
Abstract
Understanding the evolution of Earth and potentially habitable Earth-like worlds is essential to fathom our origin in the Universe. The search for Earth-like planets in the habitable zone and investigation of their atmospheres with climate and photochemical models is a central focus in exoplanetary science. Taking the evolution of Earth as a reference for Earth-like planets, a central scientific goal is to understand what the interactions were between atmosphere, geology, and biology on early Earth. The Great Oxidation Event in Earth's history was certainly caused by their interplay, but the origin and controlling processes of this occurrence are not well understood, the study of which will require interdisciplinary, coupled models. In this work, we present results from our newly developed Coupled Atmosphere Biogeochemistry model in which atmospheric O2 concentrations are fixed to values inferred by geological evidence.
Applying a unique tool (Pathway Analysis Program), ours is the first quantitative analysis of catalytic cycles that governed O2 in early Earth's atmosphere near the Great Oxidation Event. Complicated oxidation pathways play a key role in destroying O2, whereas in the upper atmosphere, most O2 is formed abiotically via CO2 photolysis.
The O2 bistability found by Goldblatt et al. (2006) is not observed in our calculations likely due to our detailed CH4 oxidation scheme. We calculate increased CH4 with increasing O2 during the Great Oxidation Event. For a given atmospheric surface flux, different atmospheric states are possible; however, the net primary productivity of the biosphere that produces O2 is unique. Mixing, CH4 fluxes, ocean solubility, and mantle/crust properties strongly affect net primary productivity and surface O2 fluxes.
Regarding exoplanets, different “states” of O2 could exist for similar biomass output. Strong geological activity could lead to false negatives for life (since our analysis suggests that reducing gases remove O2 that masks its biosphere over a wide range of conditions). Key Words: Early Earth—Proterozoic—Archean—Oxygen—Atmosphere—Biogeochemistry—Photochemistry—Biosignatures—Earth-like planets. Astrobiology 16, 27–54.
1. Introduction
I
Numerous processes—including, for example, biology, radiative transfer, and photochemistry, but also interactions with the surface—have a strong impact on the abundance of atmospheric molecular oxygen (O2). Oxygen-bearing molecules play a direct role in Earth's geological, geochemical, and geobiological systems. Furthermore, O2 plays an indirect role in making Earth “habitable” because it leads to the formation of the biosignature molecule ozone (O3), which is formed from O + O2 + M → O3 + M (where “M” denotes a background gas that removes excess vibrational energy) in the stratosphere. O3 efficiently absorbs UV radiation, which is harmful to many forms of life. Next to other important greenhouse gases, such as water vapor (H2O) and carbon dioxide (CO2), O3 is important for Earth's energy balance because O3 (besides being radiatively active itself) and other products of O2 photochemistry determine the lifetimes of atmospheric greenhouse gases, such as methane (CH4), but also other trace gases such as carbon monoxide (CO) and hydrogen sulfide (H2S). Hence, the evolution of O2 on Earth is one of the fundamental aspects that have determined the evolution of Earth's surface environment, climate, and biosphere.
1.1. O2 in modern Earth's atmosphere
The present-day atmosphere contains about 1.2·1021 g = 3.75·1019 mol of O2 (Lasaga and Ohmoto, 2002). Its abundance is regulated by the established sources and sinks of the oxygen cycle. About 99% of photosynthetically produced organic carbon is approximately in balance with the reverse process, namely, respiration (Prentice et al., 2001), which results in a net primary productivity (NPP) of about 45 petagrams (1015 g) C per year (Prentice et al., 2001). A small amount of the remaining organic carbon (about 0.2%, Prentice et al., 2001) escapes oxidation during aerobic respiration via sedimental burial at the ocean floor, and the O2 that remains is then liberated into the atmosphere (1 mol of organic carbon corresponds to 1 mol of O2). This process constitutes the major source of atmospheric O2. There is an additional source from the burial of other redox-sensitive chemical compounds like pyrite (FeS2) and ferrous iron (FeO), whereas the burial of sulfate minerals in sediments removes a small portion of O2. Sedimental burial is estimated to account for 0.3–0.8 Pg O2/yr (Holland, 2002); this process would require 1.5–4 million years to produce the present mass of atmospheric O2. There also exists a much smaller modern-day source of ∼0.4·10−3 Pg/yr (Jacob, 1999), which arises via H2O photolysis followed by escape of atomic hydrogen from the atmosphere. Although this is only a minor source in the modern atmosphere, it might have been much faster for early Earth (Kasting and Catling, 2003). The main O2 atmospheric modern-day sinks include the following: (1) weathering, estimated to remove ∼0.5 Pg/yr (Holland, 2002); (2) metamorphic reactions that occur on hot volcanic rock, estimated to account for ∼0.08 Pg/yr (Catling and Claire, 2005); and (3) reactions with reduced gases in the atmosphere, estimated to remove about 0.03–0.06 Pg/yr, although this value is highly uncertain (Catling and Claire, 2005).
1.2. Atmospheric O2 in Earth's history
The O2 concentration has risen from its prebiotic abundance of less than 10−13 present atmospheric level (PAL) (Kasting and Donahue, 1981) during the Archean eon 2 up to about 1% PAL by about 2.3 Gyr ago and may have exceeded 15% PAL by about 2.1 Gyr (e.g., Knoll and Holland, 1995; Holland and Yang, 2001) (see Fig. 3). Realizing the nature of the Great Oxidation Event (GOE) is central to understanding the development of life on Earth and, hence, atmospheric biosignatures on Earth-like, potentially habitable extrasolar planets. Geological indices that imply an anoxic Archean atmosphere include the mass-independent fractionation (MIF) of sulfur isotopes (e.g., Farquhar et al., 2000; Pavlov and Kasting, 2002) and the existence of banded iron formations (BIFs) (e.g., Cloud, 1968; Holland, 1984; Isley and Abbott, 1999). On the other hand, an oxidized Archean scenario was proposed, for example, by Ohmoto (1997) based on paleosol data. Although cyanobacteria were already present on Earth by about 2.5 Gyr (Summons et al., 1999), for reasons not clear the recorded rise in O2 took place at least 200 million years later. The O2 rise may also have changed Earth's climate, triggering a so-called snowball Earth (Kopp et al., 2005) possibly due to responses in the photochemistry and/or negative impacts on methanogenic bacteria leading to a decrease in the warming due to CH4 (e.g., Zahnle et al., 2006). Note, however, that there are additional possible cooling mechanisms, for example, a change in volcanic gases (see e.g., Catling and Claire, 2005). Canfield (2005) and Holland (2006) reviewed the geological evidence and current understanding of oxidative transitions in Earth's atmosphere. Typically, Earth-system box models (without detailed atmospheric photochemistry and climate calculations) are applied to investigate the rise in O2. For example, Kump et al. (2001) suggested that oxidation events in the Paleoproterozoic corresponded to a mantle overturning event, where previously oxidized mantle was brought to the surface. This would lead to less-reduced gases being emitted by volcanoes and allow atmospheric O2 to rise. Lasaga and Ohmoto (2002) applied a box model to investigate negative feedbacks of atmospheric O2, which may account for its reasonably constant abundance since the Phanerozoic era. For example, lowering O2, according to their results, would (1) strongly lower the weathering rate and (2) increase oceanic anoxicity and, hence, increase organic carbon burial via less oxidation of organic carbon in the ocean column. Effects (1) and (2) are both negative feedbacks that would stabilize O2 in the atmosphere. Holland (2003), however, questioned their model assumptions, for example, in deriving the dependence of burial rates on oxygen abundance in seawater. Catling and Claire (2005) summarized the various theories proposed to account for the rise in O2. Briefly, these include (1) an increase in the burial rate of organic carbon and other redox-sensitive compounds, (2) an increased supply of nutrients that stimulate photosynthesis, (3) a change in volcanic gas composition, or (4) a higher hydrogen escape rate from the top of the atmosphere. Claire et al. (2006) applied a zero-dimensional (0-D) box model to calculate the GOE and suggested a lowering in CH4 as O2 increases, whereas Goldblatt et al. (2006) suggested a bistable state (oxic and non-oxic) of the atmosphere whereby O2 abundances would respond very strongly to small changes in surface fluxes of O2. To explain this behavior, they proposed a positive feedback mechanism in which an initial increase in O2 abundances leads to more O3 and, hence, stronger UV shielding that reduces the O2 photolytic sink and allows O2 to build up.
1.3. Contribution of this work
Until now, 0-D box models were used to model Earth's atmospheric O2 evolution by evaluating the important biogeochemical processes. In these studies, the atmosphere was included in a simplified way in order to investigate CH4 oxidation as in the works of, for example, Claire et al. (2006) and Goldblatt et al. (2006). On the contrary, there are numerical studies in which the atmospheres of early Earth and Earth-like extrasolar planets have been assessed at different time epochs by applying both one-dimensional (1-D) global mean atmospheric column models as well as three-dimensional (3-D) general circulation models (see, e.g., Kienert et al., 2012; Charnay et al., 2013; Leconte et al., 2013; Wolf and Toon, 2013, 2014; Kunze et al., 2014; Wordsworth et al., 2011).
In the case of the 1-D global mean atmospheric column models, investigations were performed by using either climate (see, e.g., Kasting et al., 1993; von Paris et al., 2008; Goldblatt et al., 2009; Kitzmann et al., 2010, 2011a, 2011b; Wordsworth et al., 2010; Wordsworth and Pierrehumbert, 2013), photochemistry (see, e.g., Kasting et al., 1997; Zahnle et al., 2006; Segura et al., 2007; Haqq-Misra et al., 2011), or coupled climate-photochemistry calculations (see, e.g., Kasting et al., 1984; Pavlov et al., 2001, 2003; Segura et al., 2003, 2005; Grenfell et al., 2007a, 2007b; Kaltenegger and Sasselov, 2010; Grenfell et al., 2011, 2012; Rauer et al., 2011; Roberson et al., 2011), thereby neglecting biogeochemical cycles. However, since biogeochemical processes strongly influence the abundance of atmospheric constituents such as O2 and CO2, it is important to add these processes. Additionally, performing time-dependent integrations is a central scientific goal. However, this is currently only achievable for box-model studies (e.g., Claire et al., 2006; Goldblatt et al., 2006). Unfortunately, performing long integrations over millennia with state-of-the-art column models such as our scheme is not possible.
In this study, we therefore developed a Coupled Atmosphere Biogeochemical (CAB) model with detailed photochemistry and climate calculations as well as biogeochemical surface processes that have an impact on atmospheric O2. Therefore, the approach in our article, which is similar to other atmospheric model studies (e.g., Kaltenegger and Sasselov, 2010; Roberson et al., 2011), is to “insert” Earth's evolution, for example, via O2 surface concentrations from proxy data and via solar luminosity changes, as a boundary condition in the CAB model. Our main motivation thereby is to focus on understanding the associated atmospheric climatological, chemical, and biological responses. Thus, we model the atmosphere and biosphere of early Earth analog planets consistently by including the effect of the fainter Sun, increased volcanic and metamorphic outgassing, and the impact of the biosphere on the atmosphere. The photochemistry atmosphere module calculates atmospheric O2, CO2, and molecular nitrogen (N2) abundances self-consistently by accounting for their in situ atmospheric sources and sinks. For early Earth analog planetary atmosphere scenarios, we apply a snapshot-like procedure by using the Earth system as a reference. We are able to reproduce the modern Earth atmospheric composition and temperature structure. We further calculate atmospheric surface fluxes required to maintain specified O2, CO2, and N2 abundances in the modern-day atmosphere. For these surface fluxes, we calculate with the biogeochemical module the corresponding NPP of the biospheres, which are needed to maintain the specified O2 surface volume, vmrs.
As a case study, we investigated atmospheric chemical responses of O2 in an early Earth analog atmosphere at 10−6 PAL O2 by applying the Pathway Analysis Program (PAP) developed by Lehmann (2004).
In the following sections, the CAB model (Section 2) and the PAP algorithm (Section 3) are described in detail. The CAB model is validated in Section 4. Section 5 gives an overview of the simulated early Earth analog planet scenarios, whereas in Section 6 the results are presented and discussed. Section 7 gives an overview of the sensitivity studies performed with the CAB model. We present our main conclusions in Section 8.
2. Coupled Atmosphere Biogeochemical (CAB) Model
To study how the atmospheric, geological, chemical, and biotic systems interact, we developed a CAB model. It is composed of two submodules: (1) atmospheric climate and chemistry and (2) biogeochemistry. Both submodules are coupled via the stagnant boundary layer model (Liss and Slater, 1974; Kharecha et al., 2005). The overall outline of the CAB model is given in Fig. 1. In the following sections, each submodule and their coupling will be briefly described.
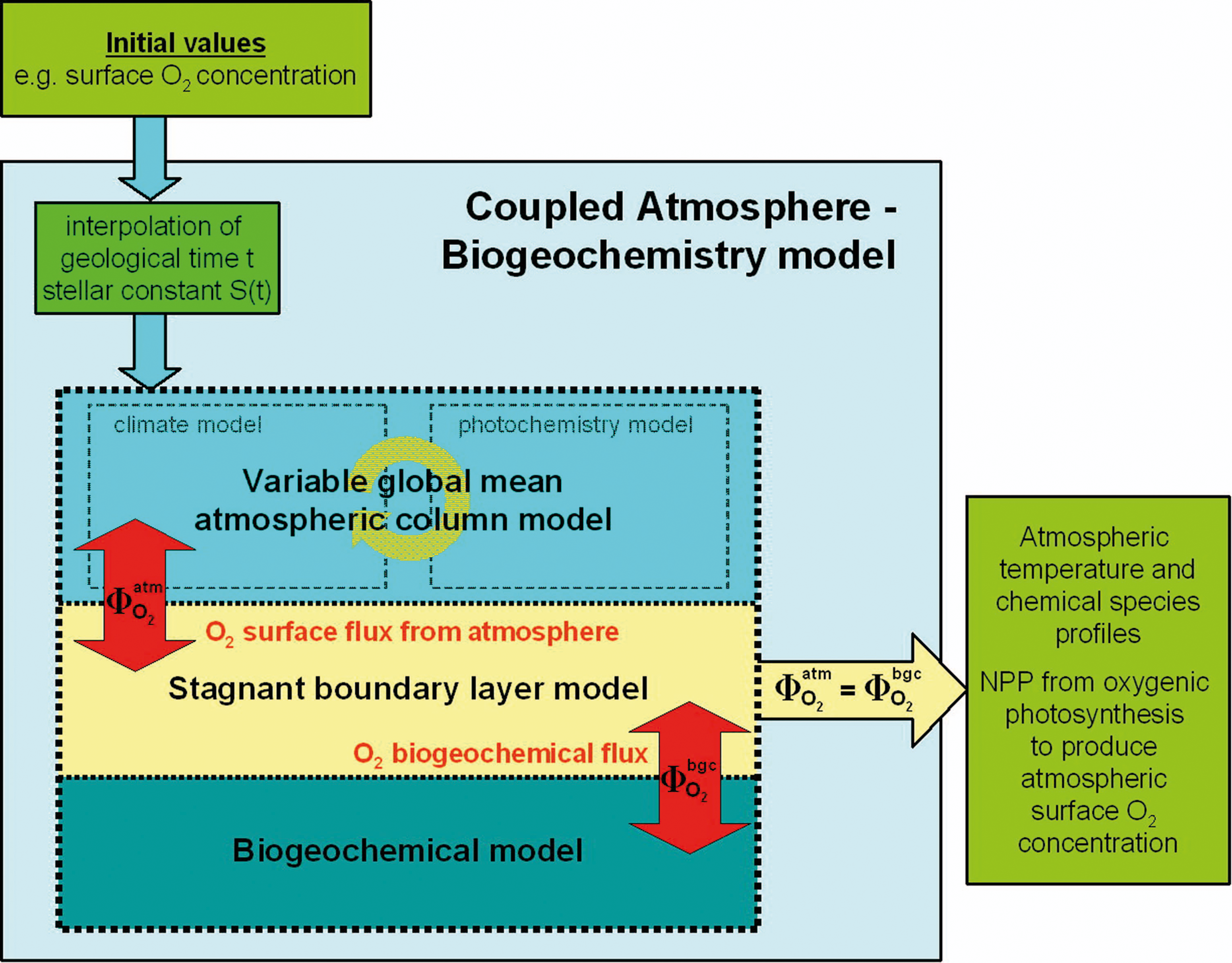
Outline of the CAB model.
2.1. Atmosphere module description
The “variable” 3 1-D global mean cloud-free, steady-state atmospheric column module is based on the 1-D model, which was described in detail by Kasting et al. (1984), Segura et al. (2003), Grenfell et al. (2007a, 2007b), Rauer et al. (2011), and Grenfell et al. (2011).
The climate module calculates temperature and water profiles from the planetary surface up to the lower mesosphere by solving the radiative transfer equation. Convective adjustment is applied as a common procedure (see, e.g., Manabe and Wetherald, 1967; Kasting, 1988; Mischna et al., 2000; Pavlov et al., 2000) if necessary. That is, the radiative temperature gradient,
The chemistry module features more than 200 chemical reactions and calculates concentration profiles for 54 chemical species including the hydrogen oxide (HO
x
), nitrogen oxide NO
x
, and chlorine oxide (ClO
x
) families and sulfur-containing species, and it solves the chemical reaction network as a coupled set of continuity equations by using the backward Euler method to calculate the concentration profiles of chemical species. Below the tropopause, H2O is given by the climate module, whereas above it is calculated self-consistently by the chemistry module according to its in situ atmospheric sources and sinks. The chemistry module accounts for wet deposition of chemical species as an atmospheric sink in the lower atmosphere by using solubility constants given by Giorgi and Chameides (1985). In the newly developed CAB model, CO2 is also removed via rainout by introducing an effective Henry's law constant of 3·10−2 M atm−1 (Jacob, 1999). At the model boundaries, several different boundary conditions are applied. Natural biogenic and source gas emissions of CH4, chloromethane (CH3Cl), CO, and nitrous oxide (N2O) are included at the lower boundary of the model, whereas for all other species (except O2, CO2, and N2, see below) dry deposition is applied. At the upper boundary, an effusion flux for O and CO is used. In the CAB model, the concentrations of O2, CO2, and N2 are now treated as altitude dependent, that is, similar to all other chemical species, and are calculated throughout the atmosphere according to their in situ chemical sources and sinks. For all scenarios, the surface volume mixing ratio (vmr) of these chemical species is held fixed, and their corresponding atmospheric surface fluxes
We have introduced further boundary conditions and input parameters that will be described in more detail in the following sections.
