
Abstract
We investigated the formation mechanisms of the nucleobases adenine and guanine and the nucleobase analogues hypoxanthine, xanthine, isoguanine, and 2,6-diaminopurine in a UV-irradiated mixed 10:1 H2O:NH3 ice seeded with precursor purine by using ab initio and density functional theory computations. Our quantum chemical investigations suggest that a multistep reaction mechanism involving purine cation, hydroxyl and amino radicals, together with water and ammonia, explains the experimentally obtained products in an independent study. The relative abundances of these products appear to largely follow from relative thermodynamic stabilities. The key role of the purine cation is likely to be the reason why purine is not functionalized in pure ammonia ice, where cations are promptly neutralized by free electrons from NH3 ionization. Amine group addition to purine is slightly favored over hydroxyl group attachment based on energetics, but hydroxyl is much more abundant due to higher abundance of H2O. The amino group is preferentially attached to the 6 position, giving 6-aminopurine, that is, adenine, while the hydroxyl group is preferentially attached to the 2 position, leading to 2-hydroxypurine. A second substitution by hydroxyl or amino group occurs at either the 6 or the 2 position depending on the first substitution. Given that H2O is far more abundant than NH3 in the experimentally studied ices (as well as based on interstellar abundances), xanthine and isoguanine are expected to be the most abundant bi-substituted photoproducts. Key Words: Astrophysical ice—Abiotic organic synthesis—Nucleic acids—Origin of life—RNA world. Astrobiology 17, 771–785.
1. Introduction
A
Nucleic acids can be made of the two types of nitrogenated cyclic aromatic hydrocarbons or N-heterocycles: purine and pyrimidine. Nitrogenated heterocycles have long been searched for within the gaseous interstellar media, in dense and diffuse clouds, but their presence has never been confirmed (Simon and Simon, 1973; Kuan et al., 2004). Pyrimidine (C4H4N2), a nitrogenated monocyclic molecule, has not been observed in the interstellar media in the gas phase either, despite an extensive search (Charnley et al., 2005). Yet the formation of pyrimidine cation via ion-molecule reactions has been shown to occur in the gas phase (Hamid et al., 2014). Formation of purine, a pentamer of HCN, by the combination of five hydrogen cyanide molecules has been predicted to be very exothermic and is therefore unlikely to happen in isolation (Roy et al., 2007).
Nitrogenated cyclic molecules have been, however, identified in meteorites (Folsome et al., 1971; Stoks and Schwartz, 1979; Callahan et al., 2011). The nucleobases adenine, guanine, cytosine, and uracil were extracted from the well-known Murchison and Murray carbonaceous chondrites (Stoks and Schwartz, 1979) and have been proved to be of extraterrestrial origin (Hayatsu, 1964; Hayatsu et al., 1975; Shapiro, 1999; Nelson et al., 2001; Martins et al., 2008). Together with nucleic acids, amino acids (Kvenvolden et al., 1970), and other organic compounds of biological interest, have been found in the meteoritic samples (Cronin and Pizzarello, 1997; Dworkin et al., 2001). Whether they are synthesized in the gas phase and delivered to the surface of the grains or were synthesized on the cold grain surfaces due to photoprocessing is not known. It has been shown, however, that once delivered on the surfaces of grains, these biomolecule precursors can be further photoprocessed into larger, more complex molecules. A plausible pathway for their formation is via the photoprocessing by cosmic radiation of the ices accumulated on the surfaces of grains. These ices are known to contain volatile organic molecules such as H2O, CH3OH, CO, CO2, NH3, CH4 (Gibb et al., 2004; Dartois, 2005), as well as larger molecules such as PAHs and nitrogenated heterocycles (Sandford et al., 2004).
Materese et al. (2017) studied the photoprocessing of purine in astrophysical ices in the laboratory. In their experiments, they deposited a mixture of H2O, NH3, and purine (relative abundances of approximately 1:0.1:10−3) on a substrate cooled to ∼15 K. At the same time, the growing ice was irradiated with UV light (Lyman α at 121.6 nm, i.e., 10.2 eV, and a broadband continuum centered at 160 nm) in order to simulate astrophysical photoprocessing. After 1–2 days of irradiation, samples were analyzed using gas-chromatographic (GC-MS) techniques. In a companion paper, Materese et al. (2017) presented their experimental findings. These authors found that 2-aminopurine, 6-aminopurine (adenine), 2,6-diaminopurine, hypoxanthine, xanthine, isoguanine, and guanine are formed in identifiable quantities. Previously, it was found that irradiation of pyrimidine in analogously prepared ices produced uracil, cytosine, and thymine (though only small quantities of the latter after elevated doses of radiation), and their formation mechanisms were studied (Nuevo et al., 2009, 2012, 2014; Bera et al., 2010; Sandford et al., 2015).
