
Abstract
Bacteria are important organisms for space missions due to their increased pathogenesis in microgravity that poses risks to the health of astronauts and for projected synthetic biology applications at the space station. We understand little about the effect, at the molecular systems level, of microgravity on bacteria, despite their significant incidence. In this study, we proposed a systems biology pipeline and performed an analysis on published gene expression data sets from multiple seminal studies on Pseudomonas aeruginosa and Salmonella enterica serovar Typhimurium under spaceflight and simulated microgravity conditions. By applying gene set enrichment analysis on the global gene expression data, we directly identified a large number of new, statistically significant cellular and metabolic pathways involved in response to microgravity. Alteration of metabolic pathways in microgravity has rarely been reported before, whereas in this analysis metabolic pathways are prevalent. Several of those pathways were found to be common across studies and species, indicating a common cellular response in microgravity. We clustered genes based on their expression patterns using consensus non-negative matrix factorization. The genes from different mathematically stable clusters showed protein-protein association networks with distinct biological functions, suggesting the plausible functional or regulatory network motifs in response to microgravity. The newly identified pathways and networks showed connection with increased survival of pathogens within macrophages, virulence, and antibiotic resistance in microgravity. Our work establishes a systems biology pipeline and provides an integrated insight into the effect of microgravity at the molecular systems level. Key Words: Systems biology—Microgravity—Pathways and networks—Bacteria. Astrobiology 16, 677–689.
1. Introduction
G
In this study, we established a systems biology pipeline and reanalyzed the global gene expression of P. aeruginosa (Crabbe et al., 2010, 2011) and S. typhimurium (Wilson et al., 2007, 2008) from four seminal studies under simulated microgravity and spaceflight conditions. We identified a large number of new cellular and metabolic pathways in a statistically significant way. We extracted the distinct patterns in global gene expression under microgravity and examined the functional and regulatory nature of genes with similar expression patterns at the network level. The aim of this study was to understand the effects of microgravity on bacteria at the molecular systems level.
2. Materials and Methods
A flowchart of the systems biology pipeline is shown in Fig. 1.
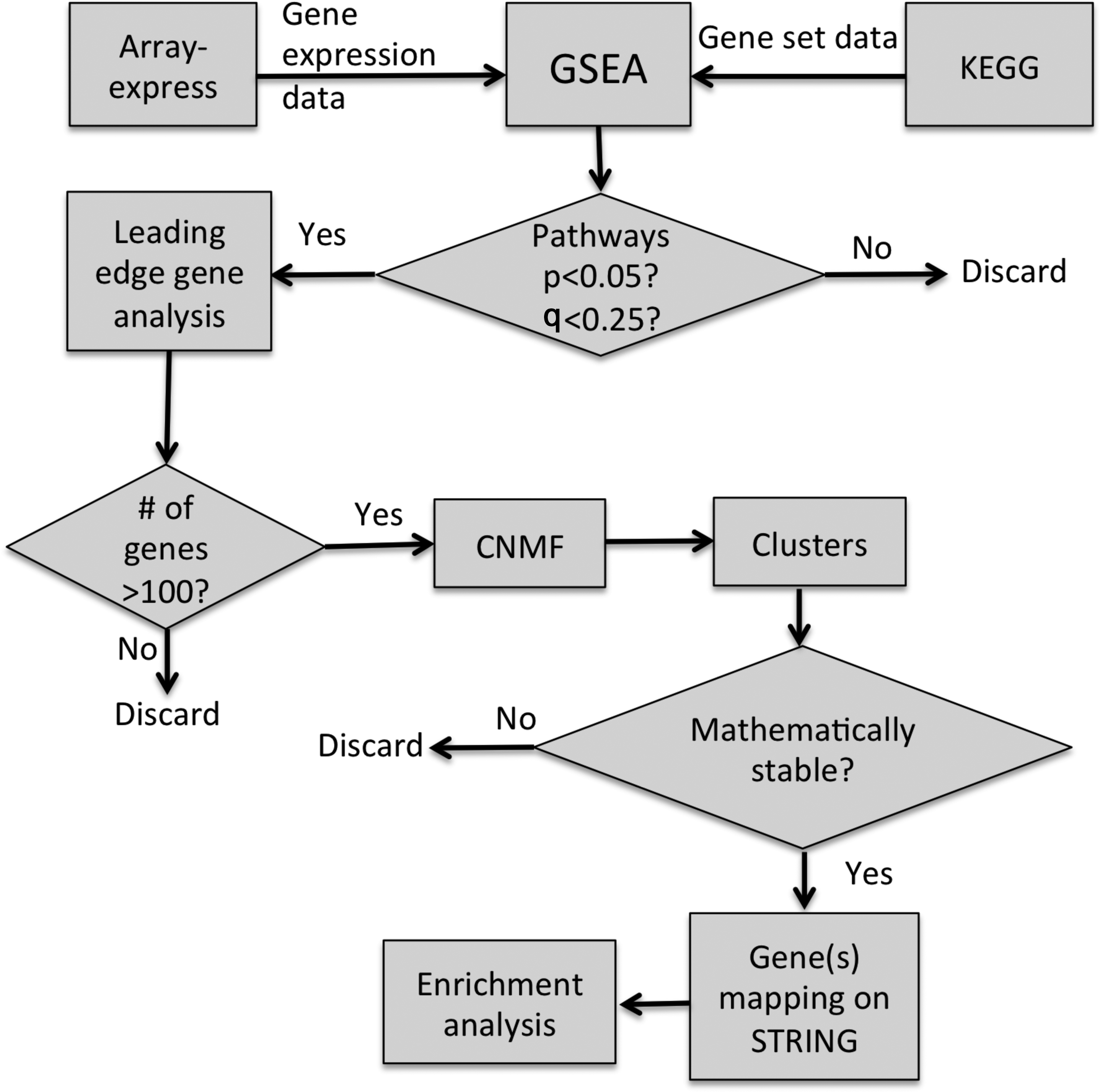
Flowchart of the systems biology pipeline.
