowledgments
We are indebted to NASA and the Templeton World Charity Foundation Inc. for financial assistance.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Materials
High-resolution ESI mass and 1H and 13C NMR spectra of each purified compound are available online at
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.