In the present study, we used electronic structure calculations to explore the pathways by which doped purine could be aminated and hydroxylated in the irradiated NH3:H2O experimental ices. We investigated the physical processes that may occur under the experimental conditions, the intermediates that may be generated, and the possible reactions that they may undergo. The results constitute reaction pathways leading from irradiated reactants to a range of products, independent of experimental input. We then compared the computational results with those obtained from the experiments. With the experiments characterizing many (though not all) key products of photoprocessing of purine in astrophysical ices, our computational results may help unravel the preferred reaction pathways and the preference for some products over others. We consider the roles played by the physical conditions such as UV irradiation and condensed phase environment in this transformation in order to gain insight into the nature of the photochemical transformations that occur on cold grain surfaces or small Solar System objects such as asteroids and comets.
2. Methods
Structures and relative energies of the reactants, intermediates, and products were calculated using density functional theory, the most widely used class of electronic structure methods. Specifically, geometry optimizations were carried out by using the B3LYP functional (Lee et al., 1988; Becke, 1993) with the cc-pVDZ basis set. Higher-accuracy relative energies of the structures were calculated with ωB97M-V, a range-separated hybrid meta-GGA functional (Mardirossian and Head-Gordon, 2016) with the cc-pVQZ basis set. ωB97M-V has been established as very accurate for noncovalent interactions, as well as thermochemistry, barrier heights, and more across the main group elements. Moreover, as a range-separated functional, it reduces the self-interaction error (Chai and Head-Gordon, 2009; Kronik et al., 2012) and thus is suitable for the description of radical cations.
To model the effect of the extended ice environment, we used the conductor-like solvent model C-PCM (Barone and Cossi, 1998) from the class of polarizable continuum model (PCM) (Tomasi et al., 2005). Specifically, we employed an implementation (Lange and Herbert, 2010a, 2010b) that ensures continuous potential energy surfaces, as a function of molecular geometry changes. In this model, the bulk is represented as a polarizable medium characterized by its dielectric constant ɛ. It is expected that the solvent dielectric will better represent the condensed phase and enable us to study the effect of the solvent. In the PCM calculations, the geometries were first re-optimized with the solvent model, again by using B3LYP/cc-pVDZ. Single-point PCM energies were then calculated at the ωB97M-V/cc-PVQZ level of theory. We used ɛ = 78.39 for the dielectric constant of water for all calculations (Weast, 1989).
All calculations were done using the Q-Chem 4 quantum chemistry program package (Shao et al., 2015).
3. Results
Under the direct influence of UV irradiation, organic molecules undergo a number of physical and chemical changes. Ultraviolet irradiation of purine will generally lead to electronic excitation into excited states, simultaneous excitation of vibrational and rotational modes, and ionization to create cations and free electrons. The excess energy in excited states and/or ions will potentially lead to isomerization and photofragmentation. Irradiation of the ices may also produce various reactive species—ions and radicals—such as OH and NH2 in H2O and NH3 ices, respectively, or both in a mixed molecular ice. It can also produce electronically excited solvent molecules that either dissociate into fragments or release energy collisionally and radiatively.
H2O possesses a large photodissociation cross section in the UV-wavelength range (Engel et al., 1992) of the H2 UV source employed experimentally (Materese et al., 2017). NH3 possesses a non-negligible dissociation cross section and dissociates into NH2 and H around 181 nm (Suto and Lee, 1983; Nakajima et al., 1991). Furthermore, hydroxyl radicals can abstract hydrogen from NH3 and produce NH2 radicals (Stuhl, 1973; Monge-Palacios et al., 2013). In summary, hydroxyl (OH) and amino (NH2) radicals are potentially produced in the ices by the following processes:
Of the above-mentioned photoprocesses, all channels but (3) seem open under Lyman α irradiation. Radicals and ions can also be generated from purine itself via the following processes:
where [Pu/H]• designates a radical formed by the cleavage of a C–H bond on the purine ring.