2.1. Gene set enrichment analysis of expression data
Gene set enrichment analysis (GSEA) was performed with GenePattern (Reich et al., 2006), a multitool platform for genomic analysis. The GSEA module in GenePattern was run with global gene expression data and corresponding gene sets. The gene sets (ontology) for both Pseudomonas and Salmonella were obtained from the KEGG database. The expression data sets (from ArrayExpress, see the result section for details) were converted to an appropriate file format as required in GenePattern. To generate the ranked list of genes, we applied a “signal-to-noise” metric for ranking, which may be represented by (M
microg − M
1g
)/(SD
microg + SD
1g
), where M
microg and M
1g
represented the mean expression values of individual genes among experiments in microgravity and 1g conditions, respectively. The SD represented the corresponding standard deviation. In one case (Pseudomonas in RWV E-GEOD-16970), we used a “difference in class” metric (difference in mean values of gene expression) due to a lower number of samples (<3). For enrichment analysis, we applied running sum statistics in order to use a weighted Kolmogorov–Smirnov-like statistic scoring scheme (as described in Subramanian et al., 2005). In brief, the score was evaluated by walking down the ranked gene list (RL), and if genes in a gene set (GS) matched (hit) in the ranked list, the score would increase. It would decrease in the case of a nonmatch (miss).
where gj
represents the j
th gene in RL = {gi
, … , gN
}, r(gj
) = rj p = 1 and
The enrichment score was calculated as the highest deviation of P hit − P miss from zero. To estimate the p value through null hypothesis testing, genes of gene sets were randomly permuted 1000 times, keeping the total number of genes in each set constant, and GSEA was performed for each permuted gene set. Normalized enrichment score (NES) values from this permuted set were compared with the original one to estimate the p values for each NES. The corresponding false discovery rate (FDR) q values were estimated from the distribution of the p values. We ran the same GSEA on each data set three times to check the consensus among various runs.
2.2. Consensus non-negative matrix factorization—clustering of gene expression data
The consensus non-negative matrix factorization (CNMF) algorithm was run on an expression data set of leading edge genes in the NMFC module of GenePattern. Clustering was based on the expression levels on the 1g and microgravity (simulated or space) conditions. The details of CNMF can be found elsewhere (Brunet et al., 2004). Briefly, taking the gene expression data set as a positive matrix L of size M × N, with a number of clusters C, the non-negative matrix factorization (NMF) computes iteratively for matrices F and G such that L = FG with F × C and C × G sizes. Starting from a randomly initialized matrix of size F and G, the matrices are iteratively updated to minimize a divergent functional as described in Lee and Seung (1999). We set the number of iterations per clustering as 2000 times. A consensus algorithm is run on top of the NMF to evaluate the mathematical stability for the number of clusters (C). For a given number of NMF clusters, the original data are perturbed and simulated by applying an in-built resampling tool in GenePattern. This assesses a consensus among multiple runs of NMF clustering on those simulated (perturbed) data. The resultant consensus matrix (summary of the consensus) is evaluated for a cophenetic coefficient, which determines the pairwise distance from the original data set. The highest number of mathematically stable clusters is determined from the change of the cophenetic correlation coefficient as a function of the number of clusters. The point after which the coefficient value falls sharply is estimated as the maximum possible number of mathematically stable clusters.
2.3. Mapping the genes in a protein network database
The genes that belong to each stable cluster were mapped on the STRING protein association network database to extract protein-protein association (PPA) maps. The mapping was performed by simply uploading the list of genes from each cluster on the STRING database and assigned the associated organism for those genes. This resulted in PPA networks. On each network, the interaction enrichment score and associated KEGG pathways with p and q value were determined from the in-built functional tools.
2.4. Orthology analysis
The orthology analysis of genes from 15 common altered pathways between Pseudomonas aeruginosa PAO1 and Salmonella enterica serovar Typhimurium was performed by using STRING. Taking Pseudomonas as a reference, the orthologus genes in Salmonella were searched, and the degree of protein sequence conservation between these two species was reported as a heat map.