2.1.1. Volcanic and metamorphic outgassing
In addition to the already-existing volcanic production rates from SO2 and H2S in the chemistry module, we have implemented volcanic outgassing rates of the chemical species H2, CH4, CO, and CO2. The present-day fluxes of these chemical species are summarized in Table 1. Since the input of volcanic gases might have been higher in the past, for example, due to larger interior thermal gradients (Sleep and Windley, 1982; Christensen, 1985), we have introduced a heat flow parameterization, Q(t
geo), as given by Claire et al. (2006) such that the production rate from volcanic outgassing equals
Fluxes are given in teragrams (1012 g) per year.
Here
where η = 0.7 according to Sleep and Zahnle (2001). t geo = 0 represents the present day.
The additional outgassing from metamorphic processes in the crust was introduced for H2 and CH4 based on the work of Claire et al. (2006) and taking the crustal mineral redox buffer into account. In the photochemistry module, the metamorphic production rate is implemented for H2 as
and for CH4 as
Here
2.1.2. Hydrogen escape
In the photochemical module, we implemented the loss of H2 due to diffusion-limited escape at the upper model boundary by introducing an effusion flux for H2 following Walker (1977) as
where
2.1.3. Young Sun analog star β Com
The stellar input spectrum for the young Sun analog G0V-dwarf star β Com (Ribas et al., 2005) was introduced. The spectrum was derived from observations in the UV from the International Ultraviolet Explorer satellite archive (
2.2. Biogeochemical module description
The biogeochemical module applied here is based on the approach of Goldblatt et al. (2006). Therefore, we only briefly describe the main components and processes considered. Goldblatt et al. (2006) considered the atmosphere to be part of the atmosphere-ocean system and did not perform climate and photochemistry calculations. Regarding atmospheric processes, they took into account hydrogen escape and a parameterization of CH4 oxidation. All atmospheric processes in their work are treated without a temperature dependence as atmospheric temperature is not calculated. Since CH4 oxidation and hydrogen escape are already included in our 1-D global mean atmospheric column module described in Section 2.1, they are explicitly excluded from the biogeochemical modeling described here. We furthermore split the atmosphere-ocean system used by Goldblatt et al. (2006) into the atmosphere and the ocean systems. In addition to our detailed atmosphere module, described in Section 2.1, we focus on the marine environment in our biogeochemical module. This constitutes the strongest long-term source for atmospheric O2 due to the burial of organic carbon in the ocean (continental biomass contributes only on the short-term scale and, therefore, can be neglected for long-term processes).
For the biogeochemical module, we consider two reservoirs, namely, molecular oxygen ([O2]) and buried organic carbon ([Corg]). The reservoirs are quantified in units of moles. The exchange of material fluxes between theses reservoirs is given in moles per year and represents sources (F
sources) or sinks (F
sinks) for the respective reservoirs. The summation of those fluxes yields a differential equation for each reservoir according to
where [Ci ] represents the considered reservoir (or chemical species).
Several biological and nonbiological processes have an effect on the O2 reservoir, and these are described in the following text. The biological processes are summarized in Table 2. In our model, there are two types of photosynthesis—oxygenic and oxygenic whereby organic carbon is produced without the production of oxygen. Overall, the main context is as follows: Organic carbon produced from the different types of photosynthesis descends through the ocean column, and thereby different processes remove different fractions of the descending carbon that is finally incorporated into the ocean floor. The above is the theoretical foundation upon which the equations of Goldblatt et al. (2006) are based, and this can be described in more detail as follows: The NPP of the biosphere is described by the NPP from oxygenic photosynthesis, N (mol O2/yr), plus the additional input of inorganic reductants for anoxygenic photosynthesis, r (mol O2 equivalent/yr), which is represented by ferrous iron, Fe2+. Therefore, the rate of total organic carbon produced by oxygenic and anoxygenic photosynthesizers in the ocean system is (N + r) (see Table 2). From the available organic carbon, an assumed amount β = 0.002 (Betts and Holland, 1991; Prentice et al., 2001) is buried in sediments at the ocean floor, namely, β·(N + r), where β is the burial efficiency. Heterotrophic aerobic respirers then use a fraction of organic carbon remaining, namely, γ = [O2]/(d γ + [O2]) [where d γ = 3.7·1017 mol corresponding to the inhibition of aerobic respiration at the Pasteur point, ∼0.01 PAL (Engelhardt, 1974)]. Afterward, the remaining organic carbon is used by fermenters and acetotrophic methanogens that produce CH4 and CO2. From the CH4 produced, a certain fraction, namely, δ = [O2]/(d δ + [O2]), is oxidized by methanotrophs depending on the available O2 concentration where d δ = 2.7·1017 mol, which is equivalent to [O2] = 2 μM (Ren et al., 1997). M is equivalent to moles per liter. Below the critical dissolved O2 concentration of 2 μM, the rate of methane oxidation is inhibited.
Organic carbon is represented by CH2O.
In summary, Fig. 2 shows the total effect of the biosphere on the O2 concentration, which can be written as
where
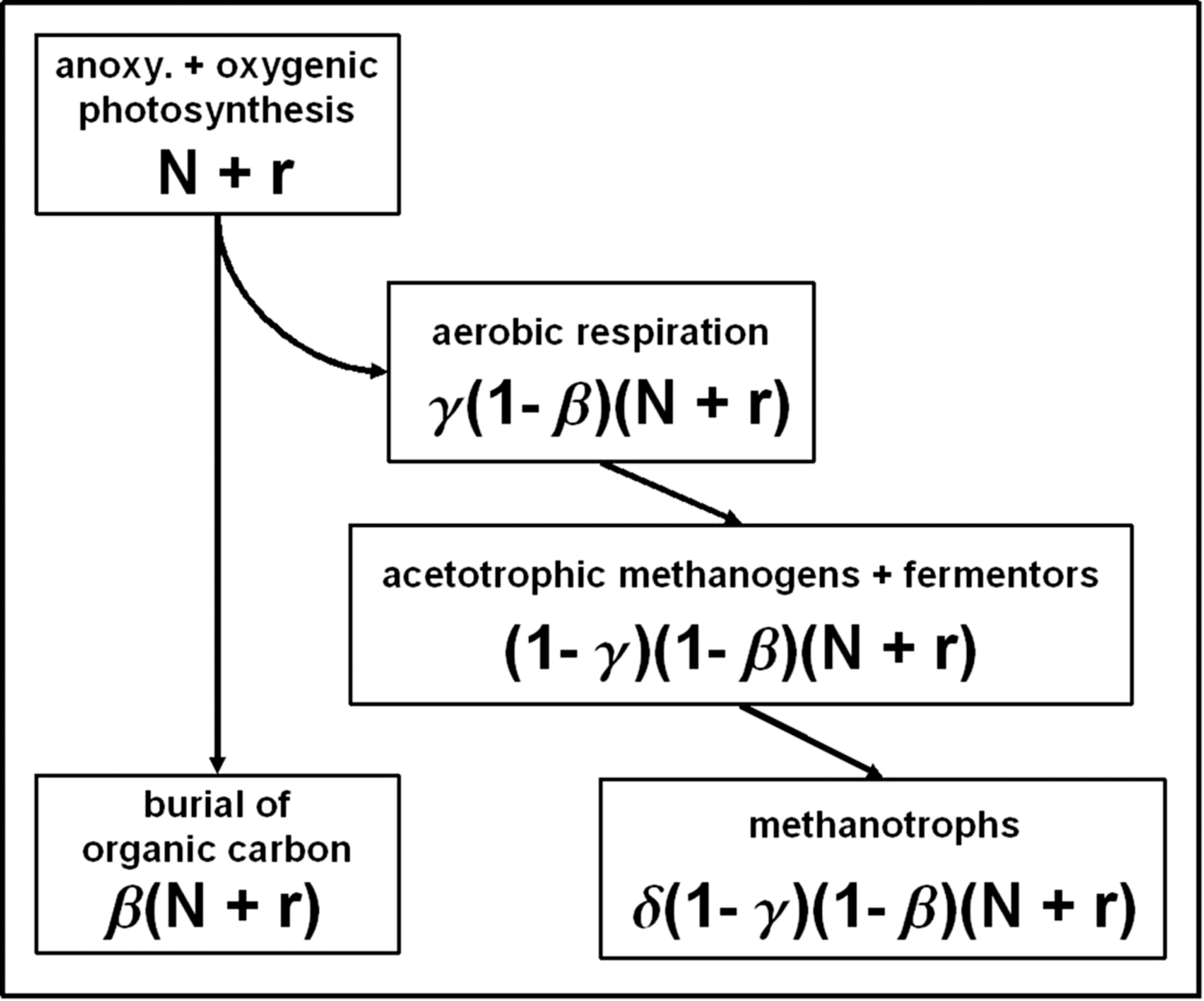
Processes in the marine biosphere after Goldblatt et al. (2006). N (mol O2/yr) is the NPP from oxygenic photosynthesis; r (mol O2 equivalent/yr) represents the input from anoxygenic photosynthesis. β is the fraction of buried organic carbon. γ resembles the fraction of aerobically respired organic carbon. δ depicts the fraction of CH4 that is oxidized by methanotrophs.
Buried organic carbon is assumed to be weathered at a rate that is determined by the rate of mechanical uplift and weathering. As in the work of Goldblatt et al. (2006), it is assumed that all organic carbon exposed by weathering is consumed by decomposers described above. The rate of weathering [Corg] is w[Corg]. The total effect of weathering of organic carbon for [O2] is given according to Goldblatt et al. (2006) by
For the evolution of the O2 reservoir in the surface ocean, Eqs. 7 and 8 are summed up to yield
In the same manner, we can find a differential equation for the buried organic carbon reservoir
If we now consider steady-state conditions for the atmosphere module, then the left-hand side of Eqs. 9 and 10 is set to zero, yielding a more simplified equation, namely,
This equation is then solved for [O2] using the Van Wijngaarden–Dekker–Brent root search method (Brent, 1971; Press et al., 1993) (also called Brent's method) yielding the number of moles of O2 depending on the input from oxygenic and anoxygenic photosynthesis N and r (see Section 2.3). This method combines linear interpolation and inverse quadratic interpolation with bisection in order to obtain the root of Eq. 11 efficiently. To derive the aqueous O2 concentration, we calculate according to Goldblatt et al. (2006)
where μ = 1.773·1020 mol is the number of moles in the present atmosphere (Goldblatt et al., 2006) and
Note that since we set Eqs. 9 and 10 to zero, the burial and weathering of buried organic carbon drop out of the calculations presented. To address these processes and their impact, however, the carbon cycle also has to be implemented, which will be part of future work.
2.3. Coupling of the atmosphere and biogeochemical module
The atmosphere and biogeochemical modules are coupled via a so-called stagnant boundary layer model (Liss and Slater, 1974; Kharecha et al., 2005). It calculates the biogeochemical O2 flux,
where v
p(O2) is the piston velocity of O2,
where
3. Pathway Analysis Program
The Pathway Analysis Program (PAP) was developed by Lehmann (2004) to automatically identify chemical pathways in arbitrary chemical reaction networks and to quantify their efficiencies by assigning rates. PAP was applied by, for example, Grenfell et al. (2006, 2013), Verronen et al. (2011), Stock et al. (2012a, 2012b), and Verronen and Lehmann (2013). The algorithm yields a list of all dominant pathways that produce, destroy, or recycle a chemical species of interest. PAP requires as input a complete list of chemical species and their concentrations, as well as their concentration changes caused by chemical reactions during a specified time interval. Furthermore, a complete list of reactions and their corresponding rates is required. Starting with individual reactions as initial pathways, longer pathways are formed step by step. For this, shorter pathways that have already been found are connected at so-called “branching point” species, whereby each pathway that forms a branching point species is connected with each pathway that destroys it. Branching point species are chosen based on increasing lifetime with respect to the pathways constructed so far. In this work, all chemical species with a chemical lifetime shorter than that of O2 are treated as branching point species. Since, in general, the chemical lifetime of a chemical species varies with altitude, the choice of branching point species adapts to the local chemical and physical conditions.
For the analysis of large and complex reaction networks, chemical pathways with a rate below a user-defined threshold rate f min are deleted to avoid long computational time. In the present study, f min = 10−13 parts per billion by volume per second (ppbv/s) was chosen, which is sufficient for finding all dominant pathways producing or consuming atmospheric O2. A PAP analysis was performed for each of the 64 vertical atmospheric column module chemistry layers. The resulting production and destruction rates of O2 from each individual pathway are integrated over the atmospheric module vertical grid and are expressed as a percentage of the total column-integrated production and destruction rate from pathways found by PAP.
4. CAB Model Validation
4.1. Atmosphere module
The CAB model was applied to modern Earth conditions to compare the modified atmosphere module presented in this work to the previous atmosphere model version discussed by Rauer et al. (2011) (denoted as RG2011), which was validated against modern Earth. Results suggest that over the calculated pressure (altitude) range a maximum relative change of the temperature profile against RG2011 between −0.5% and 0.1% is observed. The surface temperatures deviate by 0.02 K. Hence, an overall good agreement of the temperature profiles between both models was obtained.
Table 3 shows the maximum relative change in the atmosphere of relevant chemical species vmr profiles against RG2011 over altitude for O3, H2O, CH4, N2O, CH3Cl, and CO2. For O2 and N2 only small deviations are obtained. Due to the introduced hydrogen escape at the upper boundary, a maximum relative change of −90% for H2 is found (not shown) at the TOA, whereas below 40 km the change is negligible. Quite strong changes occur for sulfur-bearing species, for example, H2S (−64%), SO2 (−73%), SO (−72%) (not shown), because updated volcanic SO2 and H2S emissions were implemented, and furthermore, new volcanic emissions for H2, CO, CH4, and CO2 were introduced into the CAB model (see Table 1). Despite these changes, the biosignature species, O3 and N2O, and biosignature-related compounds, CH4 and H2O, compare very well with the previous global atmospheric column model used by RG2011, which is shown in Table 4.
4.2. Biogeochemical module
For modern Earth conditions and the present input of inorganic reductants (Fe2+) into the ocean of r = r 0 = 3·1011 mol Fe/yr (Holland, 2006), which is equal to r 0 = 7.5·1010 mol O2 equivalent/yr, the CAB model calculates a global NPP from oxygenic photosynthesis of N = 1.05 petamoles (1015 mol) O2 per year, which is of the same magnitude as the present observed value of N 0 ≈ 3.75 Pmol O2/yr (Prentice et al., 2001). The range of observed oceanic NPP varies from 2.067 to 4.167 Pmol/yr (Woodwell et al., 1978; Longhurst et al., 1995; Antoine et al., 1996), hence the lower value matches our model value within a factor of 2. The burial rate of organic carbon, β(N + r) (see Fig. 2), calculated by the CAB model is 2.1 Tmol/yr. This value is in the range of the observed value of 10 ± 3.3 Tmol/yr (Holland, 1978). Global mean burial rates are not well known, being based on measurements at individual sites. It is particularly challenging to estimate associated uncertainties; for example, the proxy data is severely limited in time and space and, hence, difficult to estimate in a global model.
5. Application to Early Earth Analog Atmospheres
The CAB model is applied to early Earth analog atmosphere scenarios before, during, and after the GOE at about 2.3 billion years ago. For all runs belonging to the control scenario, we assume
• solar input spectrum (Gueymard, 2004);
• surface pressure p surf = 1 bar (as suggested by, e.g., Som et al., 2012);
• gravity acceleration g = 981 cm s−2;
• biogenic surface fluxes of CH4 (474 Tg/yr), CO (1796 Tg/yr), N2O (13.5 Tg/yr), CH3Cl (3.4 Tg/yr) (these values are set to modern Earth conditions due to missing data), H2 deposition velocity (v dep = 2.6·10−3 cm s−1), and deposition velocities of other chemical compounds as in RG2011;
• crustal mineral redox buffer is set to QFM (
• surface vmr of CO2 of 355 ppm [e.g., Rosing et al. (2010) suggested only a moderate CO2 increase of up to 3 PAL CO2].
We use the evolutionary path of O2 by Catling and Claire (2005) as an input parameter for the O2 surface vmr (see Table 5), which is shown in Fig. 3 as a thick dashed line. It is constrained from upper and lower biogeochemical limits [for further details, see the work of Catling and Claire (2005)]. Dotted horizontal lines with upper bounds (downward arrows) and lower bounds (upward arrows) show biogeochemical constraints on the O2 partial pressure (

A possible evolutionary path of O2 in the atmosphere (thick dashed line) that satisfies biogeochemical data. For further details, see text. Data are taken and modified from Catling and Claire (2005).
The following boundary conditions are additionally varied according to the geological time t
geo: • volcanic emissions of H2, H2S, SO2, CO, CH4, and CO2 and metamorphic emissions of H2 and CH4 (following Section 2.1.1).
