
Abstract
The mineral greigite (Fe3S4) distributes widely in anoxic marine and lake sedimentary systems, with important implications for magnetostratigraphy and paleomagnetism. In living organisms, magnetotactic bacteria can synthesize greigite grains with regular sizes and morphologies. The cubic Fe3S4 structure also occurs as an integral constituent and active center in a family of iron-sulfur proteins in all life-forms on Earth. This basic biochemistry shared by all organisms implies that the Fe3S4 structure might have evolved in the first protocell. Therefore, greigite is of general interest in geochemistry, geophysics, biomineralogy, and origin-of-life sciences. However, the growth of thermodynamically metastable Fe3S4 crystals often requires strictly defined conditions because both Fe and S show variable valences and it is hard to tune their valence fluctuation. Here, we show that freshly precipitated FeS can be selectively oxidized to form greigite in the presence of α-oxo acids, even at room temperature. Based on a brief overview of the experimental findings, a metal-organic complex intermediate model has been put forward and discussed for the discriminative chemical transformation. The results not only provide a possible pathway for the abiotic formation of greigite in nature but also may help explain the biotic mineralization of greigite in magnetotactic bacteria. Moreover, in the context of prebiotic evolution, along with the synergic evolution between greigite and α-oxo acids, Fe3S4 might have been sequestered by primordial peptides, and the whole finally evolved into the first iron-sulfur protein. Key Words: Greigite—Mineralization—α-Oxo acid—Magnetosome—Iron-sulfur protein—Prebiotic evolution. Astrobiology 15, 1043–1051.
1. Introduction
G
Greigite has also drawn much attention because of its importance for magnetostratigraphy and paleomagnetism (Snowball and Thompson, 1990; Tric et al., 1991; Roberts et al., 1996; Jiang et al., 2001; Ron et al., 2007; Vasiliev et al., 2007; Chang et al., 2008), and especially over the past decade due to its inviting semi-metallic and magnetic properties (He et al., 2006; Cao et al., 2009; Ma et al., 2010; Vasilenko et al., 2010; Beal et al., 2011; Paolella et al., 2011; Feng et al., 2013; Liu et al., 2014; Liao et al., 2015). Those investigations were mostly undertaken on laboratory-synthesized samples and increased the knowledge of the magnetic properties of greigite. However, since greigite is thermodynamically metastable, the growth of pure Fe3S4 crystals often requires strictly defined conditions, toxic reagents, extreme temperatures, and in some cases time-consuming, post-synthetic processing. The most frequently used method is hydrothermal reaction between iron salts and sulfur-bearing compounds in aqueous or organic solvents at temperatures between about 125°C and 300°C. Recently, in a fascinating effort to simulate the formation of hydrothermal vents in deep Hadean ocean, White et al. (2015) fabricated FeS chimneys by injecting S2−-bearing alkaline hydrothermal fluids into Fe2+-bearing acidic solutions. They found that mixtures of mackinawite and greigite were spontaneously formed at 70–75°C after 69–72 h in the precipitates, although the yields of greigite were limited and the transformation mechanism remained unclear. On the other hand, amorphous FeS is the first iron-sulfide phase formed from aqueous S2− and Fe2+. Both FeS and Fe3S4 are apt to be oxidized and mostly end up as pyrite (FeS2, Eqs. 1 and 2) (Wilkin and Barnes, 1996; Butler and Rickard, 2000).
Therefore, the chemically selective oxidation of Fe2+, but not S2−, remains a challenge for the biomineralization of Fe3S4 at moderate temperatures (Rickard et al., 2001).
In a previous study in our laboratory, we demonstrated that FeS can participate in the reversible interconversion of α-hydroxy acids and α-oxo acids (Wang et al., 2011). At that time, the authors also found that, in the presence of H2S and α-oxo acids, the solid products of FeS can be adsorbed by a magnet, suggesting it may contain magnetic greigite components. Since α-oxo acids are intermediates of pivotal importance in metabolic processes in all living organisms (Cooper et al., 1983), we entertained the notion that such biomolecules may play a key role in the biomineralization of Fe3S4 in vivo.