3. Results
3.1. GSEA reveals cellular and metabolic pathways altered in microgravity
The GSEA algorithm (Subramanian et al., 2005) uses a running sum statistics on the global gene expression data based on known functional sets of genes (ontology) to determine whether particular cellular or metabolic pathways are more likely to be involved with microgravity in a statistically significant way. The gene expression data for P. aeruginosa and S. typhimurium under microgravity (Wilson et al., 2007, 2008; Crabbe et al., 2010, 2011) were extracted from ArrayExpress (EMBL-EBI). The data accession numbers of these studies are E-GEOD-22684 (Crabbe et al., 2011), E-GEOD-16970 (Crabbe et al., 2010), and E-GEOD-8573 (Wilson et al., 2007, 2008). Crabbe et al. (2011) examined the effect of spaceflight and RWV on Pseudomonas. Crabbe et al. (2010) examined the effect of RWV and RPM on Pseudomonas. Similarly, Wilson et al. (2007) and Wilson et al. (2008) examined the effect of spaceflight conditions on Salmonella in rich and minimal media, respectively. To our knowledge, those are the only global gene expression data in microgravity for both species, which is available in the public domain. We then classified all genes of known cellular and metabolic functions of P. aeruginosa and S. typhimurium into 102 and 103 gene sets, respectively, by using the KEGG database. Next, we performed the GSEA, which determines whether a gene set is enriched on the top or bottom of a ranked list of all genes in a gene expression data set. Genes were ranked according to difference in the mean expression between microgravity and normal gravity divided by the summation of the standard deviation of that particular gene among replicates in each of the two experimental conditions (Fig. 2A). The histogram of the ranked genes with distribution of the score is shown in Fig. 2B. Next, enrichment scores for each gene set were calculated with running sum statistics, where the score increases when genes in the gene set are encountered in the ranked list (Fig. 2C). NES values were calculated by