The main question of interest is identifying the secondary pathways by which hydroxyl and amino substituted purine derivatives are formed from the parent neutral molecules and the radicals and ions that are formed in the primary photoinduced processes discussed above. Furthermore, ionization energy of NH3 is 10.02 eV, which is low enough for Lyman α (121.6 eV, ∼10.2 eV) to ionize it to NH3
+. But Stuhl (1973) and Monge-Palacios et al. (2013) showed that hydroxyl radical abstracts hydrogen atoms from NH3 to produce NH2 easily (reaction 5 in the manuscript). As H2O is 10 times more abundant than NH3 in the experimental conditions, hydroxyl radicals would be produced in large quantities, which would produce NH2 radicals. The process would augment the primary NH2 formation process via direct dissociation. Therefore, we believe that NH2 would be the most dominant nitrogenous attacking group, although the possibilities of reactions with NH3
+ cannot be ruled out. In Fig. 1, we present a summary of the stepwise substitution of NH2 and OH groups on the purine ring. Here, we enumerate some of the main possible reaction channels. First, neutral purine can react with neutral H2O or NH3, or with the corresponding OH and NH2 radicals. Second, purine cations can likewise react with H2O or NH3, or the OH or NH2 radicals. Third, purine radicals (that have lost a H atom) can recombine with either OH or NH2 radicals. Other channels are also possible.
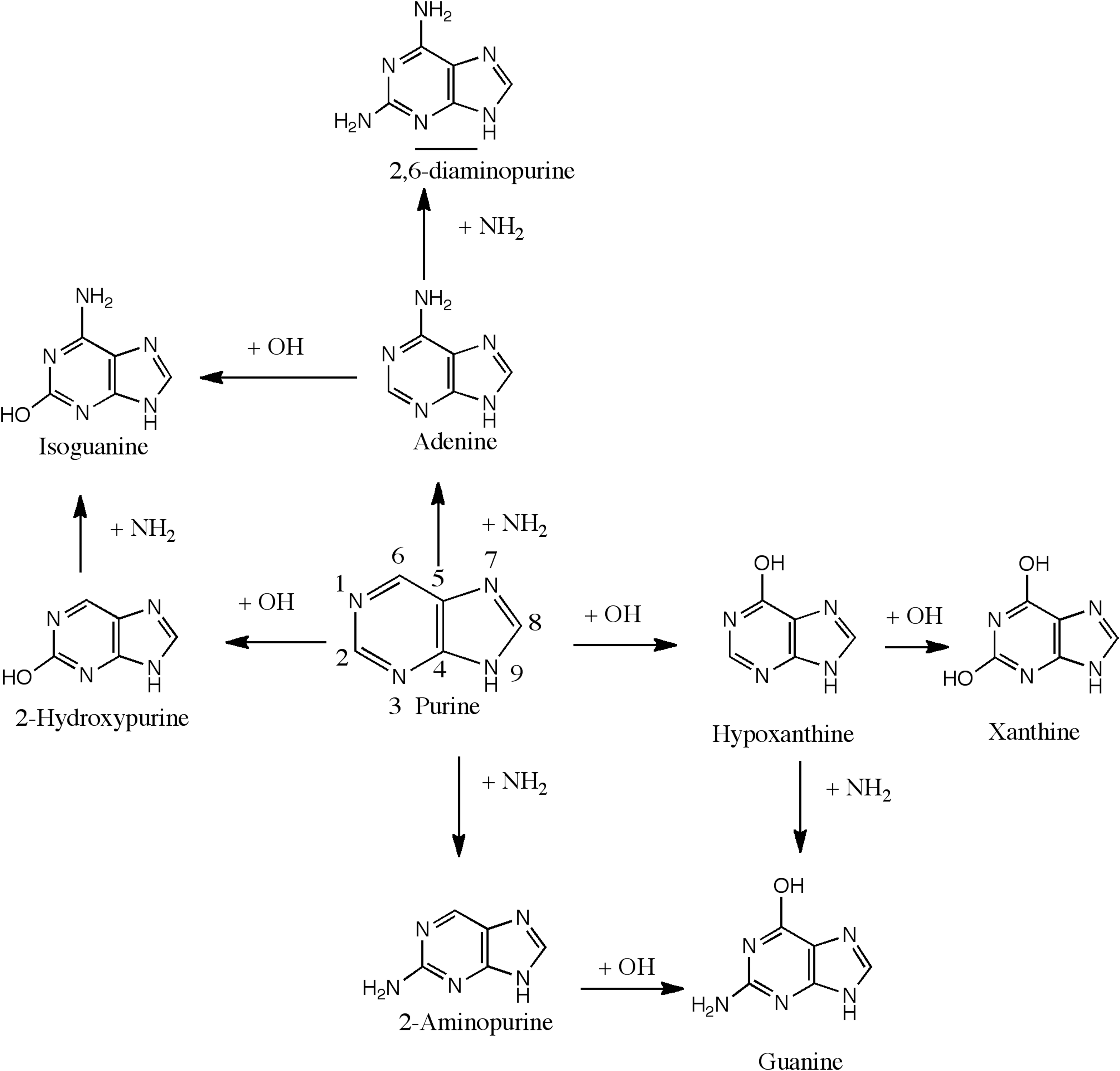
A reaction scheme indicating the amino and hydroxyl group substitutions on the purine ring. Horizontal arrows indicate hydroxyl group substitutions, while the vertical arrows indicate amino group substitutions. Quantum chemical calculations indicate that 2-hydroxypurine and adenine are the most favorable singly substituted photoproduct, and xanthine, isoguanine, guanine, and 2,6-diaminopurine are the most favorable doubly substituted photoproducts.