2. Materials and Methods
2.1. Materials
Oxo acids (>99%) were purchased from Sigma-Aldrich and used without further purification. All other chemicals were obtained in analytical grade from Sinopharm Chemical Reagent Co. Ltd, China. Ultrapure water (Millipore) was deoxygenated with high-purity argon gas (Ar, 99.999%) by bubbling it (approximately 1 L/min) through 500 mL water for 1 h and used throughout.
2.2. Experimental procedure
Reactions were performed in 20 mL headspace vials fitted with metal caps and Teflon septa. Batch experiments were prepared anaerobically inside a vacuum glove box kept under an argon atmosphere. In a typical run, a vial was charged with 1 mmol FeSO4·7H2O, 0.25 mmol keto acid, and 4 mL of water. Subsequently, 2 mL of 0.5 M Na2S solution was added dropwise for in situ precipitation of FeS. Then the vial was surcharged with 10 mL of 0.1 M H2S solution produced by H2SO4 acid decomposition of Na2S · 9H2O and sealed immediately. Standard experimental runs were carried out at 50°C for 2 days.
After the reactions, all samples were centrifuged and filtered by using 0.22 μm microvoid filter films. High-performance liquid chromatography (HPLC) was used to determine organic compounds in the supernatant liquor (Wang et al., 2012a). The resulting solid products were collected, washed with deionized water and ethanol several times to remove impurities, and dried in a vacuum oven at 60°C for 4 h. Then they were characterized by X-ray diffraction (XRD) on a PANalytical Empyrean X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å). The samples were also quantified carefully and then sealed into plastic tubes for magnetization measurement. Hysteresis loops were collected on a Physical Property Measurement System (PPMS, Quantum Design) at 300 K. A wet chemical method was employed for the determination of ferric iron in the samples according to the work of Husler et al. (2011).
3. Results
The typical XRD patterns of the samples are shown in Fig. 1. It can be found that, in the presence of H2S and the absence of pyruvic acid, the main phases of the solid products are mackinawite and pyrite; while in the presence of both H2S and oxo acids, the newly precipitated FeS is apt to selectively evolve into Fe3S4, even at room temperature (Fig. 1a). The optimal temperature is about 50–100°C. As we know, amorphous FeS is the first iron-sulfide phase formed from aqueous S2- and Fe2+. Since the poorly ordered FeS is unstable, it spontaneously transforms into mackinawite (tetragonal FeSm) (Rickard, 2006). Mackinawite is metastable with respect to pyrite. Without pyruvic acid, the reaction between FeSm and aqueous H2S produces pyrite and hydrogen (Eq. 1). The results suggest that mackinawite may have occurred as an intermediate during the formation of greigite.
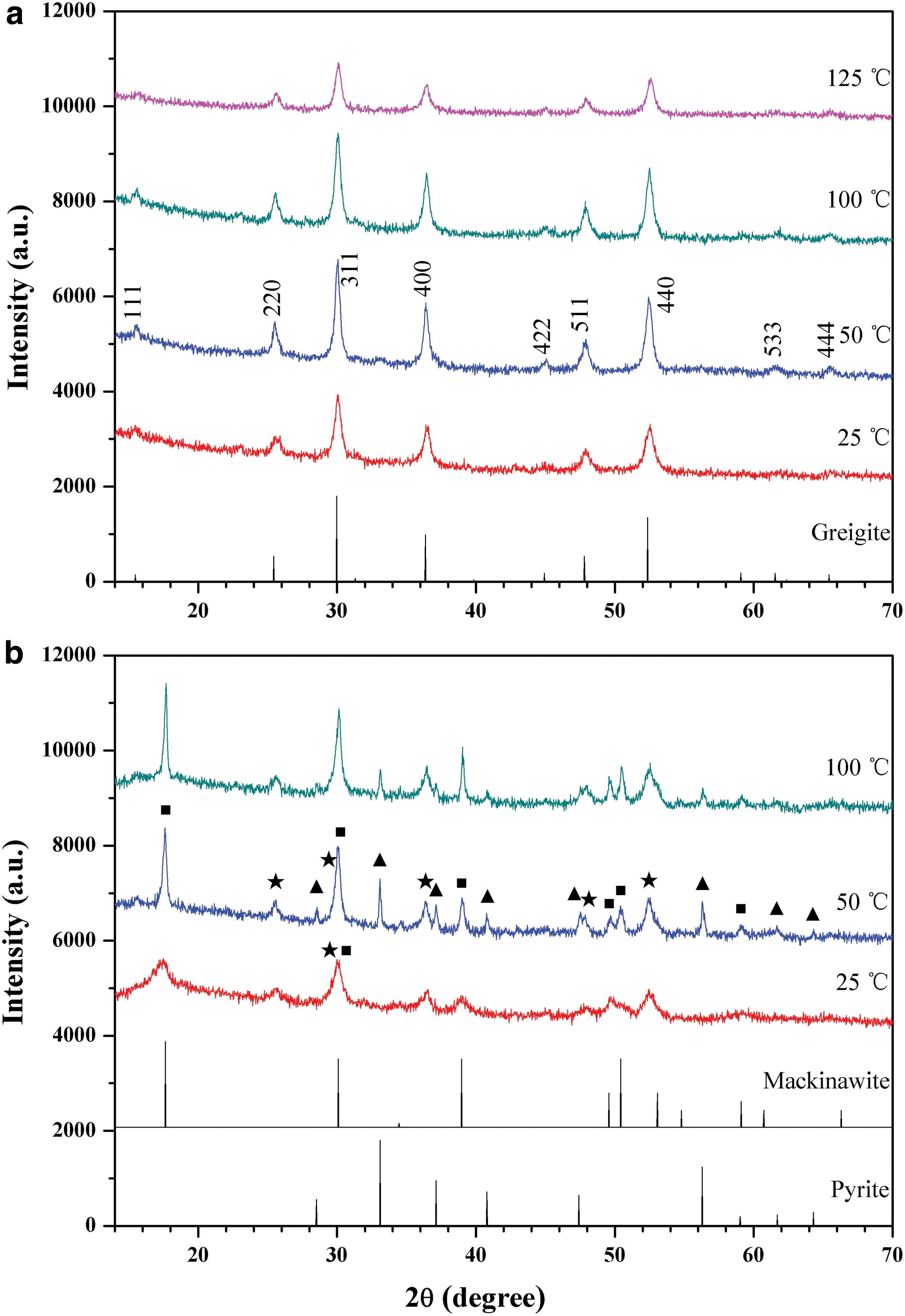
X-ray diffraction patterns of the iron sulfide products prepared at different temperatures for 2 days, in the presence (
In further experiments, a set of organic molecules was evaluated for their influence over the transformation of FeS to Fe3S4. The results are represented in Fig. 2. It can be seen that pyruvic acid (PA) and all other α-oxo acids, that is, glyoxylic acid (GA), oxalacetic acid (OAA), ketoglutaric acid (KGA), 3-methyl-2-oxovaleric acid (MOVA), and phenylpyruvic acid (PPA), can promote the above-mentioned phase transition. However, lactic acid (LA), an analogue of pyruvic acid that owns an α-hydroxyl instead of α-carbonyl, does not show such a chemical selectivity. The decarboxylated analogue of pyruvic acid, acetaldehyde (AA), also inhibits the formation of pyrite but has a very low efficiency in facilitating the formation of greigite. The diffraction peaks are very weak, indicating low yields and bad crystallinities of the products. In contrast, formaldehyde (FA) can work more efficiently, although the pure greigite phase was not obtained in this case. The order of reactivities of the organic compounds follows the trend GA∼PPA∼PA∼MOVA > KGA∼OAA > FA > AA. In light of the above findings, we recalled that in a previous pioneering study Rickard et al. (2001) reported that the products of the oxidation of FeS at 100°C by aqueous H2S switch between Fe3S4 and FeS2, depending on the additive amounts of aldehydes. However, when we repeated that work by using formaldehyde, we found that the product greigite was always accompanied by a small amount of mackinawite. In contrast, in the presence of pyruvic acid, greigite is the only phase detected by XRD, even at room temperature.