normalization of the enrichment scores based on the number of genes in each gene set. Unlike differential gene expression analysis, the p value and FDR q value are not assigned for an individual gene in the ranked gene list. The p value and FDR q value of an entire gene set are calculated in GSEA. A significance test was performed by estimating p values for these NES values by using a null hypothesis approach (Subramanian et al., 2005), and the FDR q was calculated by comparing the tails of NES distributions for each gene set. The resultant gene sets with positive and negative NES are defined as upregulated and downregulated, respectively.
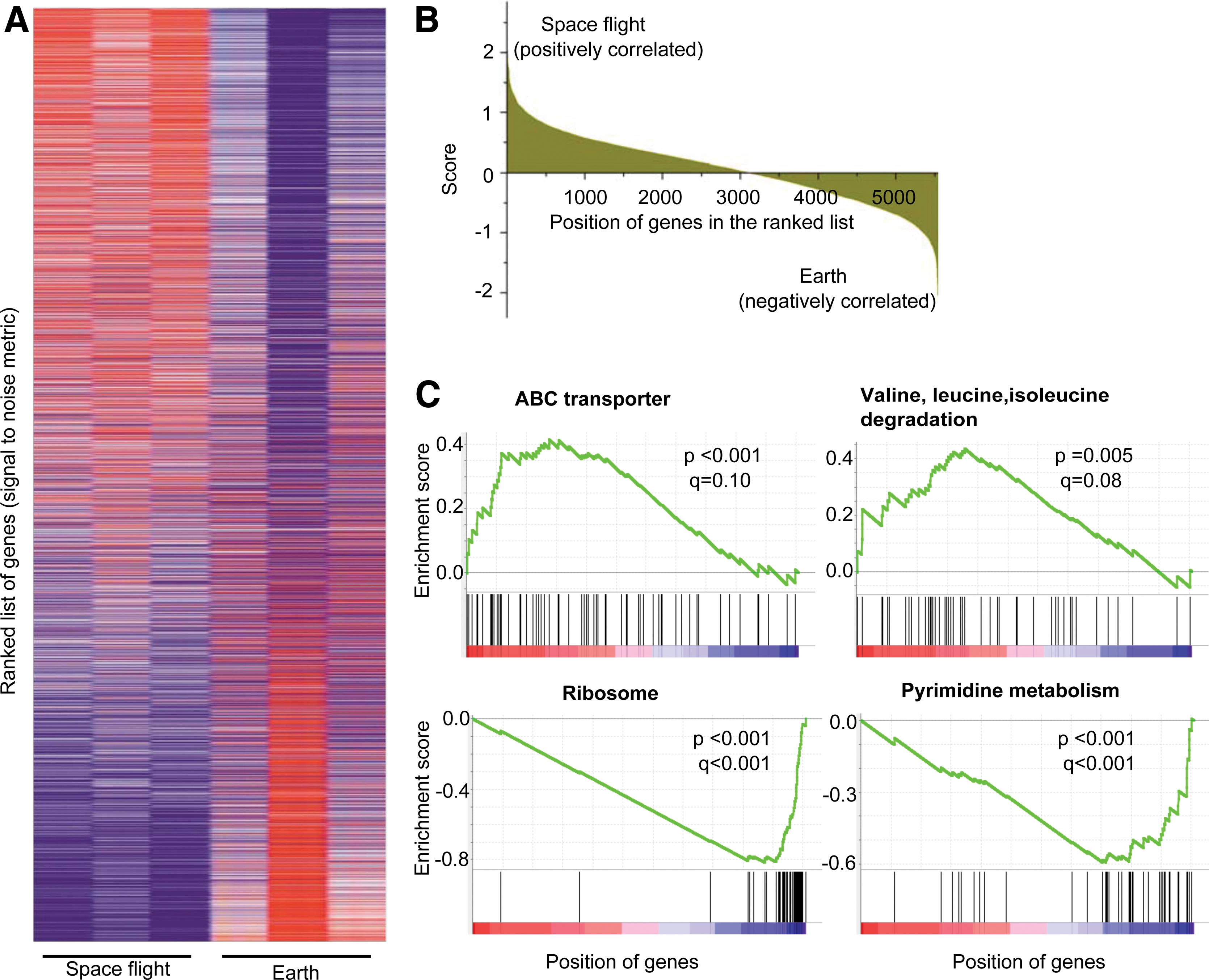
GSEA of P. aeruginosa under spaceflight conditions. (
Figure 2C shows the GSEA for two upregulated (ABC transporter and valine, leucine, isoleucine degradation) and two downregulated (ribosome and pyrimidine metabolism) pathways in P. aeruginosa under spaceflight conditions. For each set of experiments, a large number of cellular and metabolic pathways (gene sets) associated with microgravity conditions were altered (upregulated and downregulated) with statistically significant p and q values (p < 0.05, q < 0.25). The high NES values (absolute) were generally associated with high statistical significance (Supplementary Fig. S1; Supplementary Data are available online at
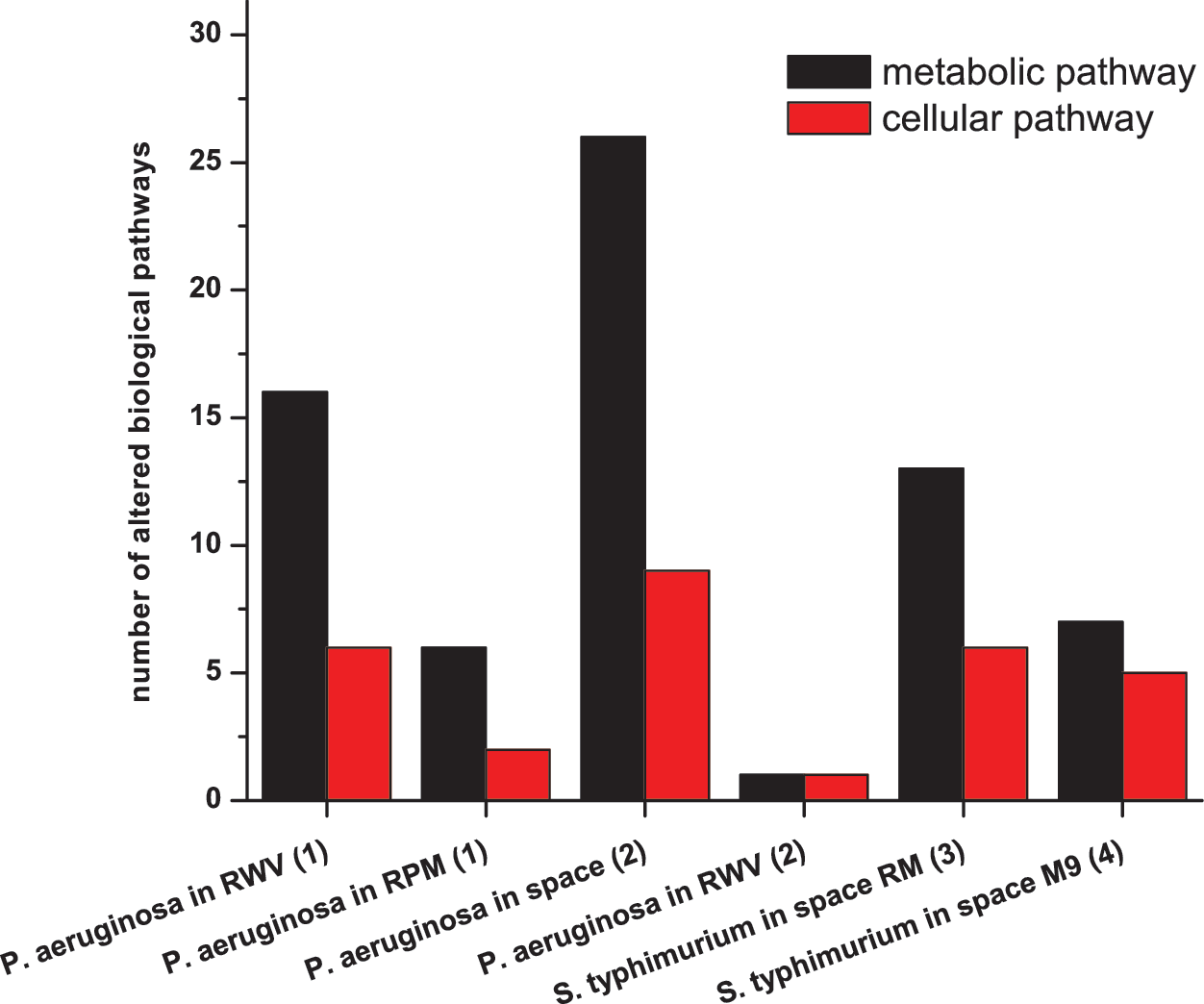
Number of altered cellular and metabolic pathways identified by GSEA on different studies. The numbers 1, 2, 3, and 4 with each experiment represent data sets analyzed from the references Crabbe et al. (2010), Crabbe et al. (2011), Wilson et al. (2007), and Wilson et al. (2008), respectively. (Color graphics available at
Pathways in italics represent new pathways found in this analysis. The boldface letters represent the pathways found common between at least two studies in our analysis. Six pathways (ribosome, flagellar assembly, chemotaxis, TCA cycle, oxidative phosphorylation, and nitrogen metabolism), which are not in italics, suggest the already-known pathways.