Neutral–neutral reactions (8 and 9) between H2O (or NH3) and purine are expected to be several orders of magnitude slower than reactions involving radicals or cations, because of their high activation barriers, particularly in cold ice matrices. Thus, it is reasonable to assume that the chemistry in these ices is going to be dominated by the reactions of OH and NH2 radicals with ionized, radical or neutral purine, and its derivatives.
We studied the neutral purine and hydroxyl radical (and amine radical) pathways (10 and 11) and found that the addition of the hydroxyl (or the amino) radical to purine, leading to radical intermediates, is endothermic due to the loss of aromaticity. This result, in our view, makes channels 10 and 11 unlikely relative to some of the other possibilities discussed below. The overall reactions, after H atom loss to yield neutral substituted products, are exothermic.
Purine cations are likely to be formed in the irradiated mixed molecular ices (primary channel 6) as the UV lamp possesses enough energy to ionize purine. Purine cation can undergo an addition reaction with nucleophilic H2O or NH3 (secondary channels 12 and 13). This reaction is exothermic in the entrance channel, as well as barrierless, so this initial step is likely to proceed. Consider reaction with the water molecule as an example:
Furthermore, this reaction is likely under a relatively low radiation flux because it requires only one primary photoproduct to proceed. Completion of the reaction involves elimination of a proton and an H atom (or conceivably H2
+•) to make the final hydroxylated or aminated products from the initial intermediates. Therefore, channels 12 and 13 involve multiple steps as seen in Fig. 2. One of these steps, the loss of the H atom from the intermediates, is associated with a large reaction barrier and therefore will be unlikely to occur directly. This barrier would be lowered and possibly entirely removed if the H atom is later abstracted by another radical, R (H or OH or NH2):