6. Results
6.1. Atmosphere modeling
6.1.1. Climate responses
Figure 4 shows the temperature profiles of a selected number of calculated early Earth analog atmosphere runs in comparison to the modern Earth profile calculated by the CAB model. For the modern Earth p-T profile (t geo = 0 Gyr, solid black), the temperature decreases with decreasing pressure from T surf = 288 K at the surface to about T ≈ 212 K at the tropopause at p ≈ 0.1 bar. The distinct temperature maximum at the stratopause is caused by radiative heating due to the absorption of UV radiation by O3. At the stratopause with a pressure of p ≈ 10−3 bar, a maximum temperature of T ≈ 262 K is reached.
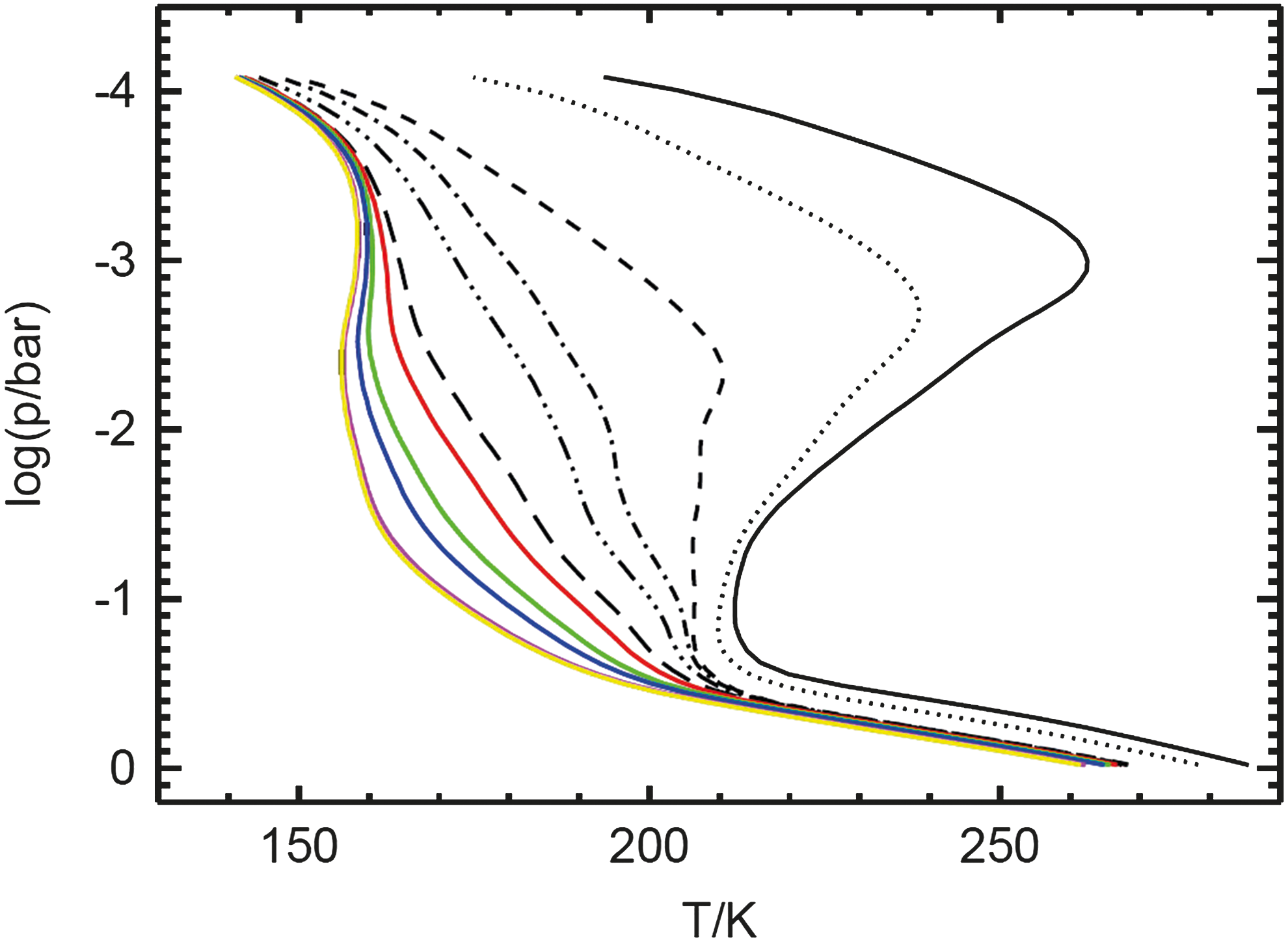
Selected p-T profiles of early Earth analog atmosphere control scenario runs calculated by the CAB model. Notation is as follows: solid black: 1 PAL O2 (modern Earth), dotted: 2.5·10−1 PAL (t
geo = 0.82 Gyr), short dashed: 10−1 PAL (t
geo = 2.18 Gyr), dash-dot: 2.5·10−2 PAL (t
geo = 2.22 Gyr), dash-dot-dot-dot: 10−2 PAL (t
geo = 2.22 Gyr), long dashed: 2.5·10−3 PAL (t
geo = 2.23 Gyr), red: 10−3 PAL (t
geo = 2.24 Gyr), green: 2.5·10−4 PAL (t
geo = 2.25 Gyr), blue: 10−4 PAL (t
geo = 2.28 Gyr), magenta: 10−5 PAL (t
geo = 2.59 Gyr), yellow: 10−6 PAL (t
geo = 2.69 Gyr). (Color graphics available at
Going forward in geological time t geo, the O2 content, thereby also O3, and the solar radiation input at the TOA increase. For the 10−6 PAL O2 atmosphere run at t geo = 2.689 Gyr ago, the solar flux incident at the TOA was decreased to about 81% of the modern Earth value (“faint young Sun”). Furthermore, in the low O2 atmosphere the incoming radiation is less absorbed by molecules such as O3 (total column: 15.8% of downward shortwave radiation is absorbed compared to 25.5% for modern Earth) due to smaller atmospheric chemical abundances of O3 (see Fig. 7) and other greenhouse gases. A diminishing of the temperature inversion is observed for the low O2 atmosphere runs as a result of the low O3 concentrations (see Fig. 7). Altogether, this leads to a increase in surface temperature (see Fig. 5) by 24 K (2.689 Gyr ago at 10−6 PAL O2: T surf = 264 K, today at 1 PAL O2: T surf = 288 K).

Surface temperature for calculated Earth-in-time atmosphere runs. Additionally indicated as a horizontal line is the freezing temperature of H2O of T = 273.15 K. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
Note that in the case of the low O2 atmosphere runs before 1.85 Gyr ago, global surface temperatures are below 0°C, implying no habitable surface conditions. Results in Fig. 4 are only applicable for modern Earth CO2 concentrations since we have kept CO2 at modern Earth concentrations in order to analyze the atmospheric responses on reducing surface O2 concentrations firstly. However, see also sensitivity run G in Section 7 with 5 PAL CO2. Further note, however, that it was recently shown by 3-D general circulation models used to investigate early Earth—for example, in the works of Wolf and Toon (2013) and Kunze et al. (2014)—that, even with global mean surface temperatures below the freezing point of water, areas with liquid surface ocean water could still exist, which implies habitable conditions.
Since the thermal radiation scheme RRTM is only valid for atmospheres that are quite close to modern Earth conditions (see, e.g., Segura et al., 2003; von Paris et al., 2008; Rauer et al., 2011), we compared the thermal fluxes of RRTM with thermal fluxes computed by the line-by-line radiative transfer code SQuIRRL (Schwarzschild Quadrature InfraRed Radiation Line-by-line, Schreier and Schimpf, 2001; Schreier and Böttger, 2003). For all runs considered, the maximum relative change of net TOA fluxes calculated by RRTM versus SQuIRRL amounted to a maximum value of −1.4%.
6.1.2. Photochemistry responses
UV environment
To a large extent, atmospheric chemistry is driven by the incoming TOA UV fluxes. To understand the absorbing nature of the atmosphere, UVA (315–400 nm), UVB (280–315 nm) and UVC (176–280 nm) radiation was calculated at the surface for reduced O2 concentration and incoming TOA solar flux. UVA and UVB wavelength ranges are taken from the International Standard (ISO 21348) 2007. Note that in the photochemistry module the term “surface” denotes the lowermost layer at a height of about 500 m for modern Earth. In the case of the 10−6 PAL O2 atmosphere, the lowermost layer sinks to a height of z 1 = 375 m. UVA radiation at the surface stays more or less constant for atmospheres before 2.18 Gyr. It increases from about 75 W m−2 at t geo = 2.18 Gyr ago (0.1 PAL O2) to about 90 W m−2 for modern Earth. For the considered runs, a steady decrease from more than 12 W m−2 to 2.3 W m−2 is observed for surface UVB radiation before the GOE and after the GOE, respectively. The modern Earth control run exhibits a surface UVB radiation of 2.3 W m−2, which is broadly similar to the observed value for cloud-free conditions of 1.4 W m−2 (Wang et al., 2000). Going forward in geological time, UVC radiation decreases from 2.8 W m−2 before 2.7 Gyr to virtually zero t geo = 2.25 Gyr ago. This behavior is due to increasing O2 and, hence, O3 and H2O concentrations that result in more absorption of photons in the atmosphere and more shielding of the surface from UV radiation. The impact on UVC is directly correlated with the increase in O3, whereas UVB is affected by O3 and H2O together.
Figure 6 shows the ratio surface/TOA radiation, R UV, as a measure of the radiation shielding efficiency of an atmosphere for UVA, UVB, and UVC radiation on going forward in geological time. For high R UV values, the radiation passes efficiently through the atmosphere, whereas for R UV = 0 it is totally blocked. UVA radiation passes efficiently through the atmosphere for all calculated runs. With the beginning of the GOE, UVB radiation is blocked rather strongly by the atmosphere, whereas UVC radiation is already starting to be blocked 2.6 Gyr ago. After the GOE, the atmosphere is totally opaque for UVC radiation. Possible negative impacts on organisms in the marine environment before the GOE might have been reduced due to the Urey effect (Urey, 1959), where a small quantity of O2 produced during photosynthesis of H2O absorbs a considerable amount of UV radiation. For modern Earth, UVB radiation is almost totally blocked by the atmosphere.
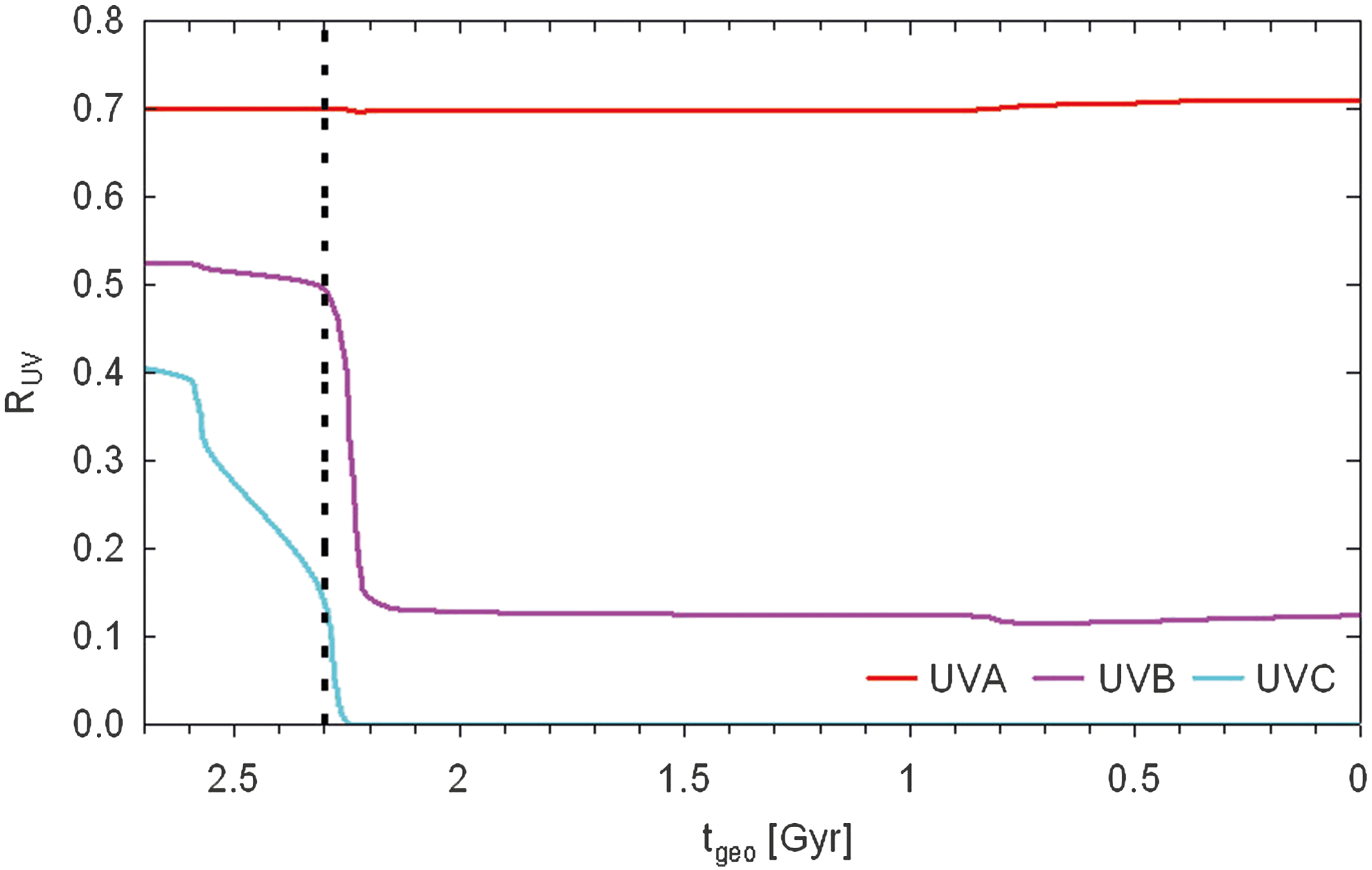
Ratio (surface/TOA) radiation fluxes, R UV, for UVA (red), UVB (magenta), and UVC (cyan) as a function of geological time t geo. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
In the following photochemical analysis, we only focus on the impact on important atmospheric constituents such as biosignatures (O3, N2O, O2) and related bioindicators (CH4, H2O) as well as CO2. “Bioindicators” are indicative of biological processes but can also be produced abiotically.
Ozone—O3
O3 is suggested to be a biosignature because it is formed in the stratosphere mainly from molecular oxygen 5 via the Chapman mechanism (Chapman, 1930), which is initiated by the photolysis of mostly biogenic O2. In the troposphere, O3 is produced via the smog mechanism (Haagen-Smit, 1952), which requires O2, volatile organic compounds, nitrogen oxides, and UV radiation. The destruction of O3 in the stratosphere proceeds via catalytic cycles that involve, for example, HO x , NO x , or ClO x (e.g., Bates and Nicolet, 1950; Crutzen, 1970), which are stored in reservoir species and can be activated by changes in, for example, temperature or UV radiation. In the troposphere, O3 is removed via wet/dry deposition, photolysis, or reactions with NO x , HO x , and unsaturated hydrocarbons.
Figure 7 shows the O3 vmr profiles for reduced O2 content of the atmosphere. Modern Earth's O3 layer (solid black) exhibits a distinct maximum at about 0.01 bar due to the mechanisms described above. Figure 7 suggests that, for reduced O2 concentrations, the O3 layer moves to higher pressures (lower altitudes), for example, for 10−5 PAL to 0.2 bar, because in consequence of a lower O2 concentration, less O3 is produced at the same altitude. This results in increased UV radiation in atmospheric layers directly below, which stimulates the Chapman mechanism and produces more O3; hence the peak of the O3 layer moves to lower altitudes. However, if O2 concentrations fall below those of CO2 ( = 3.6·10−4, left of the green line), results are consistent with CO2 photolysis now being the major source for atomic oxygen (O) and, hence, O3. The O3 peak moves upward where CO2 is photolyzed more easily. Additionally, in the upper atmosphere a second O3 maximum becomes visible, which is related to an increase in O2 vmr toward lower pressures (see Fig. 16). Note that for modern Earth this secondary O3 maximum lies in the vicinity of the mesopause (see e.g., Evans et al., 1968; Hays and Roble, 1973; Smith and Marsh, 2005) and arises due to the interplay between active-hydrogen and active-oxygen chemistry, relatively low temperatures, and a local maximum of O (Allen et al., 1984; Smith and Marsh, 2005).

O3 profiles of Earth-in-time atmosphere runs calculated by the CAB model. Notation as in Fig. 4.
The change in overall O3 column amount on going forward in geological time is shown in Fig. 8. It increases from zero before the GOE to 296 Dobson units (DU) after the GOE. At about 0.8 Gyr ago, a Second Oxidation Event occurred, resulting in an increase of the O3 column amount to 325 DU. Afterward, it decreases to the modern Earth value of 305 DU. To illustrate this decrease in O3, we have included Fig. 9, in which it can be seen that the troposphere becomes damper (going forward in time) due to evaporation because of increasing surface temperatures. However, in the stratosphere this is not always the case (compare the solid black line with the short-dashed and dotted line). A dryer stratosphere leads to less HO x and, hence, enhanced NO x , resulting in an increase in catalytic O3 loss. The dryer stratosphere is a result of a decrease in CH4 (see Fig. 13) that is driven by OH, which has a complex photochemistry. Berkner and Marshall (1965) estimated that effective UV shielding would be provided by a column amount of 200 DU, which is reached in this work at t geo = 2.22 Gyr ago (0.025 PAL O2), whereas Ratner and Walker (1972) adopted a more stringent lower limit on the O3 column amount of 259 DU [reached in this work at t geo = 2.19 Gyr ago (0.1 PAL O2)].