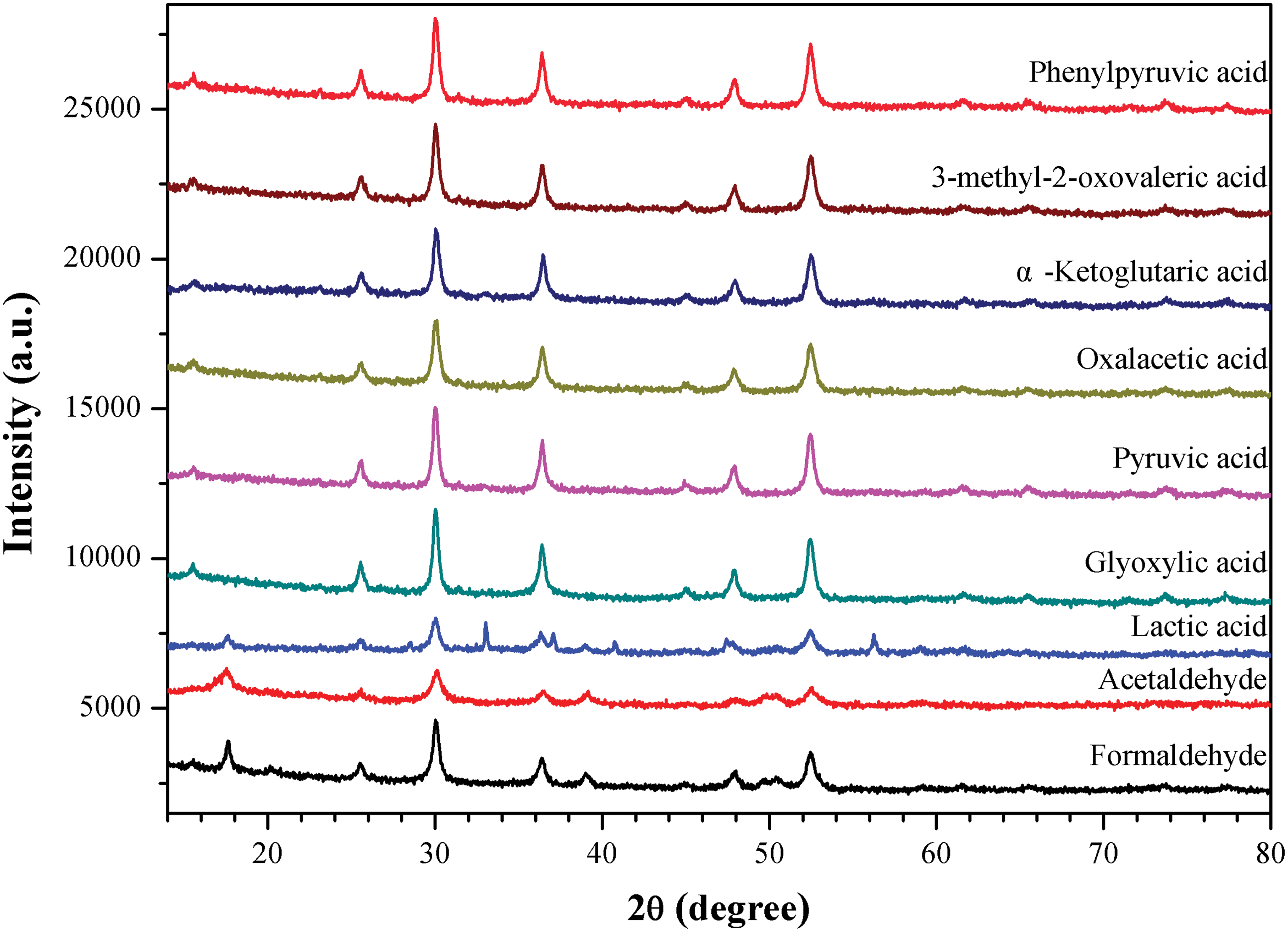
X-ray diffraction spectra of iron-sulfide minerals formed at 50°C for 2 days, in the presence of different organic molecules. The XRD profiles show that (1) the product in the case of lactic acid is a mixture of pyrite/mackinawite/greigite; (2) a molecule with a carbonyl moiety such as α-oxo acids and aldehydes can efficiently promote the transformation of FeS to Fe3S4; and (3) only in the presence of a molecule with both a carbonyl and a carboxylic group (α-oxo acids) can the pure greigite phase selectively evolve. (Color graphics available at
Otherwise, we also found that acetone, dimethylformamide, N-acetyl-phenylalanine, and glycylglycine acted similarly to lactic acid, producing a mixture of pyrite, mackinawite, and greigite (data not shown). Therefore, it appears that only the carbonyl moiety of oxo acids or aldehydes, but not amides or ketones, can give rise to the formation of pure phase of Fe3S4.