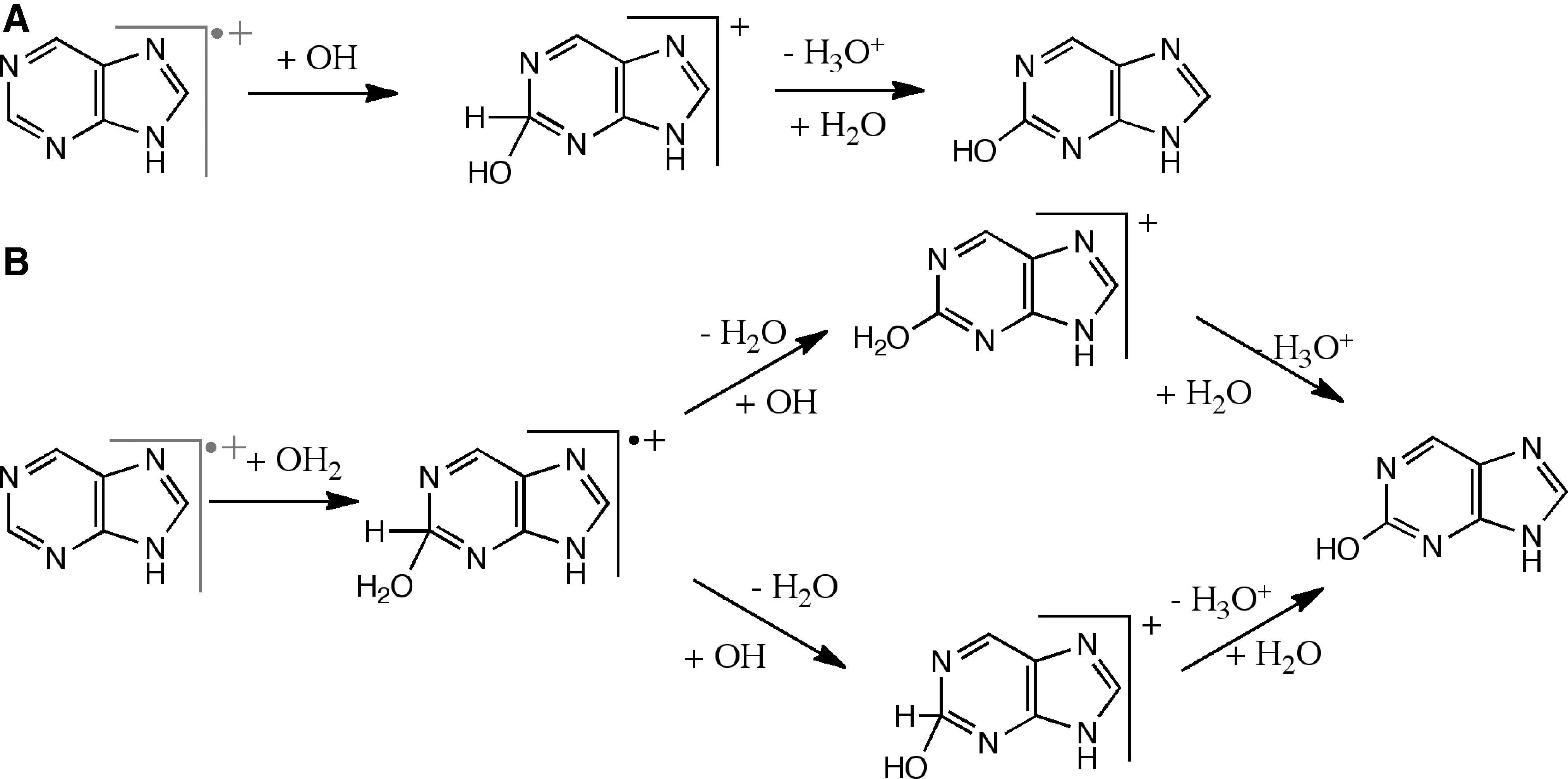
Mechanisms of OH (
Finally, proton abstraction would yield the final product in a third reaction:
Pathway A on Fig. 2 is a nucleophilic substitution pathway that proceeds via secondary reactions 14 and 15 between purine radical cation and OH radical (or NH2 radical). The ionization of an electron from purine's π-electron system leaves a hole in the aromatic system. The charge is likely to be primarily on the less electronegative carbon atoms. A radical NH2 or OH group can then attack one of the two carbon atoms (C2 or C6) on the six-membered aromatic ring, or the C8 carbon atom on the five-membered ring. Addition to the nitrogen atoms on the ring is also theoretically possible. This channel is most likely to proceed in high radiation conditions as it requires simultaneous presence of two reactive intermediates derived from the primary photochemical pathways. It has been shown that a reaction between a polycyclic aromatic hydrocarbon and hydroxyl is exothermic and barrierless in the entrance channel (Ricca and Bauschlicher, 2000).
Figure 2, pathway B shows the intermediates associated with reactions 18–20. The radical cation complex, 18, after H2O addition (Fig. 2B) loses a hydrogen atom and a proton in stepwise fashion (concerted H2 + loss is unlikely) to yield hydroxypurine. This may happen by H abstraction (19), followed by H+ loss (20); it can also occur in the reverse order. Note that there are two places from which H (or H+) can be abstracted—either from CH at the sp3 site or from H2O as seen from Fig. 2. Our calculations suggest that H is abstracted first with the help of an OH radical to form a closed-shell cationic intermediate. This intermediate then undergoes proton loss with the help of the surrounding H2O molecules, similar to pathway A.