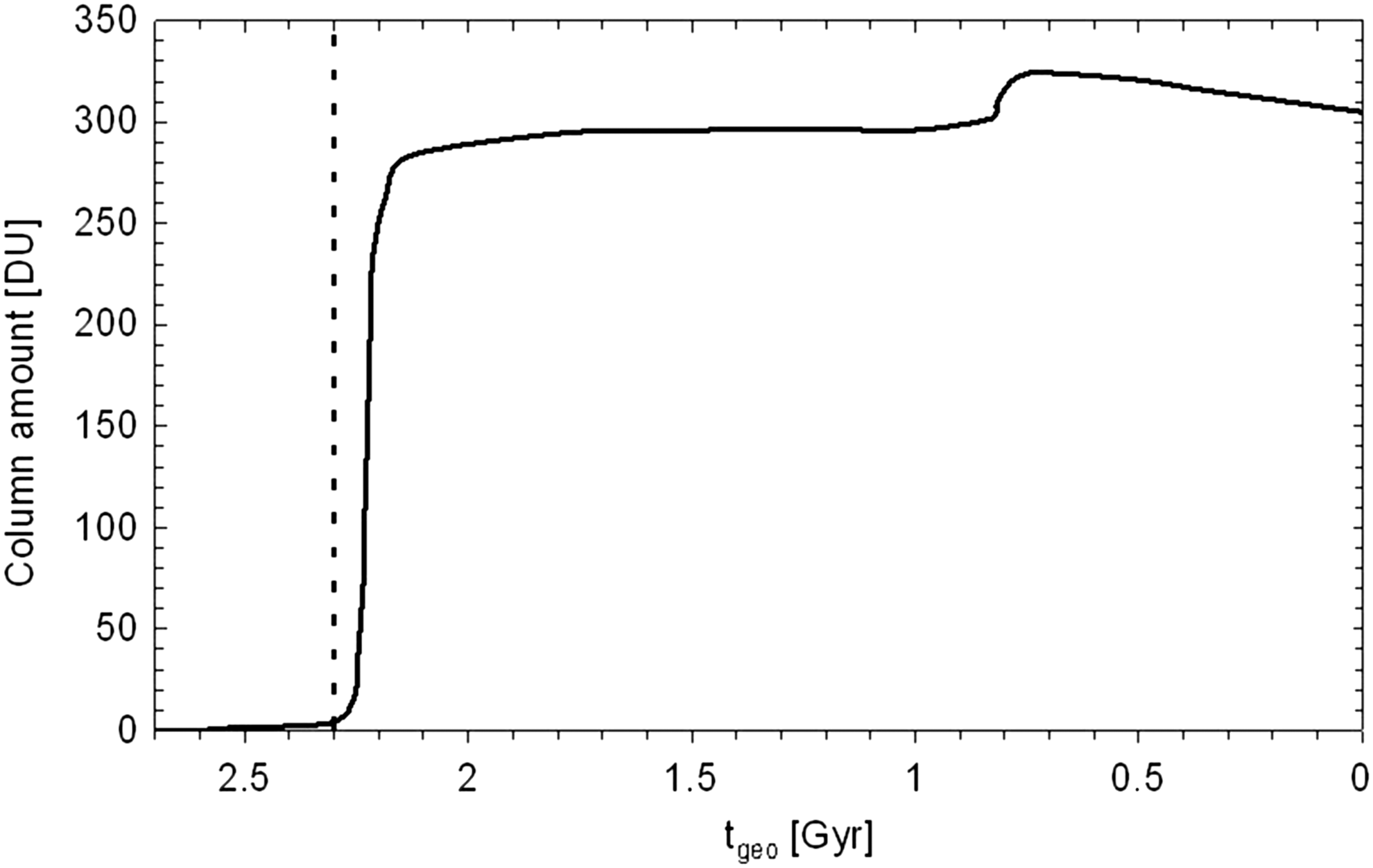
O3 column (DU) amount as a function of geological time t geo for considered Earth-in-time atmosphere runs. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
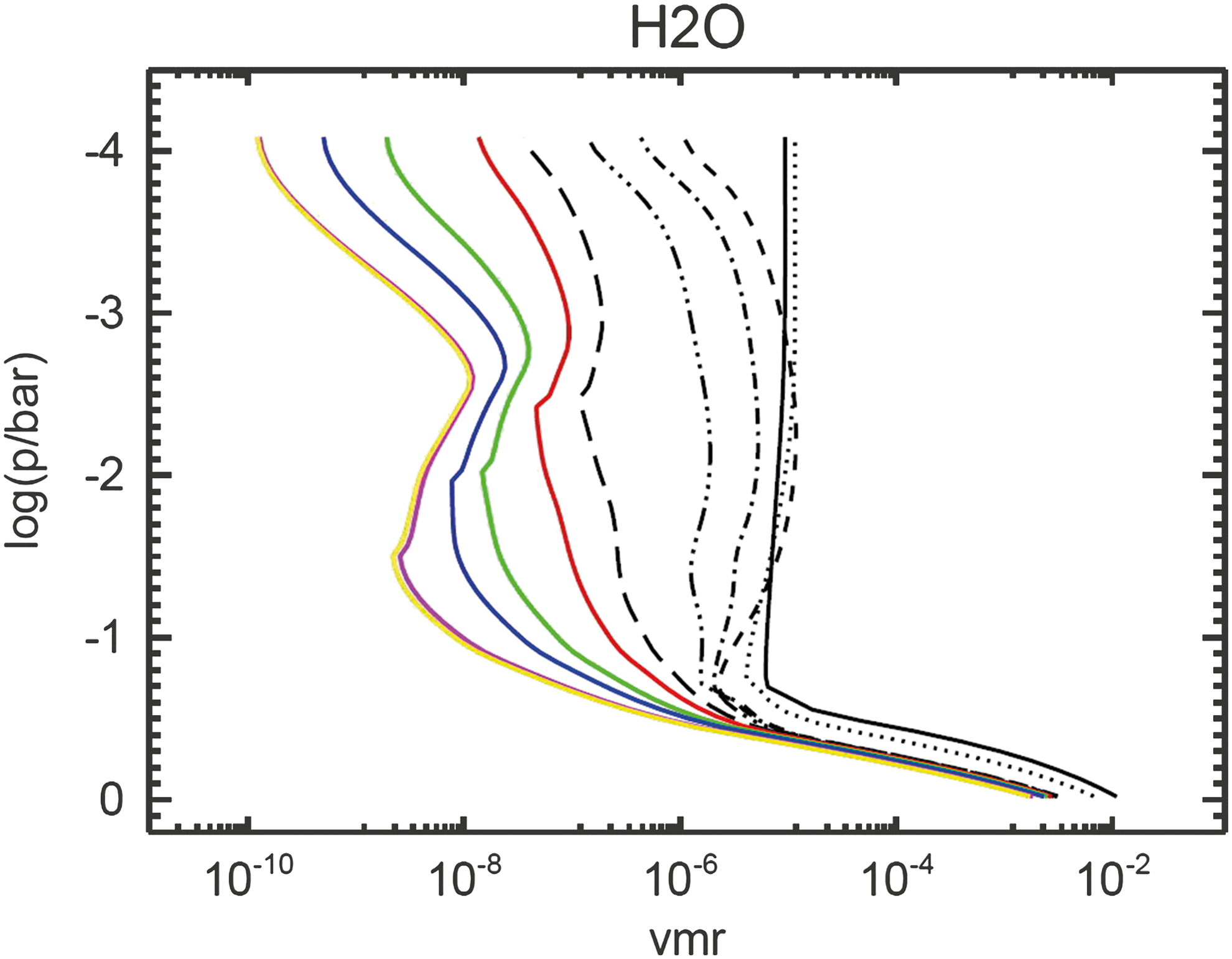
H2O profiles of Earth-in-time atmosphere runs calculated by the CAB model. Notation as in Fig. 4.
Water—H2O
In the case of modern Earth, the water vapor vmr in the troposphere is chemically inert and subject to the hydrological cycle. Above the tropopause, however, the H2O vapor concentration is determined by its chemical sources (here, CH4 oxidation) and sinks as well as transport from the troposphere and approaches an isoprofile for modern Earth.
Figure 9 shows the H2O vapor vmr profiles on decreasing ground-level O2 concentrations on going back in time. The solar flux incident at the TOA decreases, which results in a decrease in surface temperature and, hence, condensation of H2O in the troposphere. The atmosphere becomes dryer due to decreasing surface temperatures. The stratospheric H2O profile deviates from the isoprofile (found for modern Earth), and a distinct maximum develops in the early Earth analog runs. In this region, H2O is primarily produced from O2 (which increases in this region—see later) via the net reaction CH4 + O2 → H2O + H2 + CO, which is initiated by the photolysis of CH4. Furthermore, H2O is predominantly destroyed via photolysis in the low O2 atmosphere due to enhanced UV radiation resulting in the formation of H2 and CO2.
The overall H2O column amount increases on going forward in geological time as shown in Fig. 10, which can be attributed directly to the temperature behavior shown in Fig. 5. There is the caveat, however, that there could be deviations in our temperature behavior in comparison to the geological record.
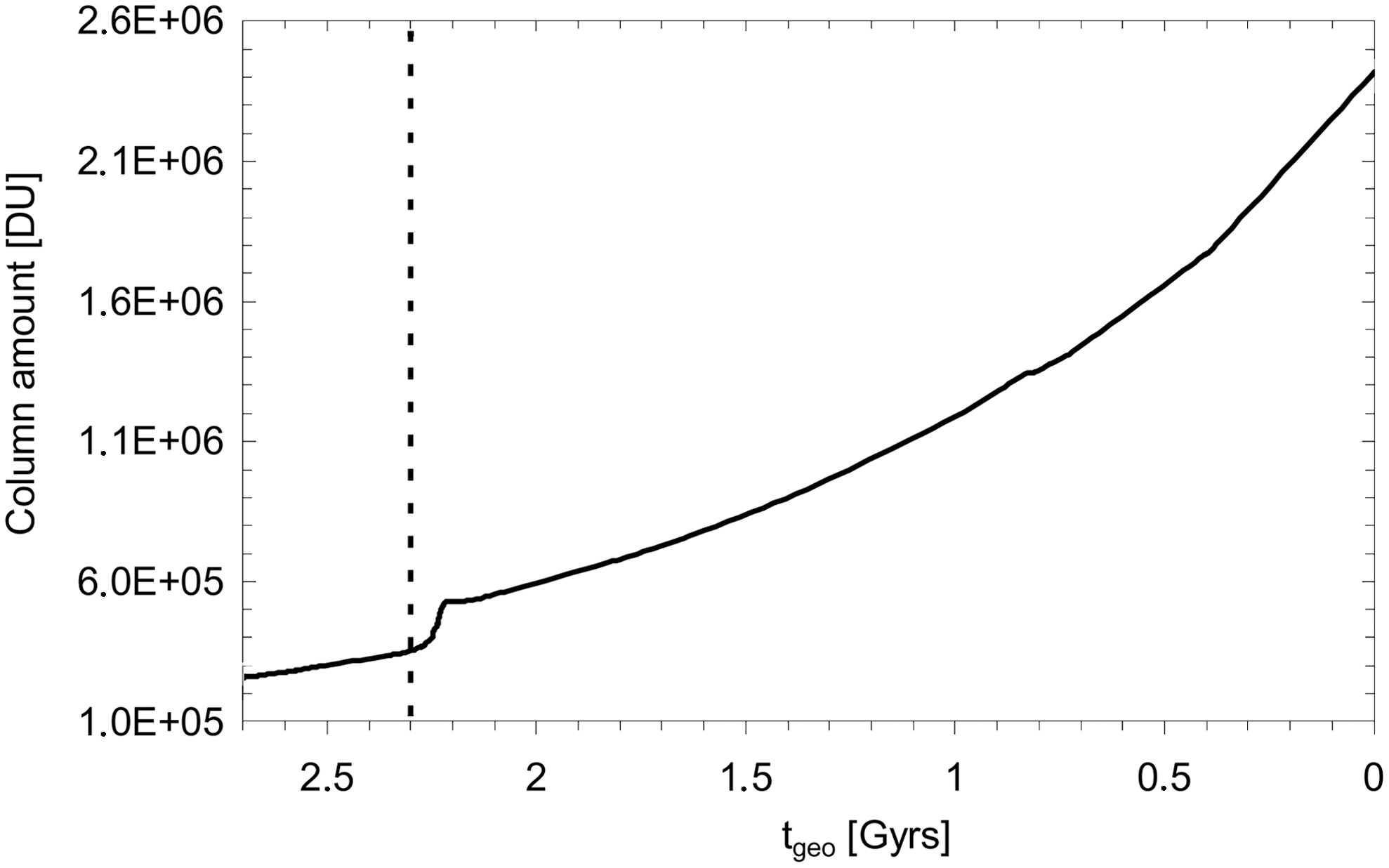
H2O column amount (DU) as a function of geological time t geo for Earth-in-time atmosphere runs considered. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
Nitrous oxide—N2O
N2O is an important biosignature. For modern Earth, it is almost exclusively produced by bacteria as part of the (de)nitrifying cycle (IPCC, 2001). It is destroyed mainly in the stratosphere via photolysis or via the reaction with excited oxygen (O(1D)).
Figure 11 shows the N2O vmr profiles for decreased ground-level O2 concentrations. N2O destruction via photolysis becomes more efficient in the atmosphere due to increased UV radiation for atmospheres with lower O2 surface vmrs. This results in decreased N2O concentrations and, hence, lower column amounts (shown in Fig. 12) in the geological past. In situ inorganic production of N2O is negligibly small.
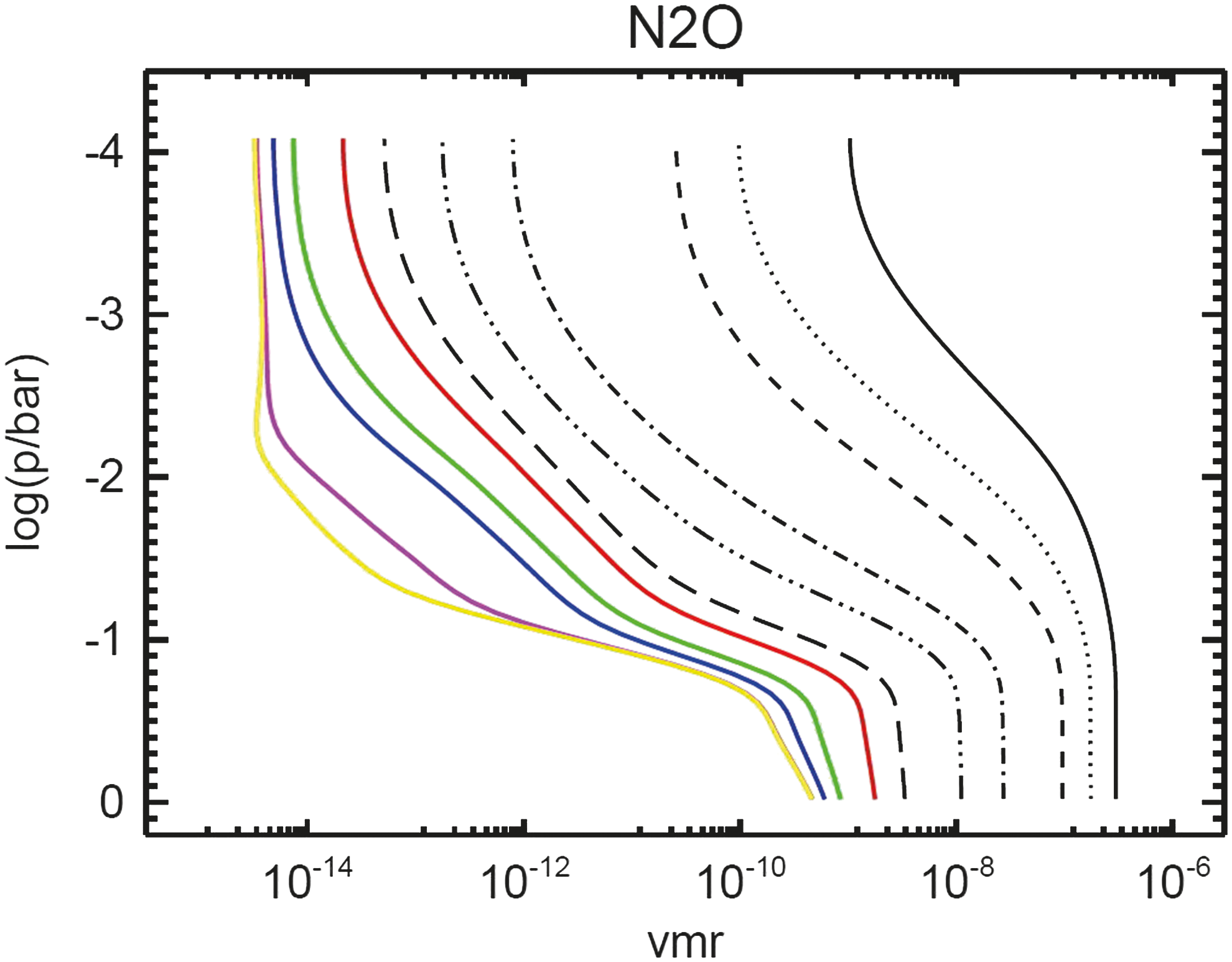
N2O profiles of Earth-in-time atmosphere runs calculated by the CAB model. Notation as in Fig. 4.

N2O column amount (DU) as a function of geological time t geo for Earth-in-time atmosphere runs considered. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
Methane—CH4
Atmospheric CH4 is a bioindicator since in addition to biogenic sources some geological sources exist. CH4 is destroyed in the atmosphere mainly by the reaction with hydroxyl (OH) and in the upper stratosphere by photolysis.