3.2. Comparative analysis suggests involvement of new and overlapping pathways in microgravity
For P. aeruginosa under simulated microgravity, a previous study (Crabbe et al., 2010) suggested seven altered functional categories, namely, AlgU and Hfq regulon, quorum sensing, motility, stress, anaerobic metabolism, and ATP synthesis. Similarly, in spaceflight conditions (Crabbe et al., 2011), the suggested categories are Hfq regulon, anaerobic metabolism, virulence factor, ribosome synthesis, and dehydrogenation of succinate. Two studies on S. typhimurium under space conditions (Wilson et al., 2007, 2008) showed involvement of Hfq regulons, ribosome, iron utilization, biofilm, virulence, motility, and several genes without any assigned pathways. Our analysis showed a higher number of altered pathways, which include a large number of cellular and metabolic pathways that have not been reported previously. The new pathways found in this study are tabulated in italics in Table 1. We observed that in the work of Crabbe et al. (2011) five KEGG pathways were altered. In our analysis, with the same data set, we found 37 altered pathways, which included all five pathways indicated by Crabbe et al. (2011). It was interesting to find that, in all four cases, metabolic pathways outnumbered the cellular processes.
However, GSEA runs only for the genes that are assigned to functional classes. Therefore, unclassified proteins (around 50% of the total genes including hypothetical proteins) in KEGG are not enriched in our analysis. Though the Hfq gene was found to be downregulated in the RNA degradation pathway for both species in our results, there is no functional class assigned for Hfq regulon genes in KEGG. When we added two gene sets—Hfq regulon genes upregulated (480 genes) and Hfq regulon genes downregulated (334 genes) for Pseudomonas (Sonnleitner et al., 2006), which were used by Crabbe et al. (2010) and Crabbe et al. (2011) to identify the Hfq regulon genes in microgravity—GSEA enriched the downregulated and upregulated Hfq regulons in space with p < 0.001 and q ≤ 0.1. However, these two gene sets are broad in nature and are not rigorously curated or specific, and were therefore excluded from further analysis. For Salmonella in space, the Hfq regulons were divided in the multiple functional categories (Wilson et al., 2007). KEGG does not classify any of those functions except ribosomal proteins, and our analysis showed downregulation of ribosomal proteins as was the case in the work of Wilson et al. (2007).
Next, to discern common pathways related to microgravity, if there is some association, we compared the number of overlapping pathways among different data sets (Fig. 4). It was observed in our analysis that the number of overlapping pathways between P. aeruginosa and S. typhimurium in space was much higher than the number between P. aeruginosa in space and simulated microgravity (RWV and RPM) conditions. Ribosome, RNA degradation, protein export, flagellar assembly, methane metabolism, toluene degradation, oxidative phosphorylation, TCA cycle, glycolysis, purine metabolism, and pyrimidine metabolism were among the common altered pathways between P. aeruginosa and S. typhimurium in space. We tested the degree of protein sequence conservation in pathway-specific orthologous genes for all the 15 common altered pathways between the two species (Supplementary Fig. S2). The results show a high degree of sequence conservation. The effect of spaceflight on both the species, therefore, may have a common cellular origin.