Previously, while studying similar mechanisms involving the pyrimidine (C4H2N2) molecules, we found the ionic pathway to be the most important and efficient one (Bera et al., 2010; Sandford et al., 2015). Nucleobases are known to undergo a number of bond-breaking and structural transformations due to ionizing radiation (Bera and Schaefer, 2005; Lind et al., 2006). Henceforth, in this paper we will be discussing the results involving the cationic pathways (coming from 14 and 15 directly, or from 12 and 13 indirectly). We further discuss the likelihood of the ionic mechanism in mixed molecular ices over other types of mechanisms in Section 4.
Dehydrogenated purine radicals (Pu/H) generated by CH or NH bond breaking in purine can undergo reaction with OH or NH2 radicals to make hydroxylated or aminated products (16 and 17). We found that these types of reactions can directly produce the products and are exothermic. However, because they require the simultaneous presence of two reactive species produced from the primary radiative processes, they will only be important when the concentration of such species becomes similar to the parent species. Of course, H atoms generated from H2O and NH3 can react with Pu/H to regenerate neutral purine under high radiation exposure. Those H atoms can also react with purine neutral to produce hydrogenated purine.
OH radical could further oxidize products of oxidation of purine and lead to the formation of doubly oxidized products. This could occur following ionization of the monohydroxy product (as in channel 21), or without (channel 22):
The above ion-radical and radical-molecule reactions are generally fast and typically barrierless in the entrance channel. It was shown earlier that the proton loss from the protonated intermediates in aqueous media is spontaneous (Bera et al., 2016).
We explored these mechanisms to discern whether the reactions between these reactants leading to the functionalized products are favorable and whether the formation of adenine and guanine is favored over other plausible products. We also explored the catalytic activity of the H2O, NH3, and mixed molecular ices. These quantum chemical data were used to interpret the laboratory experimental results.
In Fig. 1, we present a summary of the stepwise substitution of NH2 and OH groups on the purine ring leading to important singly and doubly substituted products. The horizontal arrows refer to the hydroxyl (OH) group substitution, and the vertical arrows refer to the amino (NH2) group substitution on the ring. After the first OH substitution on the six-membered ring, oxidized purines such as hypoxanthine (6-hydroxypurine) and 2-hydroxypurine (also known as purinol) are produced. An additional hydroxyl substitution at the C2 position would lead to xanthine (2,6,-dihydroxypurine). Both hypoxanthine and xanthine have been detected in the experimental samples produced from the UV irradiation of H2O:purine and H2O:NH3:purine mixed molecular ices (Materese et al., 2017). An NH2 group substitution to hypoxanthine leads to the formation of guanine, while an NH2 group substitution to 2-hydroxypurine leads to the formation of isoguanine. An NH2 group substitution either to the 2 or the 6 position first on the purine ring gives 2-aminopurine or adenine (6-aminopurine), respectively. A second NH2 group addition on adenine on the six-membered purine ring leads to the formation of 2,6-diaminopurine. A hydroxyl group addition to adenine gives isoguanine, and hydroxyl group addition to 2-aminopurine produces guanine. 2,6-Diaminopurine, guanine, and isoguanine were all detected in the experimental samples after irradiation of purine in mixed molecular ices (Materese et al., 2017). In the following sections, we explore these reactions stepwise via the reaction mechanisms described above.