Figure 13 shows the CH4 profiles for reduced ground-level O2 concentration. Note, however, that we fixed the biological surface flux for all scenarios considered. But see also sensitivity run H in Section 7, which considers a doubled flux. For increasing O2 concentrations, CH4 increases as well, reaches a maximum at 0.1 PAL O2, and then decreases toward higher O2 values. The corresponding column amounts are given in Fig. 14. The maximum in CH4 column amount at t geo = 2.18 Gyr (0.1 PAL O2 vmr) is directly linked with a minimum concentration of OH and, hence, minimum destruction of CH4 at the same geological time. For modern Earth, standard OH sources are, for example, H2O + O(1D) → OH + OH [where O(1D) arises mainly from O3 photolysis]; and sinks are, for example, CO + OH → CO2 + H and OH + HO2 → H2O + O2. Note that the first sink reaction allegedly destroys OH. Most of the H atoms formed react quickly via the reaction H + O2 + M → HO2 + M followed by, for example, HO2 + O3 → OH + 2O2, which reforms OH; thus the reaction between CO and OH does not lead to an efficient OH loss. The second loss reaction is a more permanent sink for OH. The main source of OH switches to H2O + hν → H + OH for the low O2 atmospheric run due to higher UV.

CH4 profiles of Earth-in-time atmosphere runs calculated by the CAB model. Notation as in Fig. 4.
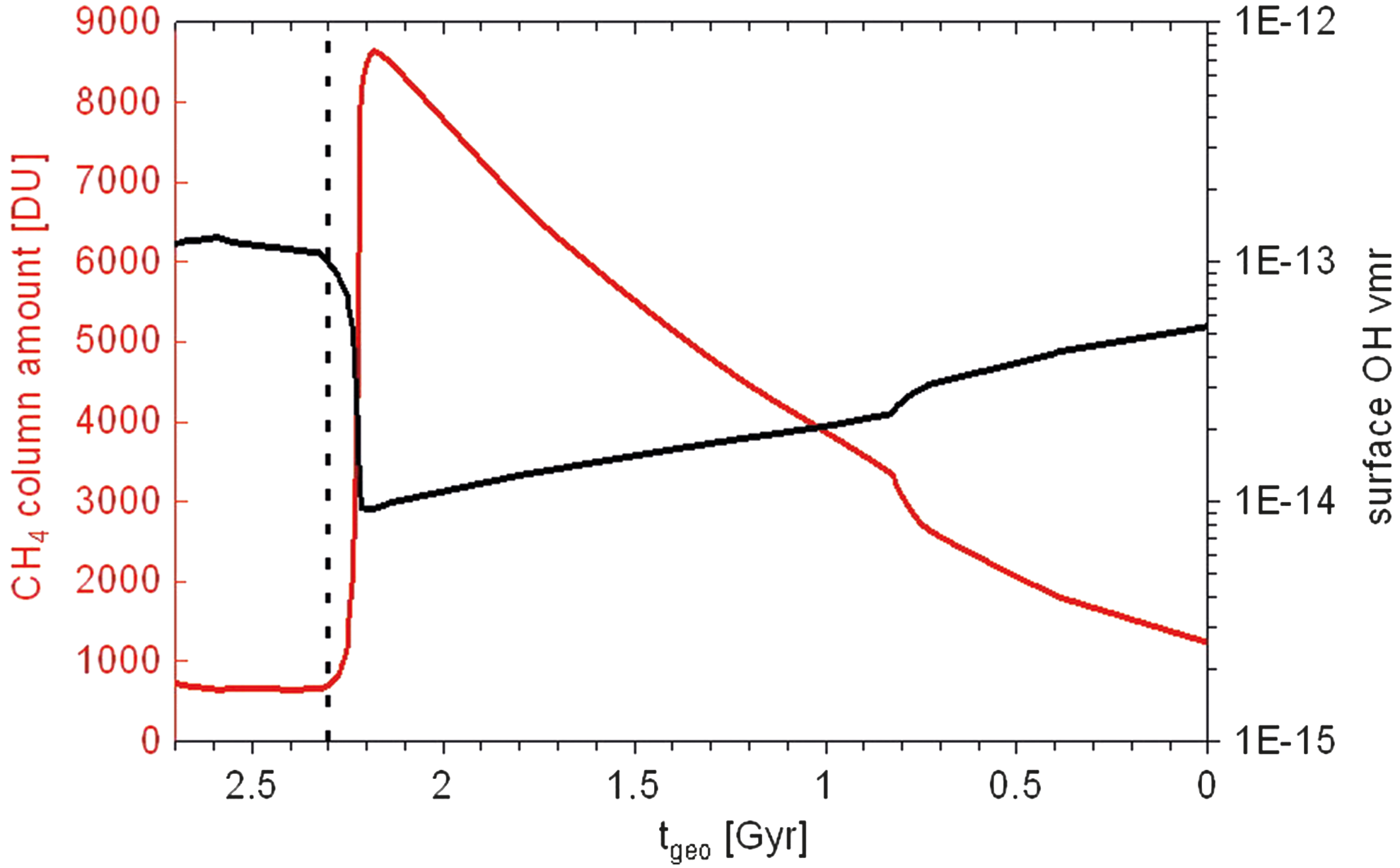
CH4 column amount (DU) (red) and surface OH vmr (black) as a function of geological time t geo for Earth-in-time atmosphere runs considered. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
Figure 14 illustrates an important result of our work; that is, we calculate an increase in CH4 concentration with increasing O2 vmrs at the GOE. Previous works (e.g., Claire et al., 2006; Zahnle et al., 2006) with much simpler chemical schemes, however, suggest the opposite effect, namely, that increasing O2 (more oxidizing conditions) would lead to stronger CH4 oxidation (into CO2 and H2O) and, hence, a reduction in CH4. This behavior is hypothesized by the authors to lead to a snowball Earth. CH4 oxidation is a complex multireaction process that usually begins by attacking OH on CH4. OH is photolytically produced, which (generally) means high OH abundances in high-UV atmospheres, all else being equal. In our study, an increase in O2 leads to an increase in O3, which blocks UV. This leads to less OH and, hence, more CH4. Our work suggests that more investigations are required in the chemical feedbacks between CH4 and O2 on early Earth. After the GOE, O2 further increases, and CH4 is more and more destroyed via the reaction with OH, which then increases because of higher production via, for example, H2O + O(1D) → 2OH (since H2O increases as discussed). Note that in our study we assume a constant biogenic input of CH4 of 474 Tg/yr at the surface for every run considered. This value could be rather weak for early Earth (see Claire et al., 2006). Also, due to the toxic nature of O2 to CH4-producing microorganisms, the rise in O2 likely had a severe impact on the biological activity and produced atmospheric CH4, therefore resulting in a dramatic decrease in biotic CH4 production. This mechanism is not included in the CAB model and is beyond the scope of this article.
Carbon dioxide—CO2
CO2 is an important greenhouse gas that is generally well mixed in the modern Earth atmosphere.
Figure 15 shows how the CO2 vmr profiles change on decreasing the ground-level O2 concentrations. For decreases down to 0.1 PAL O2, CO2 increases in the stratosphere mainly due to the HO x catalyzed net reaction O3 + 3CO → 3CO2, which is initialized by the catalyzed photolysis of O3 and O2. For further decreases in O2, stronger photolysis of CO2 leads to decreased vmrs in the stratosphere. This implies that CO2 does not exhibit an isoprofile throughout the atmosphere for low O2 concentrations. In the case of 10−6 PAL O2, the TOA vmr of CO2 is 334 ppm in comparison to the surface vmr of 355 ppm. There is a caveat, however: CO2 profiles could depend on the rather unconstrained value of the eddy diffusion profile K(z).

CO2 profiles of Earth-in-time atmosphere runs calculated by the CAB model. Notation as in Fig. 4.
Molecular oxygen—O2
Figure 16 shows that O2 exhibits an isoprofile behavior for atmosphere runs with ground-level O2 concentrations above 10−4 PAL O2, which implies that its concentration is dominated by transport processes rather than chemical production (P) and destruction (L). For the atmosphere runs with surface O2 concentrations below 10−4 PAL O2, an anticorrelation between the vertical CO2 and O2 profiles becomes visible, implying that the destruction of CO2 provides a source of atmospheric O2 in the upper stratosphere. A detailed chemical analysis will be presented in Section 6.1.3 to gain detailed insight into the rather complex production (and destruction) processes for O2.
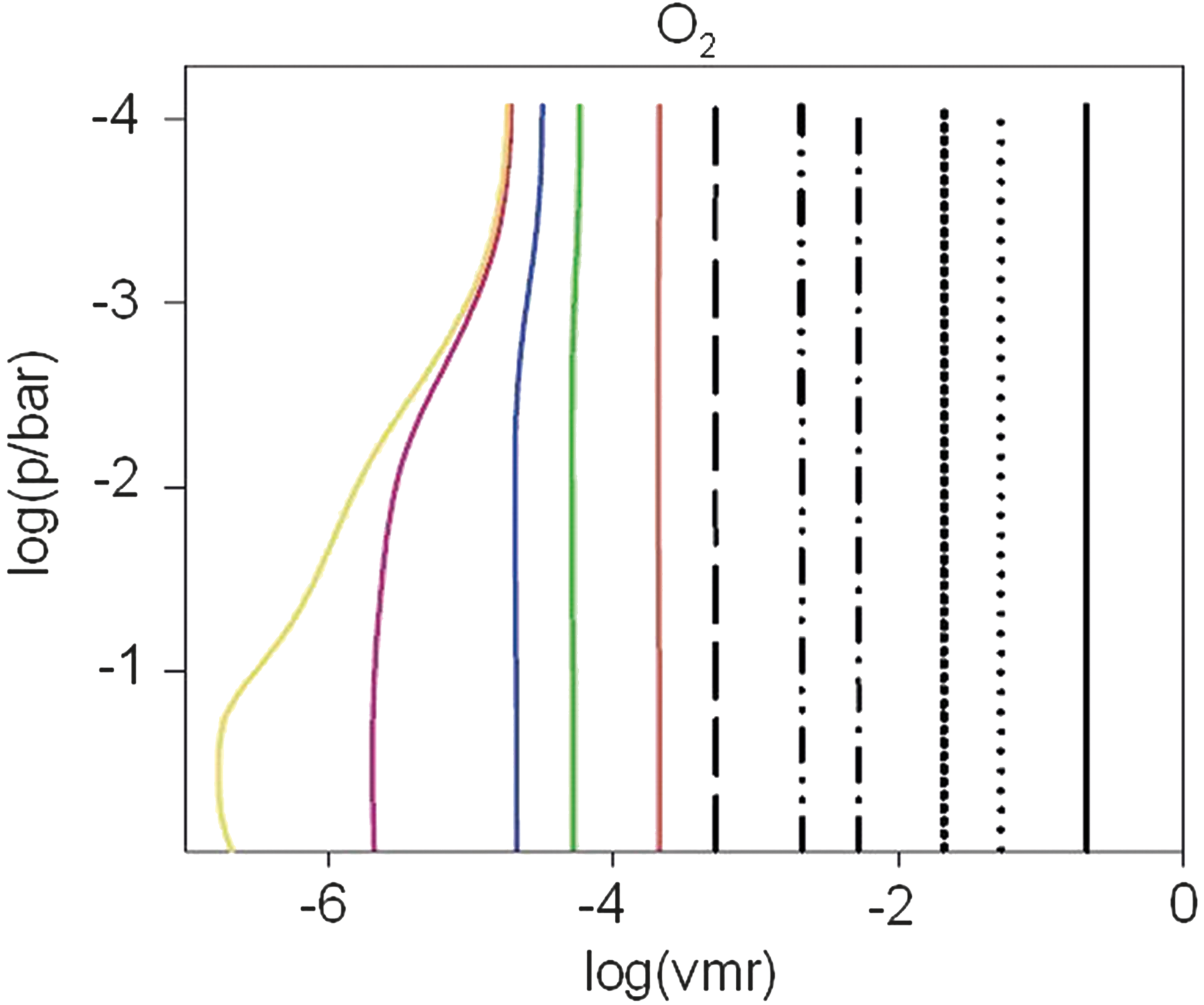
O2 profiles of Earth-in-time atmosphere runs calculated by the CAB model. Notation as in Fig. 4.
Figure 17 compares the net chemical change
of i = O2 in the atmosphere for modern Earth and low O2 atmospheres. Thereby, ni is the number density of the chemical compound i. For modern Earth, O2 is only produced immediately below and above the O3 layer maximum in the stratosphere. In general, O2 is mostly destroyed in the atmosphere except for the lowermost atmospheric layers of atmospheres with ground-level concentrations between 10−2 and 10−3 PAL. For very low O2 atmospheres, there is a net production at the uppermost layers, which is a result of increased CO2 photolysis as mentioned above.

Atmospheric net (P − L) chemical change of O2 calculated by the CAB model for modern Earth and low O2 atmospheres. Notation is as follows: solid red: 1 PAL O2 (modern Earth), dashed green: 10−1 PAL (t
geo = 2.18 Gyr), dotted dark blue: 10−2 PAL (t
geo = 2.22 Gyr), dotted purple: 10−3 PAL (t
geo = 2.24 Gyr), dot-dashed cyan: 10−4 PAL (t
geo = 2.28 Gyr), dot-dashed yellow: 10−5 PAL (t
geo = 2.59 Gyr), dot-dot black: 10−6 PAL (t
geo = 2.69 Gyr). (Color graphics available at
Figure 18 depicts the net column-integrated (global mean) (P − L) chemical change of O2 calculated by the CAB model on going forward in geological time. Additionally, in the upper right of Fig. 18 the column-integrated production and destruction rates of O2 are shown for comparison. This illustrates that O2 is generally destroyed within the atmosphere for all runs considered. Hence, this indicates that a positive O2 flux of the same amount into the atmosphere is required in order to achieve steady state and maintain O2 levels above zero. It is striking that, although the surface O2 concentration varies by several orders of magnitude, the associated O2 surface flux,
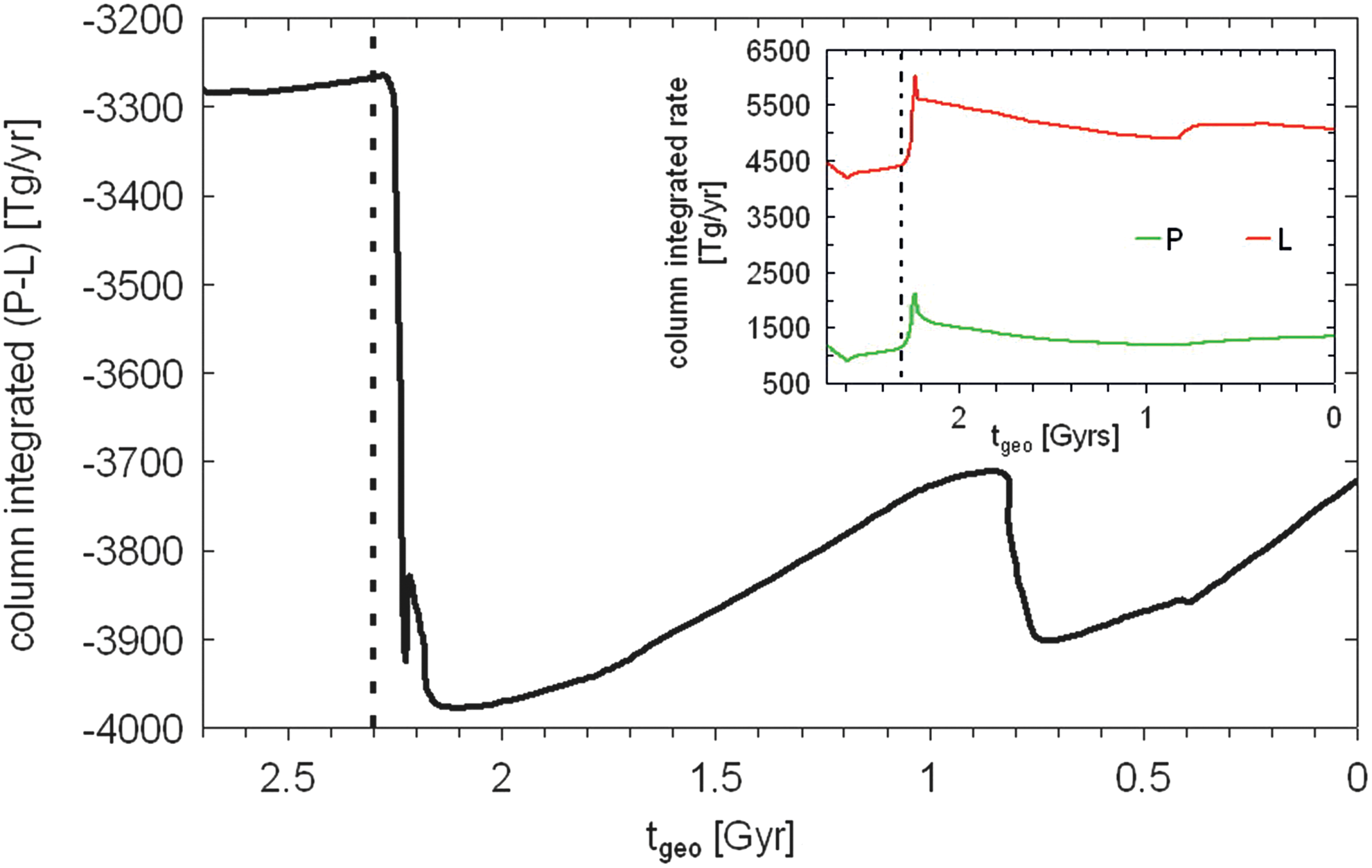
Column-integrated net (P − L) chemical change of atmospheric O2 calculated by the CAB model as a function of geological time t
geo for Earth-in-time atmosphere runs considered. The beginning of the GOE at t
geo = 2.3 Gyr is indicated as a vertical dashed line. In the upper right panel the column-integrated P (green) and L (red) rates are given as a function of geological time t
geo for comparison. (Color graphics available at
Comparison to previous work
Generally, it is not easy to compare results with those of previous works in the literature regarding early Earth since either, for example, only stand-alone photochemical models (i.e., without coupled climate calculations) have been presented (e.g., Kasting and Donahue, 1980; Kasting, 1982; Kasting et al., 1985; Zahnle et al., 2006) or coupled climate-chemistry calculations performed using an isoprofile assumption for O2, CO2, and N2 (e.g., Segura et al., 2003 6 ). Furthermore, previous workers have assumed increased CO2 vmrs (see, e.g., Kasting et al., 1984) and CH4 surface fluxes (see e.g., Pavlov et al., 2003) to be present in the early Earth atmosphere.