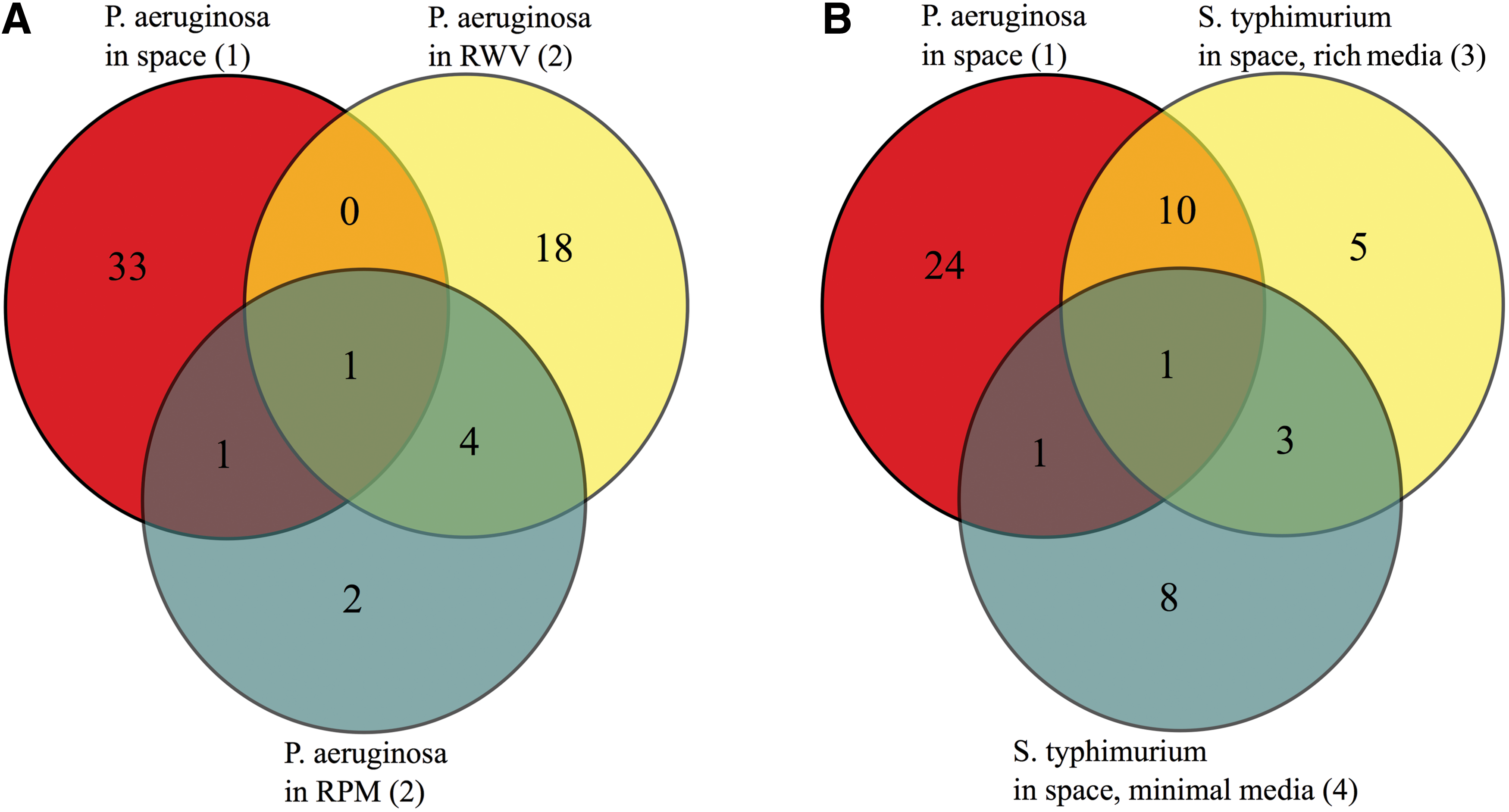
Venn diagrams among the experiments represent the common altered pathways in microgravity. (
We found that the valine, leucine, and isoleucine degradation pathway was common among space, RWV, and RPM for P. aeruginosa (Fig. 4A). In contrast, several of the pathways, which were upregulated in simulated microgravity for P. aeruginosa, were downregulated in space for both the species (Table 1). The gene expression of flagellar assembly, protein export, ribosome, RNA degradation, TCA cycle, and oxidative phosphorylation was increased in RWV and RPM, but was decreased in spaceflight conditions.
3.3. CNMF clusters the leading edge genes based on the gene expression patterns
Next, we performed a leading edge subset analysis to identify the significant genes (leading edge genes) that contributed the most for a pathway to be statistically significant. The mapped genes before the peak value of enrichment scores in Fig. 2C are the leading edge genes. Those genes for all experiments were tabulated in Supplementary Table S2. To evaluate whether a set of patterns in leading edge gene expression exists, we performed an unsupervised CNMF clustering method (Lee and Seung, 1999; Brunet et al., 2004) for the experiments, where the combined number of leading edge genes among altered pathways was more than 100. The parts-based local representation is a unique property of NMF that allows one to extract functional and more meaningful subsets from a matrix data set in comparison to other methods such as principal component analysis (PCA) and vector quantization (VQ) (Lee and Seung, 1999). NMF has also outperformed all conventional clustering methods including K-means and singular value decomposition (SVD) in extracting functional relationships in yeast gene expression data (Kim and Tidor, 2003). A consensus clustering algorithm together with NMF (CNMF) was successfully applied to classify cancer samples from global gene expression data, where other clustering methods like hierarchical clustering, self-organizing maps, and principle component analysis have failed (Brunet et al., 2004). This has been used successfully in effectively interpreting several gene expressions and other large-scale biological data (Devarajan, 2008). CNMF was applied separately for upregulated and downregulated pathways. We identified the maximum number of mathematically stable clusters in each case by plotting the cophenetic coefficient as a function of the number of clusters (k). Figure 5A shows such a correlation derived from upregulated leading edge genes of P. aeruginosa in spaceflight conditions. The cophenetic coefficient determines the mathematical stability of the clusters. Its value declined sharply after a certain number of clusters, which is indicated by an arrow (Fig. 5A). This number suggests the maximum number of mathematically stable clusters, and we can then hypothesize the possible number of patterns in gene expression. The cophenetic coefficient correlation plots for other cases are shown in Supplementary Fig. S3. Figure 5B shows the maximum number of stable clusters for all the studies. We have shown that the CNMF was able to distinguish the expression patterns between ribosomal genes and various metabolic genes by classifying them into different clusters in each and every case (Fig. 6, Supplementary Figs. S5 and S6). This validated that the unsupervised CNMF clustering was able to differentiate the gene expression patterns in a biologically meaningful way.