3.1. Step 1: First substitution
3.1.1. Amine group substitution to purine
An NH2 group can attach barrierlessly with the carbon framework of the purine cation to form three intermediates (see Fig. 3). The 2(2H)-aminopurine and 6(6H)-aminopurine cations (labeled 3b and 3a in Fig. 3) are 57.8 and 54.0 kcal/mol below the reactants, respectively. The 8(8H)-aminopurine cation (labeled 3c in Fig. 3) is the lowest-energy intermediate, 68.2 kcal/mol below the reactants, namely a bare purine cation, H2O, and an NH2 radical. In other words, the 8(8H)-aminopurine molecule has a larger proton affinity because it preserves the aromaticity of the six-membered ring compared to the proton affinities of the 2- and the 6-aminopurine when the proton attaches with the carbon framework. A proton will be lost to produce 2-, 6-, and the 8-aminopurine. Such a purely gas phase loss of the proton is energetically unfavorable, as the energies of the products are higher than the isolated reactants devoid of H2O. When a H2O molecule was explicitly included, and the proton was allowed to be abstracted by an H2O molecule, the combined energy of the products and a H3O+ falls below that of the reactants and the intermediates, as seen in Fig. 3. This situation is similar to pyrimidine oxidation, where we found that the reaction requires the presence of multiple H2O molecules that can take the proton away from the charged intermediates (Bera et al., 2010). Adenine, 6-aminopurine, is the lowest-energy product at about 73.1 kcal/mol below the reactants. 2-Aminopurine is slightly above adenine, followed by 8-aminopurine, which is an even higher-energy structure. Adenine is the most likely amine product after the first amino group addition to purine and is only synthesized in the presence of a mixed H2O-NH3 ice.
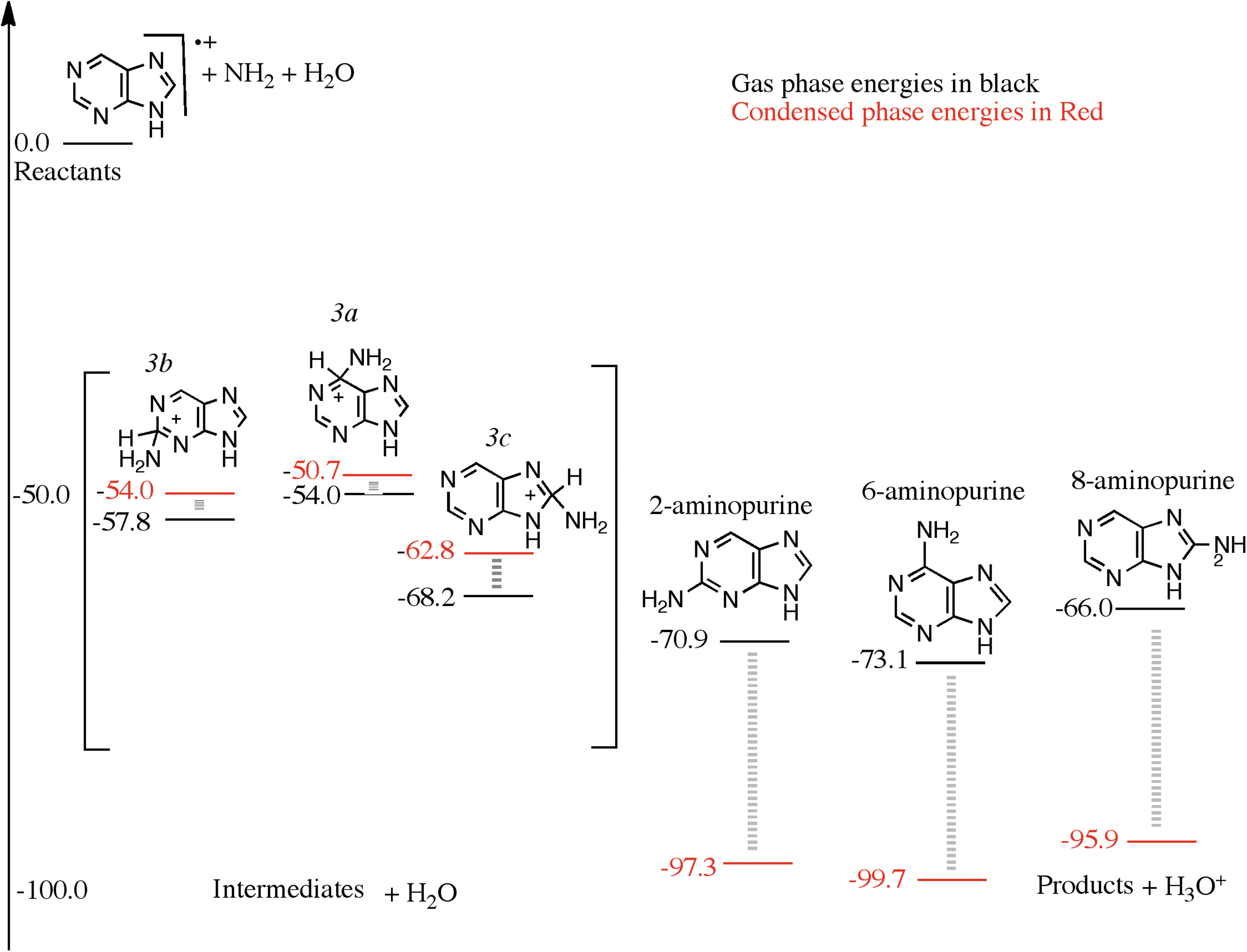
A reaction diagram for the cationic mechanism of amino group substitution to purine starting from purine cation, NH2 and H2O as reactants on the left, arbitrarily set to zero kcal/mol. The structures in the square brackets are those of the intermediates and, on the right, are those of the products. The black numbers were calculated using ωB97M-V/aug-cc-pVQZ in the gas phase, and the red numbers were calculated assuming a condensed phase environment, using a PCM.