Some key chemical reactions resulting in the production and destruction of O2 have, however, also been identified, for example, in Kasting et al. (1984), although calculated O2 profiles differ from this work. Focusing on atmospheric O3, Fig. 19 shows a comparison of the O3 column depth calculated by this study with those of Segura et al. (2003) and Kasting and Donahue (1980). To compare with the work of Segura et al. (2003), additional runs were performed with the CAB model (“Segura comp.” shown in dark green) with the initial atmospheric conditions as in the Segura et al. (2003) study. A further important result of this work is shown in Fig. 19, where it can be seen that, due to our atmosphere model improvements, we calculate a consistently enhanced O3 column amount compared to previous works in the literature. This difference calculated in O3 occurs, for example, due to an increased photolytic destruction of CO2 in the runs of this work leading to production of O2 and, hence, O3. Comparing results of the Segura comp. runs with the results of Segura et al. (2003) suggests, for example, a maximum relative change in the O3 column amount of 82% at t geo = 2.28 Gyr (10−4 PAL O2). For H2O, N2O, and CH4, qualitatively similar results were found.

Column amount of atmospheric O3 in DU as a function of geological time t
geo for Earth-in-time atmosphere runs considered in comparison to previous work. The beginning of the GOE at t
geo = 2.3 Gyr is indicated as a vertical dashed line. (Color graphics available at
6.1.3. Pathway analysis with respect to O2
For the early Earth analog runs, model calculations generally predict an increase of the O2 vmr with increasing atmospheric height, which implies an in situ atmospheric source of O2 (see Fig. 16). Furthermore, it can be seen that the CO2 profile is anticorrelated with the O2 profile in the low O2 Earth-in-time atmosphere runs (see Fig. 15). This might imply that CO2 serves as a source species for O2. Our goal is to identify how O2 is produced from CO2 in the presence of highly reactive radicals such as OH. We further focus on the O2 destruction pathways to identify the dominant reduced chemical species that are responsible for the consumption of O2. Therefore, PAP (for details, see Section 3) is applied for an atmosphere with a surface O2 vmr of 10−6 PAL O2 (t geo = 2.688 Gyr). Our study represents the first application of PAP in the context of early Earth. We consider only pathways with an individual contribution larger than 1.5% of the total production (or destruction) of O2.
In the analysis, we firstly considered all chemical species with an in situ chemical lifetime smaller than that of O2 as a branching point as described in Section 3. It turned out that CO is part of the net reactions of the dominant O2 production and destruction pathways, respectively. This indicates that, for the rates of O2 production and destruction pathways, it is relevant whether CO is treated as a branching point or not (if it is, all CO production and destruction pathways will be combined as far as possible to yield null cycles, i.e., pathways that do not have a net effect on CO; if it is not, CO production and destruction pathways are not combined). For about 40% of the atmospheric layers considered, CO was used as a branching point. To ensure a consistent treatment of all O2 production and destruction pathways at every atmospheric height, we repeated the analysis by excluding CO as a branching point.
Production pathways and their altitude dependence
Table 6 summarizes the major column-integrated chemical production pathways, P1–P7, found by PAP and their percentage contributions to the total column-integrated O2 production rate for an atmosphere with a surface O2 vmr of 10−6 PAL O2.
Only pathways that contribute >1.5% of the total column-integrated production rate are shown. For explanation of classes and subclasses see text.
The major column-integrated chemical production pathways presented in Table 6 can be categorized into two classes, as follows: • class PA: O2 is produced by pathways with the net reaction 2CO2 → O2 + 2CO ▪ subclass PA1 (P1, P2, P7): catalyzed by HO
x
species ▪ subclass PA2 (P3, P4): without the presence of HO
x
• class PB: O2 is formed by pathways with the net reaction 2O → O2
▪ subclass PB1 (P6): catalyzed by HO
x
species ▪ subclass PB2 (P5): without the presence of HO
x
The relative contributions of the pathways mentioned above to the total column-integrated production rate are given in Fig. 20. Pathways of subclass PA1 and PA2 are initiated by the photolysis of CO2, whereas class PB pathways require overall the presence of atomic oxygen. The main source for O originates from the photolysis of CO2 in the upper stratosphere, producing O(1D) and hence O, which is then transported downward by eddy diffusion, contributing to enhanced O2 vmrs in the upper stratosphere. Note that at the upper boundary of the photochemistry part of the atmosphere module an effusion flux for O and CO is implemented in order to simulate this effect from atmospheric layers above the module TOA. Pathway P2 was found by Yung and DeMore (1999) in the context of a pure CO2-dominated atmosphere.
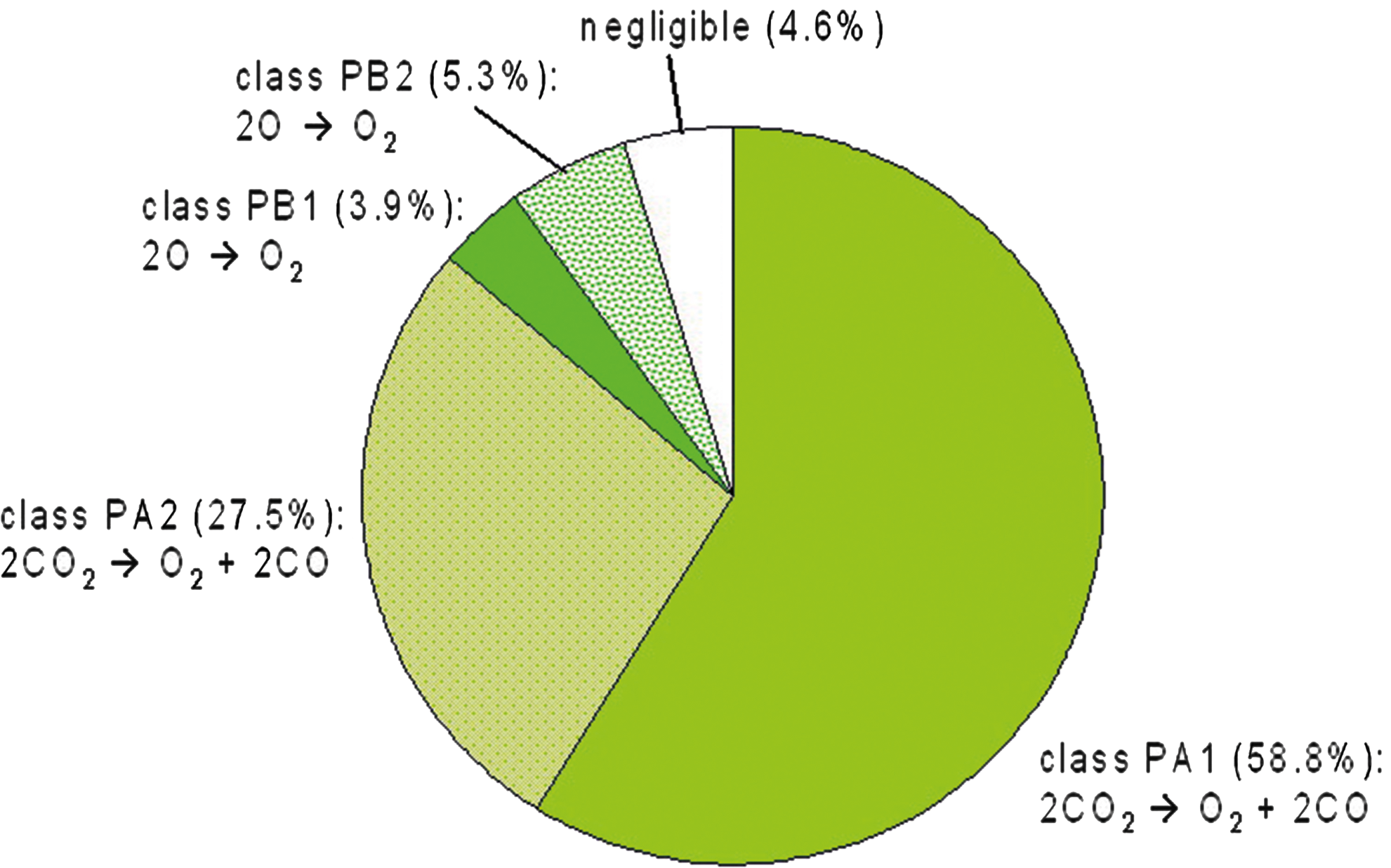
O2 production classes (see Table 6) and their contributions to the total column-integrated O2 production rate via pathways calculated by PAP for an Earth-like atmosphere with a surface O2 vmr of 10−6 PAL O2.
Figure 21 shows the altitude dependence of the production pathways of O2 for an Earth-like atmosphere with a surface O2 vmr of 10−6 PAL O2, which indicates that the production of O2 in the atmosphere is strongest in the upper stratosphere. The total production rate due to chemical pathways calculated by PAP is also shown (solid black line). About 98.7% of the total column-integrated O2 production rate is explicable via pathways found by PAP with individual contributions above 1.5%. The dominant production pathways P1 and P3 (as well as P7), which produce excited oxygen (O(1D)) from CO2 photolysis, act in the upper stratosphere where UV radiation is strongest. As the UV radiation passes through the atmosphere, it is absorbed; hence the second dominant pathway P2 (and also P4), which involves ground-state oxygen instead of O(1D), has its maximum rate at lower altitudes. Due to the enhanced O abundance in the upper stratosphere, which is provided from CO2 photolysis that produces O(1D) and is transported downward to the analyzed atmospheric layers, the pathways P5 and P6 have their maximum rate in the upper stratosphere. Note that, except for the photolysis of CO2, pathways P5 and P6 are equal to P4 and P2.
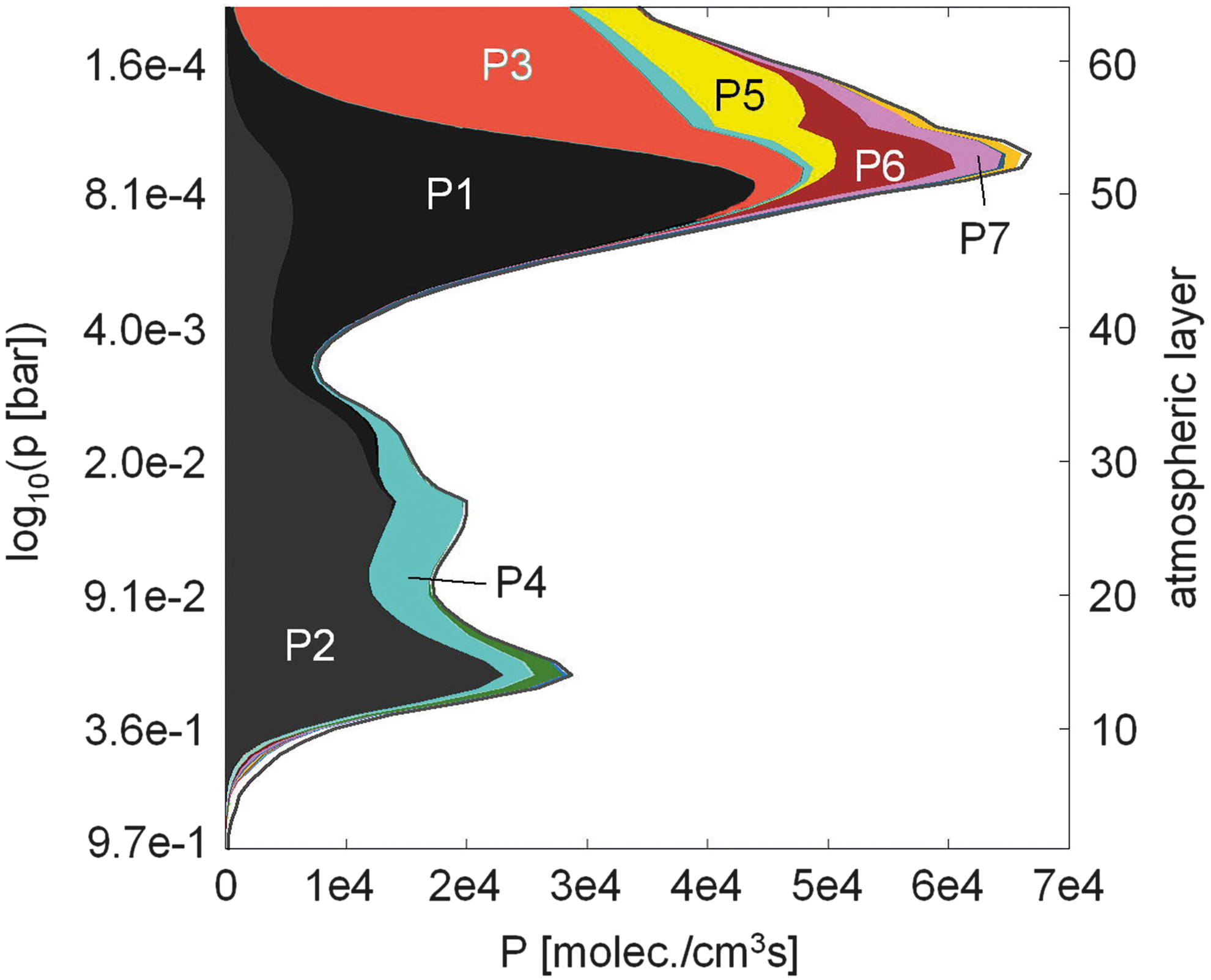
Altitude dependence of O2 production pathways P1 to P7 (see Table 6) for an Earth-like atmosphere with a ground-level O2 concentration of 10−6 PAL.
Destruction pathways and their altitude dependence
Table 7 summarizes the major column-integrated chemical destruction pathways and their percentage contributions to the total column-integrated O2 destruction rate found by PAP for an atmosphere with a surface O2 vmr of 10−6 PAL O2.
These pathways can be categorized into 4 classes: • class LA: O2 is destroyed by pathways with the net reaction O2 + 2CO → 2CO2 (CO oxidation), which are catalyzed by HO
x
and NO
x
species ▪ subclass LA1 (L1, L2, L6, L8): initialized by the oxidation of H ▪ subclass LA2 (L3, L5): initiated by the photolysis of O2
• class LB: O2 is consumed by CH4-oxidation pathways ▪ subclass LB1 (L4, L7): net reaction 3O2 + 2CH4 → 4H2O + 2CO ▪ subclass LB2 (L10): net reaction O2 + CH4 → H2O + H2 + CO • class LC (L9): O2 is consumed by pathways with the net reaction O2 + 2H2 → 2H2O (H2 oxidation), • class LD (L11): O2 is consumed by pathways with the net reaction O2 + H2O + CO → H2O2 + CO2.
Their relative contributions to the total destruction rate via pathways are given in Fig. 22. Generally, O2 is consumed forming CO2, CO, H2, H2O, and H2O2. Pathways of class LA lead to the oxidation of CO mainly via HO x . Thereby, in the subclass LA2 pathways are initiated by the photolysis of O2. The complex pathways of the subclasses LB1 and LB2 destroy O2 via the oxidation of CH4, whereas class LC leads to the oxidation of H2. Furthermore, there is a small contribution from class LD that leads to the formation of H2O2 via oxidation of CO and H2O. This is rather surprising since H2O2 is a highly reactive (oxidizing) tropospheric species (produced by self-reaction of HO2 and removed via photolysis and fast surface deposition). H2O2 features a lifetime of typically a few hours and is therefore set to be a branching point species throughout the troposphere (see Section 3). Nevertheless, L11 suggests a pathway in which H2O2 is overall produced. We interpret this result as follows: PAP was supplied with input from rate data for chemical reactions that occur only (in situ) in the atmosphere. It was not supplied with rates for other processes such as dry and wet deposition. H2O2 features a rapid depositional surface sink; therefore to attain an equilibrium concentration for this species in the atmospheric model, there must exist a pathway in the atmosphere that overall forms a net source of H2O2, to balance the strong surface depositional sink. It is this net source that PAP has found in the form of L11.