Figure 3 shows the magnetic susceptibilities (M) of the samples. The weight percentage (Wt %) of greigite in the solid products has also been calculated based on the wet chemical analysis results. Since no other Fe3+-bearing crystal phase was identified by XRD (Figs. 1 and 2), we ascribed all ferric ions in the products to greigite. As shown in Fig. 3, the yields of greigite are nearly 95% in the presence of α-oxo acids, especially phenylpyruvic acid and glyoxylic acid. With different organic additives, the M and Wt % values display a similar trend, which agrees very well with the XRD data in Fig. 2.
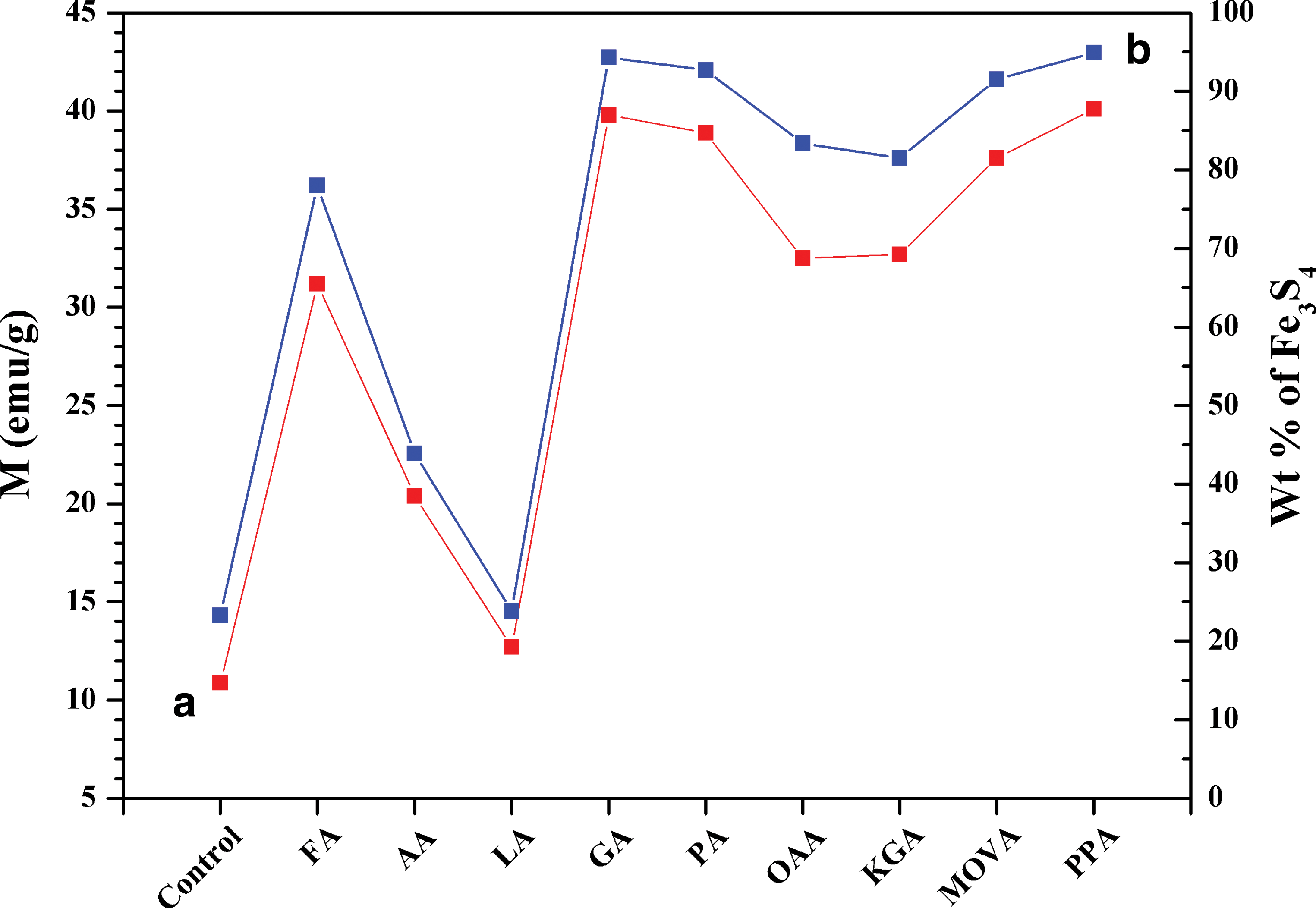
(
Chemically, the formation of Fe3S4 requires that the FeS-Fe is oxidized to Fe3+ but the FeS-S is not, while in the FeS to FeS2 transformation the FeS-S is oxidized but the FeS-Fe remains unchanged. For the production of greigite, therefore, it is of key importance to tune the valence fluctuation of iron ions (Liao et al., 2015). If a ferrous salt is chosen as the initial iron source, a weak inorganic or organic oxidant, such as elemental sulfur (Wilkin and Barnes, 1996; Beal et al., 2011; Paolella et al., 2011), polysulfide anions (Wada, 1977; Dekkers and Schoonen, 1996; Wilkin and Barnes, 1996; Hunger and Benning, 2007), an appropriate amount of oxygen (Wilkin and Barnes, 1996; Vasilenko et al., 2010), or cysteine (Wilkin and Barnes, 1996; He et al., 2006; Cao et al., 2009; Liu et al., 2014), is indispensable. In our experiments, a small quantity of Fe3S4 was produced in the absence of pyruvic acid. This may be ascribed to the rhombic sulfur (Eq. 3) (Wilkin and Barnes, 1997) formed during the acid decomposition processes of FeS (Rickard et al., 2006) or Na2S.
In the presence of pyruvic acid, however, greigite was selectively evolved, and other impurity phases like pyrite and mackinawite were exclusively inhibited.
As to the reaction mechanism, we at first entertained the notion that a redox reaction between FeS and α-oxo acids might have played a role in the transformation of FeS to Fe3S4. For instance,