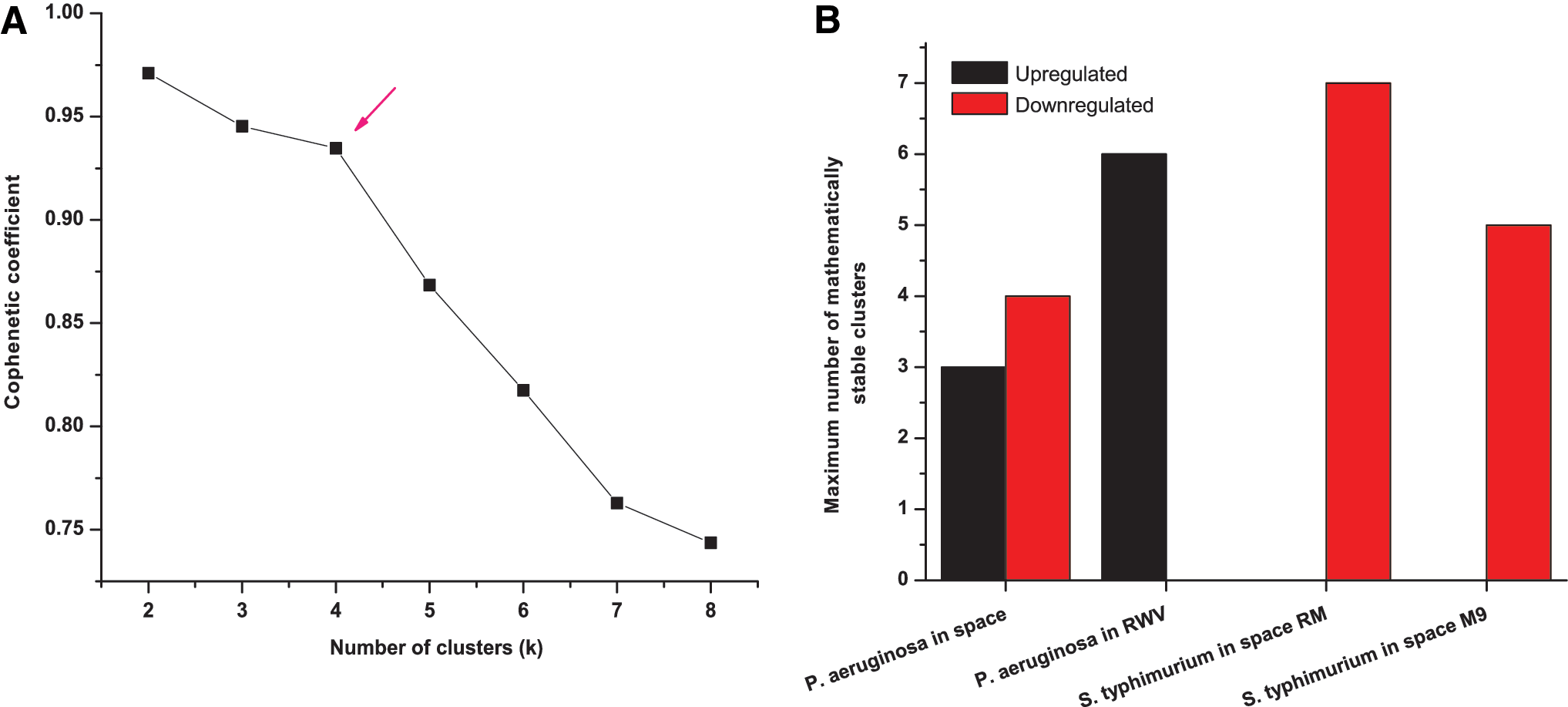
CNMF clustering on leading edge genes. (

Protein-protein association networks of P. aeruginosa from leading edge genes of an individual CNMF cluster. Each node represents a protein, and the line connecting the nodes (edge) represents the functional association. The relative thickness of each line signifies the confidence level of such association within a network. The functional annotations of different parts of the network are marked on the figure. (
3.4. Mapping of the patterned gene expression reveals the protein networks affected by microgravity
To understand the plausible regulatory and functional connections among genes, pathways, and microgravity, we mapped the genes of distinct CNMF cluster(s) with the STRING protein network database (Franceschini et al., 2013). Figure 6 shows the PPA networks for one upregulated and two downregulated clusters of P. aeruginosa in spaceflight conditions. The network maps for all other studies are shown in Supplementary Figs. S4–S7. It was observed that the clusters showed either highly interactive protein-protein networks (p < 10−8) or almost no interactions (Table 2 and Figs. 6 and S4–S7). The interaction networks were further enriched with KEGG pathways by using a built-in tool within STRING. The built-in tool requires at least nine interactions to assign any function. Five or higher numbers of interactive proteins and the enrichment p value < 0.001 were taken for further analysis. The details of the PPAs of all clusters with underlying functions are tabulated in Table 2. We observed the network connections between benzoate degradation and butanoate metabolism, between ABC transporters and two-component systems, between purine and pyrimidine metabolism and flagellar assembly, and between flagellar assembly and protein exports in various networks in P. aeruginosa (Tables 1 and 2 and Fig. 6). We have shown network connections between glycolysis and TCA cycle in S. typhimurium and between oxidative phosphorylation and flagellar assembly pathways for both P. aeruginosa and S. typhimurium. Interestingly, all those connections have been observed in other studies (Saier and Ramseier, 1996; Tobisch et al., 1999; Kapatral et al., 2004; Macnab, 2004; Maurer et al., 2005; Metzler-Zebeli et al., 2010; Dintner et al., 2011; Rooks et al., 2014) with several bacterial species not related to microgravity.