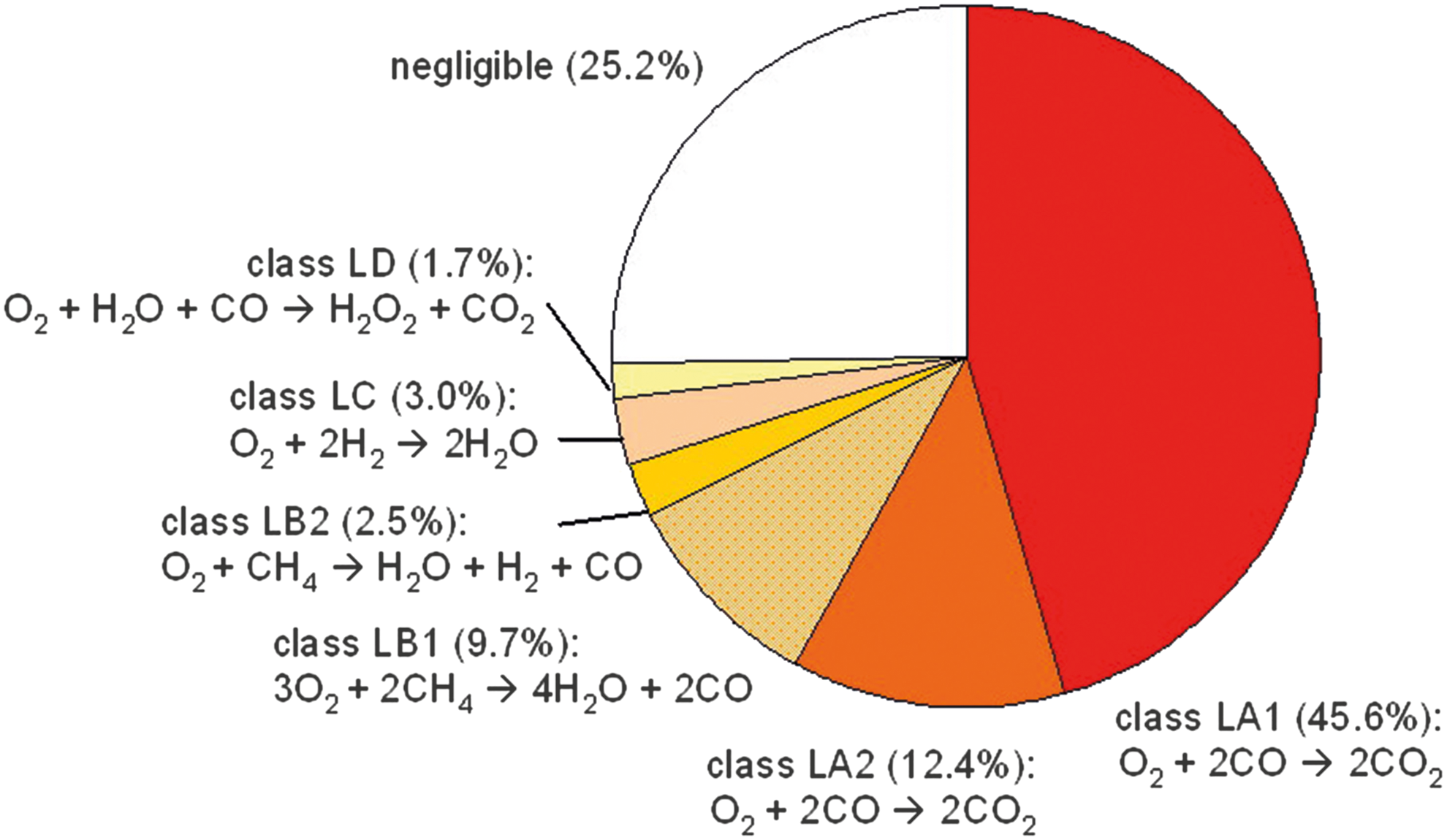
Several pathways shown in Table 7 have also been found by previous workers in the context of martian atmospheric chemistry, for example, L1 by Parkinson and Hunten (1973) and Yung and DeMore (1999), L2 by Stock et al. (2012a), L3 by McElroy and Donahue (1972), L6 by Nair et al. (1994) and Yung and DeMore (1999), and L8 by Sonnemann et al. (2006). This implies that pathways that are important for the CO2-dominated martian atmosphere may also play a key role in an Earth-like atmosphere with low O2 and 1 PAL CO2 abundances.
Figure 23 shows the altitude dependence of the O2-consumption pathways for an Earth-like atmosphere with a surface O2 vmr of 10−6 PAL O2. The total destruction rate due to chemical pathways calculated by PAP is also shown (solid green line). About 84.1% of the total column-integrated O2 destruction rate is due to pathways with individual contributions above 1.5%. Especially, in the troposphere the destruction of O2 is composed of a large number of pathways, which individually contribute less than 1.5% to the total column-integrated destruction rate. Figure 23 suggests two regimes for the destruction of O2 in the atmosphere, as follows:
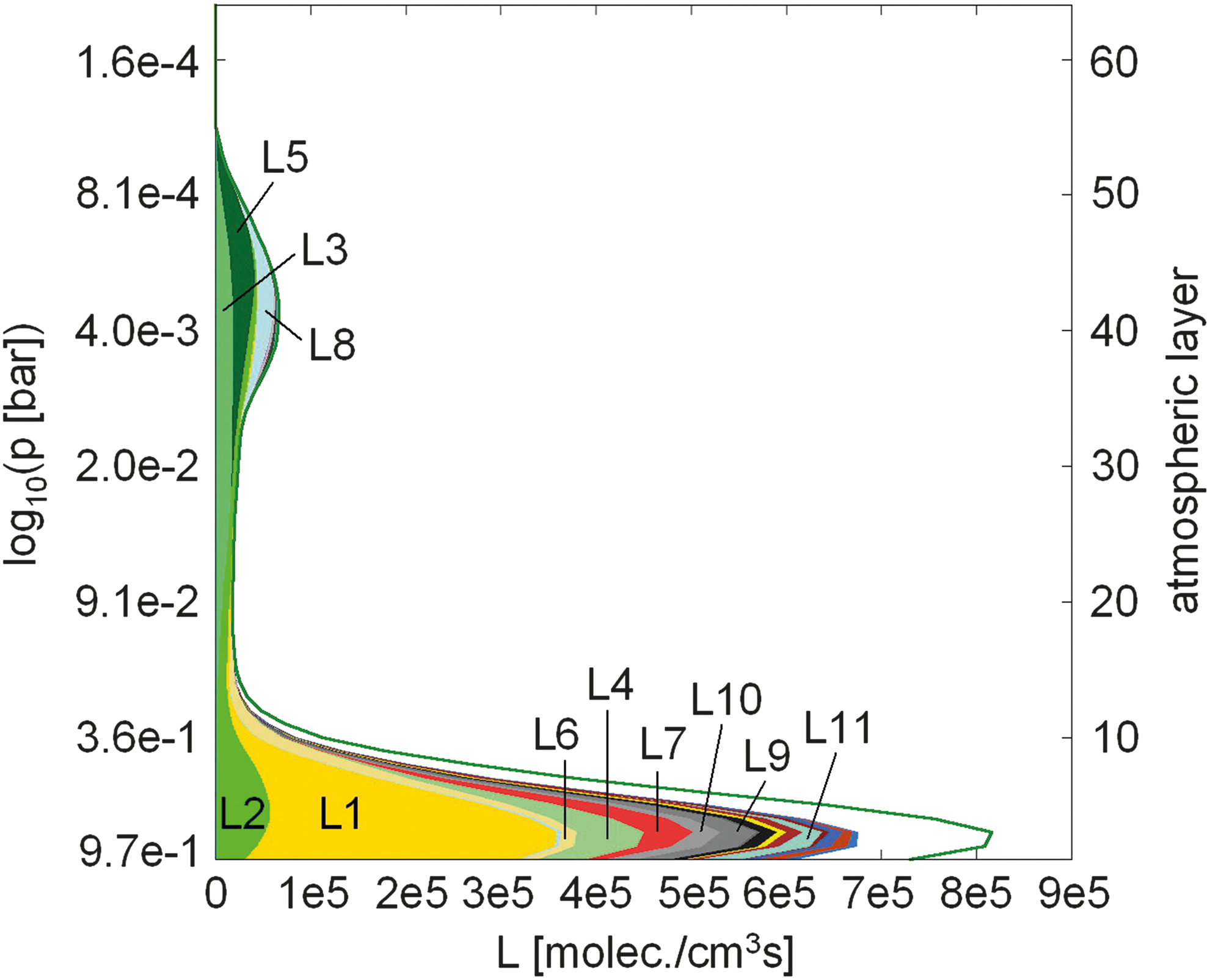
• The main consumption of O2 in the troposphere occurs via the oxidation of reduced gases such as CO, CH4, and H2 [but only to a very small amount (<1.5%) by reduced sulfur such as H2S] and is composed of a large number of different pathways. The dominant pathway L1 leads to CO-oxidation catalyzed by HO x (CO is provided by diffusion from the upper atmosphere, where it is strongly produced from CO2 photolysis), whereas the complex CH4 oxidation pathways L4, L7, and L10 have smaller contributions. Note that our results may change depending on the CH4 concentration and, hence, biological CH4 flux inserted at the surface. The minor pathway L9 results in the oxidation of H2, which is delivered by volcanic emissions from the surface and via diffusion from the upper atmosphere. H2 is destroyed in situ in the lower atmosphere. There is an additional small contribution from pathway 11 that results in the oxidation of CO in the presence of H2O.
• A minor contribution (L3, L5, and L8) to the destruction pathways originates in the stratosphere. At these altitudes O2 consumption during CO oxidation is most efficient due to a maximum in HO2 abundance (not shown), whereby pathways L3 and L5 are initiated by the photolysis of O2.
In summary, for an Earth-like atmosphere with low surface O2 concentrations (10−6 PAL O2), atmospheric O2 is produced in the upper stratosphere mainly from the in situ photolytic destruction of CO2 (whereas a small contribution relies on the delivery of O by atmospheric diffusion). O2 is mainly destroyed in the lower atmosphere by complex pathways resulting mainly in the formation of H2O and CO2 via the oxidation of CO, CH4, and H2. Thereby, there is a minor contribution from pathways resulting in the formation of H2 and H2O2. In both the production and destruction of O2, CO plays a key role. Note that the abiotic in situ production rate of O2 in the atmosphere from CO2 photolysis as well as the O2 destruction rate is comparatively smaller than the O2 flux originating from the surface biosphere.
6.2. Biogeochemical modeling
The CAB model calculates how much input from oxygenic photosynthesis, N (NPP), is needed to maintain the surface flux
Assuming three constant inputs from anoxygenic photosynthesis [r = 7.5·109, 7.5·1010 (present value, Holland, 2006), and 7.5·1011 mol O2 equivalent/yr (Archean upper limit, Holland, 2006)], Fig. 24 shows the resulting N as a function of geological time t geo.
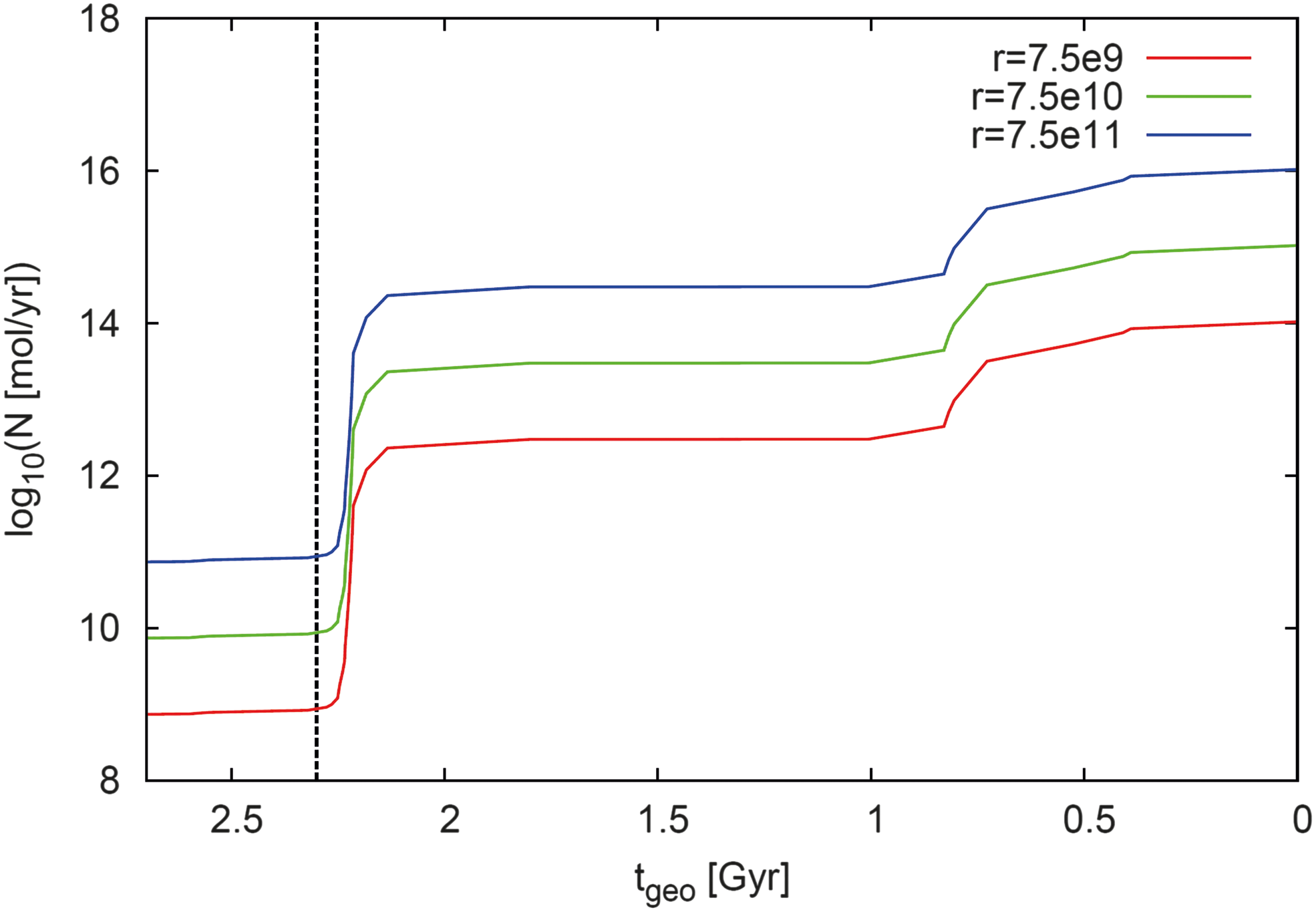
Calculated input from oxygenic photosynthesis, N, in mol O2 per year for a constant value of anoxygenic photosynthesis, r, as a function of geological time t
geo: r = 7.5·1010 mol O2 equivalent/yr is the present value (Holland, 2006) (green); r = 7.5·1011 mol O2 equivalent/yr is an upper limit for the Archean (Holland, 2006) (blue); and for comparison calculations were performed for a constant value of r = 7.5·109 mol O2 equivalent/yr (red). The beginning of the GOE at t
geo = 2.3 Gyr is indicated as a vertical dashed line. (Color graphics available at
Before the GOE at about 2.3 Gyr, N exhibits an almost constant behavior, although O2 concentrations vary over 2 orders of magnitude (see Table 5). After the GOE, it increases modestly with increasing O2 concentrations and does not reflect the sometimes oscillating behavior found for the atmospheric O2 surface flux [see (P − L) chemical change behavior in Section 6.1.2 for comparison]. On inserting Eq. 13 into Eq. 14, one obtains
Equation 16 can now be rearranged for [O2]aq, which is calculated by the biogeochemical module (see Eqs. 11 and 12) and directly depends on N. For N, two regimes can then be identified, as follows: • Regime I (low O2 regime before the GOE): • Regime II (high O2 regime after the GOE):
The switch between both regimes can be found where
Results in Fig. 24 can also be plotted over O2 mixing ratio; see Fig. 25 suggesting that one level of NPP from oxygenic photosynthesis, N, supports only one specific O2 surface vmr. Please note that the y axis in Fig. 25 is logarithmic and in the case of low O2 atmospheres changes in N are very small but monotonically increasing.

As for Fig. 24 but over O2 vmr instead of geological time t geo.
6.2.1. Modern Earth
Regarding the NPP from oxygenic photosynthesis, N, the CAB model calculates N CAB = 1.1 Pmol O2/yr for modern Earth at 1 PAL O2, which is within a factor of 4 of the rather uncertain observed present value (N obs = 3.75 Pmol O2/yr) (see red curve in Fig. 24). The calculated organic carbon burial rate of 2.1 Tmol O2/yr is in the range of the rather uncertain observed value of 10 Tmol O2/yr (Holland, 1978), whereas the calculated organic carbon reservoir of 3.5·1020 mol is somewhat less than the present observed value of 1.66·1021 mol (Holland, 1978). Since our study assumes steady state, the organic carbon burial rate, β(N + r), is equal to the weathering rate of organic carbon wCorg.
6.2.2. Archean Earth
Modern-day oceanic NPP mostly originates at the coastlines on continental shelves because the abundances of nutrients are strongly increased due to river input. However, due to the lack of knowledge about the length of the continental boundary over time, we used the continental area instead to estimate the Archean NPP, although this assumption might be crude. If we assume that the continental area was reduced [see Taylor and McLennan (1991) for evolution models of the continental crust over Earth's history] and the burial efficiency was probably higher in the past, then the NPP would have been reduced to 1% of the modern-day value. This estimate is calculated by assuming the following: • only 3% of the surface of Earth was covered by continents at 2.45 Gyr ago (before the GOE) (Taylor and McLennan, 1991; Pesonen et al., 2003), which equals about 10% of today's continental coverage. If one assumes that the NPP is proportional to the continental area, then this leads to a reduction of NPP by a factor of 10. • the absolute organic carbon burial rate of about 10 Tmol/yr is rather constant over time because the fraction of volcanic carbon that is buried as organic carbon (about 20%) has not changed strongly since at least 3.0 Gyr [see Schidlowski et al. (1983) but also Shields and Veizer (2002) for carbon isotope data from the Precambrian]. Modern burial efficiency of organic carbon in the ocean is about 0.2% (Berner, 1982; Betts and Holland, 1991), resulting in modern NPP of 5 Pmol/yr. Since the Archean burial efficiency was likely similar to that of the modern, euxinic Black Sea of about 2% (Arthur et al. 1994), the NPP should have been further decreased by a factor of 10.