Equation 4 shows the anaerobic oxidation of Fe2+ to Fe3+ with the parallel reduction of pyruvic acid to lactic acid. Stoichiometrically, if 1 mmol of FeS is used at the beginning, the final yield of greigite is about 90%, and 0.6 mmol of Fe2+ is oxidized (Eq. 5), and 0.3 mmol of pyruvic acid is reduced, producing 0.3 mmol of lactic acid.
In our experiments, however, only 0.25 mmol of pyruvic acid was added, and about 0.03 mmol of lactic acid was detected by HPLC. In the case of phenylpyruvic acid (0.25 mmol), nearly 0.18 mmol remnant of pyruvic acid was found in the solution.
Therefore, there should exist an alternative mechanism in the oxidation process. We would expect that the carboxylic anion may be adsorbed onto the surface of the iron sulfide mineral through electrostatic interactions. During this process, α-oxo acid approaches FeS to form a metal-organic complex that contains a five-membered chelate ring (Fig. 4) (cf. Kubala and Martell, 1982). In the case of aldehyde, a similar structure may also occur, which is supported by an additional hydrogen bond of the carbonyl-H to surface sulfur. Once the above complex structures form, they will polarize the carbonyl group, thus creating an electron sink. The 3d electron of Fe2+ ion is pulled toward the powerful electron-drawing carbonyl group to produce a pseudo Fe3+ ion. Subsequently, the cubic close-packed S array in mackinawite is retained in greigite (Krupp, 1994; Fig. 4). The pseudo Fe3+ ions in the mackinawite lattice drive rearrangement of Fe to form the new phase. The redundant electrons tend to flow into two well-trenched sinks: α-hydroxyl acid (the reduced product of α-oxo acids) and hydrogen gas (the reduced product of protons) (Wang et al., 2011). In the latter case, the α-oxo acid molecules play a catalytic role since they only participate in the electron transfer reaction but remain unchanged in chemical composition at the end of the mineral transformation.