4. Discussion
In previous studies (Wilson et al., 2007, 2008; Crabbe et al., 2010, 2011), metabolic pathways were rarely reported, whereas in this analysis metabolic pathways outnumbered the cellular pathways. Those newly identified pathways may have implications in understanding the observed phenotypes of bacteria in microgravity such as virulence and pathogenesis (Wilson et al., 2007; Rosenzweig et al., 2010), antibiotic resistance (Leys et al., 2004), biofilm formations (McLean et al., 2001), and the development of a metabolic model for synthetic metabolic engineering in space. A substantial overlap among reduced metabolic pathways was found between chemical induction of virulence in Salmonella (Kim et al., 2013) and in our results for both Salmonella and Pseudomonas in microgravity. Those overlapping pathways include arginine and proline metabolism; purine metabolism; glycerophospholipid metabolism; glycine, serine, and threonine metabolism; alanine, aspartate, and glutamate metabolism; pentose phosphate pathway; methane metabolism; pyruvate metabolism; and cysteine and methionine metabolism.
Further, our results help explain the higher survival rate of intracellular Salmonella in macrophages in microgravity than occurs in normal gravity during the early hours of infection. But the survival rates are similar after 2 h (Nickerson et al., 2000). During the macrophage infection, the downregulation of chemotaxis and flagellar machinery genes, ribosome genes, purine and pyrimidine metabolism, pentose phosphate pathway, glycolysis and fatty acid biosynthesis, LPS synthesis, and DNA replication were observed in intracellular Salmonella (Erikson et al., 2003; Raghunathan et al., 2009). The downregulation of the majority of those pathways in Salmonella was observed under spaceflight conditions, as is indicated in our results (Table 1), which might explain the higher survival rate in early hours. However, in later hours, intracellular Salmonella, during the normal course of infection (1g), would attend the same metabolic states. Thus, the survival rate is similar. Interestingly, Pseudomonas in space showed downregulation of most of those pathways as indicated in intracellular Salmonella. Though there is no comprehensive study about the status of metabolic pathways in intracellular Pseudomonas during infection, we can hypothesize, according to our results, that a similar metabolic signature may occur. The increase in the benzoate degradation pathway of the pathogen was a link between increased adrenergic stress of the host and increased virulence of gut pathogens in the murine model (Rooks et al., 2014). Interestingly adrenergic stress was found to be activated in humans during spaceflight (Strollo et al., 1998), and our results show upregulation of benzoate degradation in Pseudomonas in microgravity. Similarly, microgravity-induced upregulation of ascorbate and aldarate metabolism, downregulation of TCA cycle, and oxidative phosphorylation in Salmonella in our analysis may be linked with increased virulence (Yimga et al., 2006; Lamichhane-Khadka et al., 2013) and antibiotic resistance (Karatzas et al., 2008). All these examples suggest that, instead of a new mechanism, known molecular pathways are responsible for the change in the bacterial behavior in microgravity.
The identification of common molecular pathways across the data sets we have analyzed is another important aspect of this study. In spite of the difference in media, growth phase, physical forces (e.g., fluid shear and hydrostatic pressure), aeration, and temperature, we found a greater match on pathways between Pseudomonas and Salmonella in space than has been the case for a single species (P. aeruginosa) between space and simulated microgravity. Nonzero particle motion and small shear stress associated with RWV and RPM are absent in space (Tsao et al., 1994). Further, space radiation and the specially designed cell culture vessels (Freed and Vunjak-Novakovic, 2002) might also influence the overall outcome in global gene expression. These may explain the indicated discrepancies and suggest a cautious use of the results from Earth-based simulated microgravity.
On the other hand, this work may serve as a framework for metabolic engineering and synthetic biology in space. Different species of Pseudomonas have been considered as cellular chassis for metabolic engineering application including the production of 2-methyl citric acid (Ewering et al., 2006), isobutyric acid (Lang et al., 2014), and a plethora of secondary metabolites (Gross et al., 2006). The alteration of several molecular pathways in Pseudomonas as a response to microgravity needs to be considered when designing synthetic metabolic circuits for space application, as the performance of synthetic gene circuits is a strong function of the cellular context (Bagh et al., 2008; Leonard et al., 2008). In addition, our results may help in the development of correct models and engineered strains for space synthetic biology. This includes the production of
In this study, we have reported several underlying functional connections within a PPA network originated from a set of genes with a similar expression pattern in response to microgravity. To the best of our knowledge, no such network has been reported for bacteria. The interface among multiple pathways within a PPA network may help in our understanding of how a small gene network may influence several molecular pathways in microgravity. Genes with similar expression patterns, but without any known PPA network, might imply novel regulatory modules in response to microgravity.
5. Conclusions
Taken together, this work represents an integrated picture of the effect of microgravity on proteobacteria at the molecular systems level. The systems biology pipeline was able to extract the involvement of a large number of new cellular and metabolic pathways associated with microgravity, which were obscured in traditional analysis. This study is the first to show how bacterial gene network motifs are influenced in microgravity. The newly identified pathways and networks explain enhanced survival of pathogens in macrophages, the connection between metabolic pathways and virulence, antibiotic resistance, and the common cellular response, all in microgravity. This network-level understanding may be helpful in the development of new space medicines for fighting microbial disease and correct models for synthetic metabolic engineering in space by targeting appropriate gene networks and pathways. As our systematic pipeline is scalable and applicable in a low fold change regime, and identifies specific pathways directly, it may serve as a general systems biology pipeline with which to study other organisms in microgravity.
Footnotes
Acknowledgments
This work was partially funded by IBOP, Department of Atomic Energy, Govt. of India.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.