For the time of the Fe deposition found from the Hamersley BIF (2.69–2.44 Gyr ago), the blue curve in Fig. 24 has to be considered, yielding a value of N CAB = 8.4·1010 mol O2/yr, which is less than the estimated value of N archean = 0.01·N obs = 3.75·1013 mol O2/yr that was derived for the discussion points above. This large discrepancy is likely related to uncertainties, for example, in ocean circulation, length of continental shelves, and so on. Unfortunately, along with these uncertainties, burial efficiency is highly indeterminate for the Archean. Furthermore, note that there exist differences between our model output and the proxy data. In the case of the O2 concentrations, we use the upper limits (for sulfur MIF, detrital siderite) or the lower limits (for Beggiatoa, animals, charcoal). Some initial test runs (not shown) varying the O2 concentration in the Archean by a factor of 10–100 affected the NPP by only about 10%.
6.2.3. Comparison to previous work
It is difficult to compare the results of this study to previous box-model work by, for example, Claire et al. (2006) and Goldblatt et al. (2006), because they do not consider a full coupled atmospheric column model with climate and photochemistry. In Claire et al. (2006) and in the transient runs by Goldblatt et al. (2006), the rise of atmospheric O2 is accompanied by a collapse in atmospheric CH4 due to the O2-CH4 feedback as already described in Section 6.1.2.
Furthermore, Goldblatt et al. (2006) found a bistability in atmospheric O2 as a result of the nonlinear behavior of their CH4 oxidation parameterization. In their Fig. 2b, a certain range in input from oxygenic photosynthesis for a constant r can support two different atmospheric O2 concentrations, for example, a low oxygen state (before the GOE) and a high oxygen state (after the GOE) due to the O2-O3-UV feedback. However, such bistability behavior is not observed in our work since we treat the oxidation of CH4 as part of the atmosphere modeling, which is decoupled from the biogeochemical modeling. It was shown (see Figs. 24 and 25) that, for atmospheres before t geo = 2.25 Gyr ago (with surface O2 concentrations below 10−4 PAL), rather small changes in N result in different O2 vmrs; hence the atmosphere-surface system is very sensitive to changes in input from oxygenic photosynthesis.
7. Sensitivity Results
Sensitivity studies were performed to derive the key parameters and boundary conditions that have the strongest impact upon the behavior of the O2 surface flux calculated by the atmospheric module of the CAB model and the corresponding biosphere response. The following scenarios were considered: • Scenario A: here the assumed evolutionary path of O2 is set to be constant to 10−5 O2 vmr before 2.5 Gyr ago. S(t) and volcanic and metamorphic outgassing remain as for the control scenario. • crustal mineral redox buffer: ▪ Scenario B: more reduced: ▪ Scenario C: more oxidized: • heat flow: ▪ Scenario D: 2Q
▪ Scenario E: 0.5Q
• Scenario F: eddy diffusion profile is set constant to K(z) = 105 cm2 s−1 (Kuhn and Atreya, 1979) [control run: K(z) from Massie and Hunten (1981)] • Scenario G: surface CO2 concentration: 5 PAL CO2 [sediment data place an upper limit of 10–100 PAL, see Rye et al. (1995), Sheldon (2006), Hessler et al. (2004), but see also Rosing et al. (2010) for a more conservative limit] • Scenario H: doubled CH4 surface flux (control scenario: 474 Tg CH4/yr) • Scenario I: incident stellar radiation: young Sun analog star β Com (see Fig. 26 for comparison of stellar spectra of β Com and the Sun) • Scenario J: Pasteur point: lowered to 10−5 PAL O2 (Stolper et al., 2010) • Scenario K: solubility of O2 in ocean water: Henry's law constant

Normalized stellar spectrum of β Com (green) in comparison to the spectrum of the Sun (red) by Gueymard (2004). (Color graphics available at
In the case of Scenario A (not shown), only a minor increase in O2 surface flux before 2.5 Gyr ago compared to the control scenario (with a maximum relative change of 0.6%) is found. Hence, atmospheric chemistry is not strongly affected compared to the scenario discussed in the previous sections (control run). The effect upon the biosphere response is a little larger. To maintain a constant O2 vmr of 10−5 at the surface, N increases by about 12% compared to the control scenario because of the slightly enhanced O2 surface flux.
Figure 27 shows how the atmospheric O2 surface flux responds for the other sensitivity runs. A larger O2 surface flux is needed to maintain the O2 surface vmr in the cases of Scenario B (QFM-1), Scenario D (2Q), and Scenario H (doubled CH4 flux) because of the increased amount of reduced gases emitted at the surface.

Calculated atmospheric O2 surface flux as a function of geological time t geo for a number of different parameter studies compared to the control scenario described in Section 5: Scenario B (QFM − 1), Scenario C (QFM + 1), Scenario D (2Q), Scenario E (0.5Q), Scenario F (const. K(z)), Scenario G (5 PAL CO2), Scenario H (2 CH4 flux) and Scenario I (β Com). The parameter studies concerning the Pasteur point (Scenario J) and the ocean solubility (Scenario K) have the same atmospheric O2 surface flux as the control scenario. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
In the case of Scenario F (const. eddy diffusion), the constant K(z)-profile leads to a stronger O3 layer (not shown) with an O3 column amount of about 430 DU at t geo = 2.18 Gyr ago (0.1 PAL O2—cf. 263 DU for control run). Furthermore, the O3 surface vmr of 1.2·10−7 is higher than that in the control scenario (2.61·10−8) (not shown). At 0.1 PAL O2, eddy diffusion is greatly increased compared to modern Earth above 10 km, which leads to a more efficient transport of chemical species toward lower pressure; hence species such as CH4 and N2O are more quickly removed. HO x and NO x are weaker, resulting in enhanced O3 abundances. Hence, the overall increased O3 abundances result in increased O2 surface fluxes after the GOE because the production of O3 is an effective sink for O2. In the case of Scenario I (β Com), a slightly increased O2 surface flux is calculated, which is also a direct consequence of the increased O3 abundance in such an atmosphere. Due to enhanced UV radiation emitted by the star below about 300 nm, the production of O3 via the Chapman mechanism, which represents a loss process for O2, is stimulated. The O2 surface flux is generally lower for Scenario C (QFM +1) and Scenario E (0.5Q) because less reduced chemical species are emitted into the atmosphere. In the case of Scenario G (5 PAL CO2), the O2 fluxes at the surface are less than that in the control scenario after the GOE, which implies that increased CO2 concentrations lead to higher production of O2 in the atmosphere because CO2 photolysis is a source for O and O(1D) and, hence, O2. Before the GOE, O2 fluxes are equal to the control scenario.
The effect upon the biosphere is shown in Fig. 28. For the different scenarios, we find:
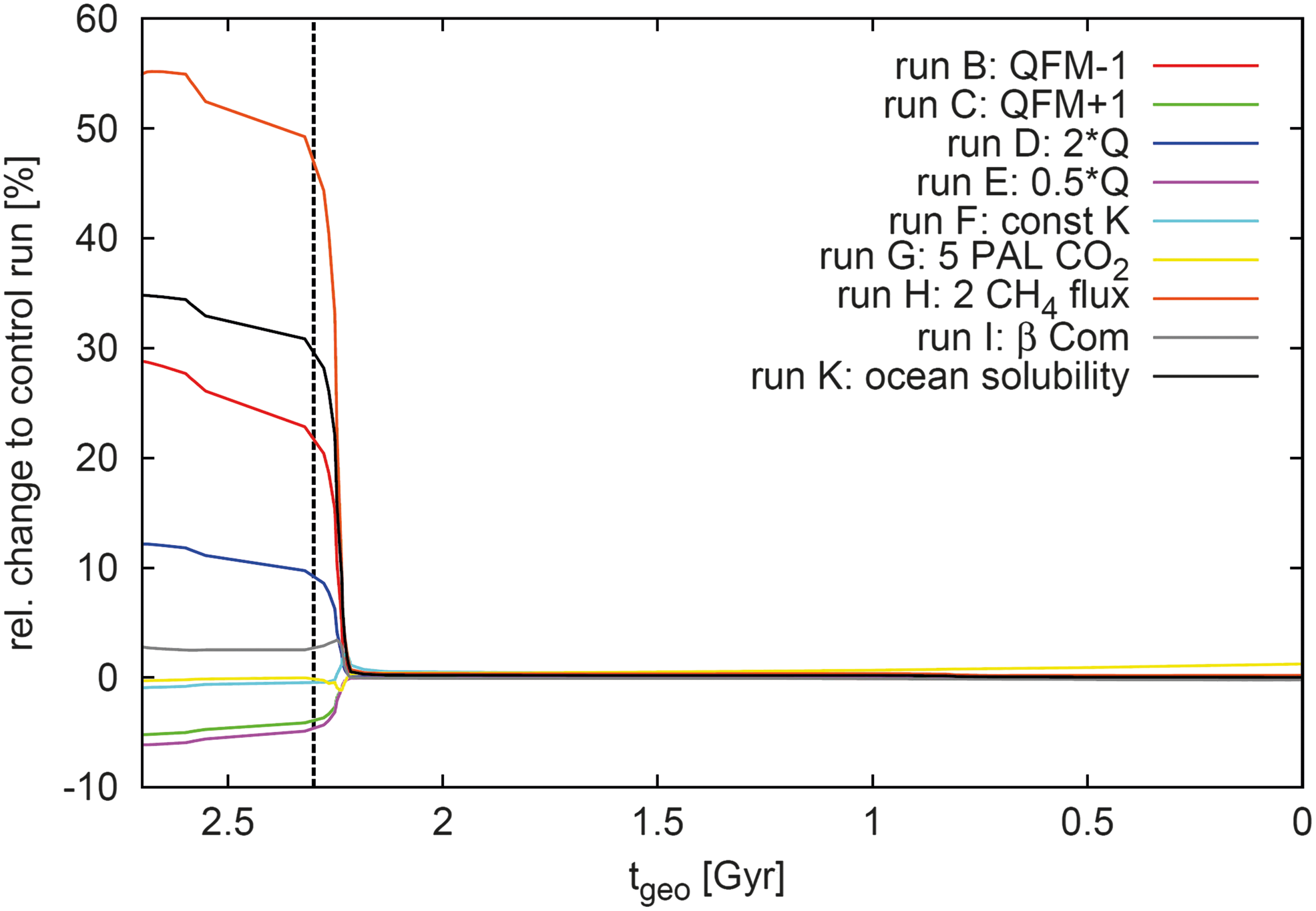
Calculated relative change in % of the input from oxygenic photosynthesis, N, for the different parameter changes compared to the control scenario as a function of geological time t geo. Scenario J is not shown in this figure. The beginning of the GOE at t geo = 2.3 Gyr is indicated as a vertical dashed line.
• Scenario B (QFM − 1, red): Due to the overall, more reduced atmospheric conditions and the associated destruction of O2, a higher O2 surface flux is required; hence a higher N is needed, which results in a relative change of about 29% at 2.7 Gyr ago.
• Scenario C (QFM + 1, green): More oxidized conditions due to less reduced gases emitted at the surface lead to a maximum decrease of 5% in N.
• Scenario D (2Q, blue): A doubled heat flow results in more reduced gases and, hence, an overall increase in N by 12%.
• Scenario E (0.5Q, magenta): A smaller heat flow yields a decrease in N by about 6%.
• Scenario F (const. K(z), cyan): There are only minor relative changes with a maximum of 2–3% at 2.22 Gyr ago.
• Scenario G (5 PAL CO2, yellow): Minor relative changes are observed with a minimum value of 2% at 2.24 Gyr; after the GOE, the relative change increases to 1–2%, which reflects the higher surface pressures and temperatures compared to the control run.
• Scenario H (2 CH4 flux, orange): A doubled CH4 flux at the surface has a strong effect on the biosphere response and results in a maximum relative change of 55% in N.
• Scenario I (β Com, gray): A higher UV radiation below 300 nm results in a larger O3 column and, hence, a higher O2 surface flux and therefore an increase in N by about 4%.
• Scenario K: (ocean solubility, black): To maintain the same O2 flux as for the control run, the aqueous concentration [O2]aq and, hence, N must increase because the solubility constant and piston velocity of O2 are lower (see Eq. 13); a maximum increase of 35% is observed.
• Scenario J (Pasteur point, not shown in Fig. 28): In the case of the reduction of the Pasteur point, O2 can be consumed by aerobic respiration down to lower aqueous O2 concentrations, which results in a stronger O2 loss; therefore the input from oxygenic photosynthesis must increase by several orders of magnitude (103). Hence, better knowledge of the Pasteur point is important.
Generally speaking, relative changes in N (except for the Pasteur point run) are very small after the GOE, although the corresponding O2 surface fluxes calculated by the atmospheric chemistry module are increased or decreased in comparison to the control scenario (see Fig. 27). This is because, in the regime after the GOE, it is the partial pressure of O2, and hence surface conditions (p surf, T surf), rather than the O2 surface flux reflecting the chemical behavior of O2 in the atmosphere (as discussed in Section 6.2), which is responsible for the biosphere response.
8. Conclusion
This is the first study to our knowledge that includes the coupling between geological, biological, and (detailed) atmospheric climate and photochemistry processes. By applying our unique global-mean atmospheric column module with interactive O2, CO2, and N2 (CAB model), which can respond to detailed atmospheric chemistry and radiative transfer, our study investigated consistently chemical and biological sources and sinks that control the evolution of atmospheric O2 on Earth and potentially on Earth-like extrasolar planets. Our new model reasonably reproduces observations of modern Earth for both atmospheric data (atmospheric chemical species, climate) and biological-geological data (NPP and burial). Our results suggest that, in the atmosphere, CO2 (we assume modern-day surface values) is not an isoprofile (as is commonly assumed in the literature) in the middle atmosphere for all early Earth analog runs, and O2 is no longer an isoprofile for runs with less than 10−4 O2 surface vmrs.
The O2 bistability found by Goldblatt et al. (2006) is not observed in our calculations due to our consistent atmospheric treatment of CH4 oxidation. Furthermore, we calculate an increase in CH4 concentration with increasing O2 vmrs during the GOE in contrast to previous works (e.g., Claire et al., 2006; Zahnle et al., 2006) with much simpler chemical schemes, which have suggested the opposite effect. Important caveats, however, include uncertainties, for example, in the modeled temperature evolution due to lacking knowledge about the evolution of greenhouse gas evolution (CO2, CH4, N2O), as well as missing processes in the model, for example, the response of methanogen activity to changing atmospheric O2(g) and temperature.
For the first time, we applied the PAP for O2 in the early Earth analog atmosphere and found that photolysis of CO2 is an important abiotic source of O2 (and CO) in the upper atmosphere. However, this process is not able to change the overall atmospheric O2 content on early Earth analog planets. In the early reducing atmosphere, O2 is mainly destroyed via oxidation of CO, CH4, and H2 in the lower atmosphere. It was found that CO is a crucial chemical species in chemical pathways that produce and destroy O2.
On applying the CAB model to the early Earth analog atmospheres, we calculated O2 surface fluxes over the geological timescales. An important result was that different types of atmospheres, that is, with different oxidizing natures, could be supported by one O2 surface flux respectively. More work is required to understand the nature of the oxidizing-reducing feedbacks in the early Earth analog atmospheres. But due to the coupling of biogeochemical processes to the atmosphere, we found that there is only one NPP from oxygenic photosynthesis that is needed to maintain the O2 surface vmr for every type of atmosphere considered. The atmospheric O2 surface flux is most sensitive to changes in the biogenic CH4 surface emission and a crustal chemical redox buffer that emits more reduced than oxidized gases (QFM − 1), but also to the assumed eddy diffusion profile. Atmospheres with enhanced CO2 concentrations lead to a decrease in the required atmospheric O2 surface flux. The NPP of the biosphere is strongly influenced by changes in the Pasteur point, biogenic CH4 surface emission, ocean solubility, and crustal chemical redox buffers that emit more reduced gases (QFM − 1); hence resolving the uncertainties in these quantities is crucial for a better understanding of early Earth analog atmospheres.
Although our main focus was on early Earth analog planets, it is nevertheless instructive to briefly discuss our results in an Earth-like exoplanetary context. Clearly, this discussion remains hypothetical and requires future work. Regarding Earth-like extrasolar planets, results suggest that different atmospheric “states” of O2 could exist even for similar biomass output. The PAP analysis suggests that atmospheres where volatile organic compounds build up atmospheric O2 are disfavored due to oxidation reactions. Furthermore, extrasolar Earth-like planets with strong interior outgassing of reducing gases can lead to so-called false negatives for O2 (e.g., PAP results show that CO- and/or CH4-containing pathways destroy O2 and, hence, mask the existence of O2-producing biospheres). Since all O2 destruction pathways found feature HO x , steamy high-UV exoatmospheres could also lead to similar false negatives in O2. Extrasolar Earth-like planets with strong mixing (e.g., due to a weak temperature inversion) have a potentially strong positive effect on atmospheric O3. Varying key parameters (e.g., interior outgassing) in low-O2-containing exoatmospheres (< 1% PAL) significantly impacts the net productivity of a potential biosphere for a given surface O2 concentration.
Footnotes
Acknowledgments
This research was supported by the Helmholtz Gemeinschaft through the research alliance “Planetary Evolution and Life.”
This study has received financial support from the French State in the frame of the “Investments for the future” Programme IdEx Bordeaux, reference ANR-10-IDEX-03-02.
M. Godolt acknowledges funding by the Helmholtz Foundation via the Helmholtz Postdoc Project “Atmospheric dynamics and photochemistry of Super-Earth planets.”
This work was partially funded by grant AyA 201232237 awarded by the Spanish Ministerio 353 de Economia y Competitividad.
The authors thank the unknown referee for his or her valuable comments.
Author Disclosure Statement
No competing financial interests exist.