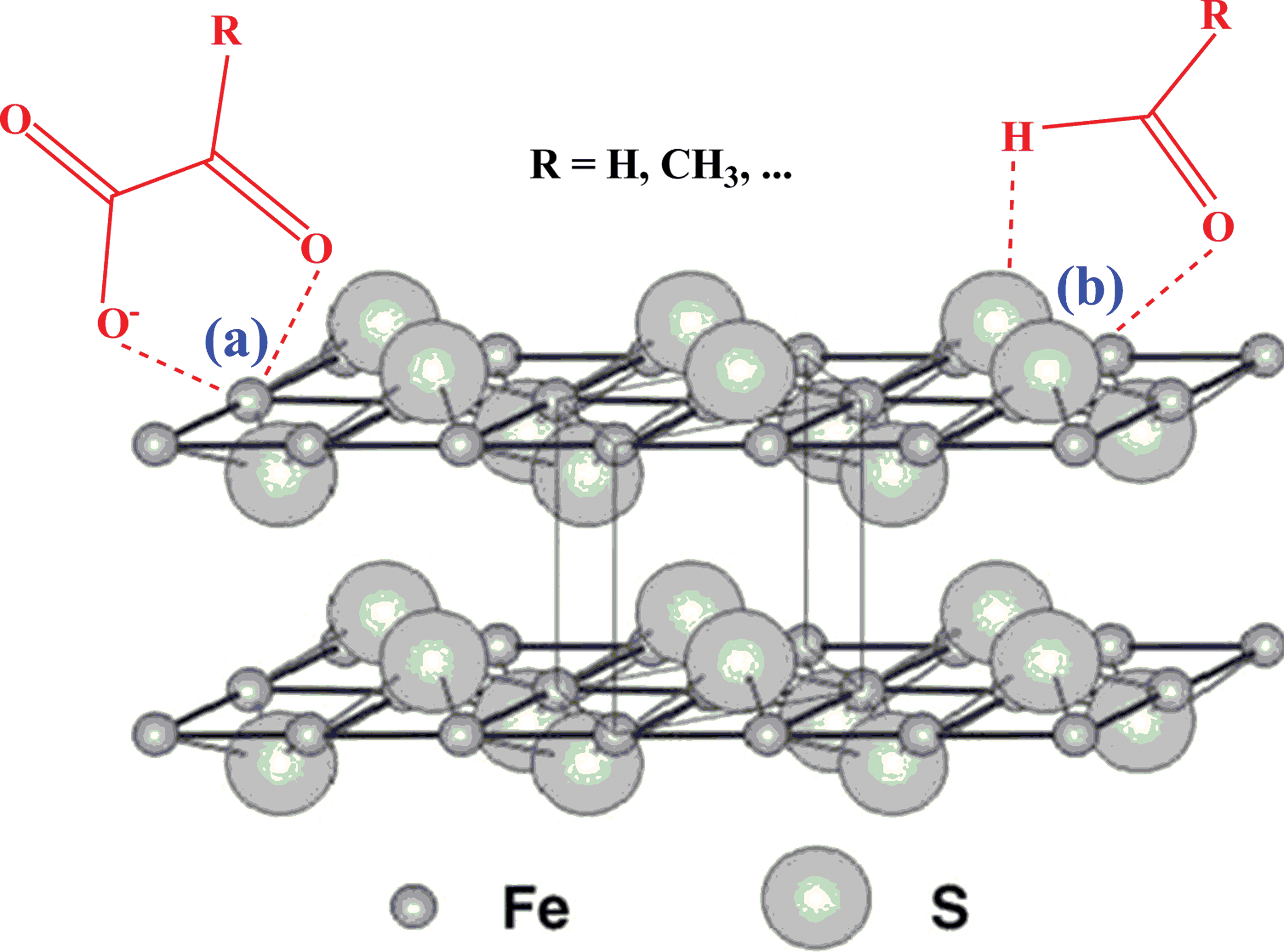
Schematic illustration of the tetragonal layer structure of mackinawite and its electrostatic interactions with α-oxo acids and aldehydes. Mackinawite (FeSm) possesses a tetragonal layer structure, where the Fe atoms are linked in a tetrahedral coordination to four equidistant S atoms, and the sheets are weakly held by Van der Waals bonding between the S atoms. Due to electrostatic and hydrogen bond interactions, α-oxo acids and aldehydes are adsorbed to the surface of FeSm to form five-membered chelating metal-organic complexes. (Color graphics available at
4. Discussion and Conclusions
In the present study, we experimentally demonstrated that α-oxo acids can promote the selective oxidation of FeS to Fe3S4 by aqueous H2S at low temperature, even at room temperature. We have also indicated and discussed a possible mechanism for this discriminative chemical transformation. Specifically, α-oxo acid and FeS may form a five-membered ring metal-organic complex in which the 3d electron of the Fe2+ ion is pulled toward the carbonyl group to produce a pseudo Fe3+ ion. The resulting pseudo Fe3+ ion then drives the rearrangement of Fe to form greigite, with the cubic close-packed S array retained in the new phase. The results suggest that organic compounds with specific functional groups can affect the chemistry of iron sulfide. Some greigite minerals in nature may develop by way of such an abiotic process. Taking into consideration that α-oxo acids are important intermediates in amino acid metabolism, we infer that this abiotic source of greigite could be a good, although not unambiguous, biosignature of past biogenic activity.
Our results also have implications for our understanding of the biomineralization of iron sulfides in vivo. It has been reported that greigite in magnetotactic bacteria forms over time from a nonmagnetic mackinawite precursor (Pósfai et al., 1998). This process is usually modulated by a family of proteins. For example, Siponen et al. (2013) analyzed the structure of a magnetosome-associated protein MamP and found that the biomacromolecule possesses a crucible-like pocket that can capture ferrous ions from the environment and make them oxidized to ferric ions. The lost electron cannot exist on its own and must be gained by a second entity, just as in the reaction in Eq. 4. Therefore, the coupled organic-inorganic reaction helps explain not only how Fe3S4 crystals develop in bacterial cells at room temperature but also why its later transformation to pyrite is inhibited.
Finally, there exists a family of iron-sulfur proteins or ferredoxin in all life-forms on Earth (Eck and Dayhoff, 1966; Hall et al., 1971; Beinert, 2000; Lill, 2009). They own Fe-S cluster active centers with high redox potential, whose structure is very similar to the cubic Fe3S4 (Cammack, 1996; Furdui and Ragsdale, 2000; St Maurice et al., 2007) (Fig. 5). As first detailed by Russell et al. (1994), this basic biochemistry shared by all organisms implies that life on early Earth might have emerged in a submarine hydrothermal sulfide mound (Russell and Hall, 1997; Russell et al., 2005; Cleaves et al., 2012). In a Hadean hydrothermal mound, the FeS niche would have acted as a flow-through chemical reactor and power plant (Nakamura et al., 2010; Yamamoto et al., 2013; Barge et al., 2015). Transition metal sulfides would have catalyzed (i) the prebiotic synthesis of organic molecules including α-oxo acids (Cody et al., 2000; Herschy et al., 2014; Yamaguchi et al., 2014; Roldan et al., 2015), (ii) the reductive amination of α-oxo acids to produce amino acid (Huber and Wächtershäuser, 2003), (iii) the condensation of amino acids to form peptides (Huber and Wächtershäuser, 1998), and (iv) the buildup of original prototypes of the prebiotic metabolism (Huber and Wächtershäuser, 1997; Huber et al., 2003; Russell and Martin, 2004; Zhang and Martin, 2006; Wang et al., 2012b, 2012c, 2013). Meanwhile, organic molecules like α-oxo acids would have selectively promoted the transformation of FeS to Fe3S4 (Fig. 5). The resulting mineral structure would then have been sequestered by the abiotic peptides (Russell et al., 2005), and the whole would finally have evolved into the first prebiotic ferredoxin.

Proposed reaction scheme diagram for the synergic evolution between Fe3S4 and α-oxo acids and the prebiotic formation of the first ferredoxin. In the Hadean submarine hydrothermal mounds, inorganic substrates would have occurred in FeS chimneys and facilitated production of biomolecules, including α-oxo acids and polypeptides, and built up an abiotic archetype for the first metabolic network; the original FeS, under the action of biomolecules, would have experienced an oriented evolution into Fe3S4 structure. The latter would then have been sequestered by antique peptides, developed ultimately into the proto-ferredoxins, and adopted by the last universal common ancestor. (Color graphics available at
Footnotes
Acknowledgments
We are grateful to Dr. Michael Russell for bringing the work of Rickard's group to our notice. Our most heartfelt thanks also go to the anonymous reviewers and editors for their constructive suggestions for the polishing of the paper. This work was funded by National Natural Science Foundation of China (No. 40902014, 51472064) and Natural Scientific Research Innovation Foundation in Harbin Institute of Technology (HIT.NSRIF 2013055).
Author Disclosure Statement
No competing financial interests exist.