Allowing this reaction step to take place in a condensed phase environment reveals interesting insights about this process. The reactants, purine cation, the hydroxyl radical, and one molecule of H2O were treated using a modified polarized continuum model with a dielectric to model the bulk water. The charged closed shell intermediates and the neutral products plus the hydronium ions were treated the same way, and their energies are given in Fig. 3. The red numbers were calculated for the reactants, intermediates, and products in the presence of the solvent. The presence of the solvent lowers the energies of the reactants, intermediates, and products but not by equal amounts. The combined energy of the reactants (purine cation, H2O and NH2 radical) was chosen as the zero in Fig. 3. The three cationic intermediates 2-, 6-, and 8-aminopurine cations are 54.0, 50.7, and 62.8 kcal/mol below the reactants, respectively. They are relatively less stabilized with respect to the reactants in the solvent than in the gas phase. This means that the reactants are slightly more stabilized by the solvent than the intermediates. The final products 2-, 6-, and 8-aminopurines are 97.3, 99.7, and 95.9 kcal/mol below the reactants, respectively, which shows that the products are much more solvent-stabilized compared to the intermediates. The relative energy lowering of the products due to solvation is approximately 30 kcal/mol. Gas phase calculations reveal that in this case only one H2O molecule is necessary to make this reaction favorable. The solvent phase calculations further establish the gas phase observations—that in the presence of H2O, the forward reaction is feasible, as shown in Fig. 3.
Finally, we note that there are certainly other intermediates that are possible, which is consistent with discussion given earlier on alternative mechanistic paths. Ammonia can form a complex with purine cation directly at the C2, C6, or C8 positions. Another radical (e.g., OH) can abstract H from either the sp3 CH bond or from NH3. The latter process yields intermediates 3a, 3b, and 3c, but the former process will produce lower-energy intermediates due to the basicity of amines. These protonated aminopurines may also be accessible by proton migration from 3a, 3b, and 3c.
3.1.2. Hydroxyl group addition to purine
In a H2O:NH3:purine mixed molecular ice, of course, the hydroxylation process is in competition with the amination in the first step. The H2O:NH3 ratio is approximately 10:1 in the experiments that have been recently carried out (Materese et al., 2017); hence, the OH radicals generated from H2O will have a higher probability to oxidize purine first. We examined the energies of the hydroxylation process by following a cationic mechanism. OH attack on the purine cation can occur at three different carbon atoms and produce 2(2H)-, 6(6H)-, and 8(8H)-hydroxypurine ion intermediates, as seen in Fig. 4 (labeled as 4b, 4a, and 4c, respectively). These intermediates lose a proton to form the products. 8-Hydroxypurine cation is the lowest-energy intermediate, 69.3 kcal/mol below the reactants, followed by the 2-hydroxypurine cation and the 6-hydroxypurine cation, 60.9 and 53.9 kcal/mol below the reactants, respectively. Allowing an H2O molecule to explicitly abstract the proton from the intermediates creates hydronium ion (H3O+) and three products: 2-hydroxypurine, 6-hydroxypurine, and 8-hydroxypurine, 77.2, 76.2, and 74.4 kcal/mol below the combined energy of the reactants, respectively (Fig. 4). The 2-hydroxypurine is the most favorable isomer if one follows a pure gas phase mechanism among the three monohydroxylated products examined in Fig. 4.
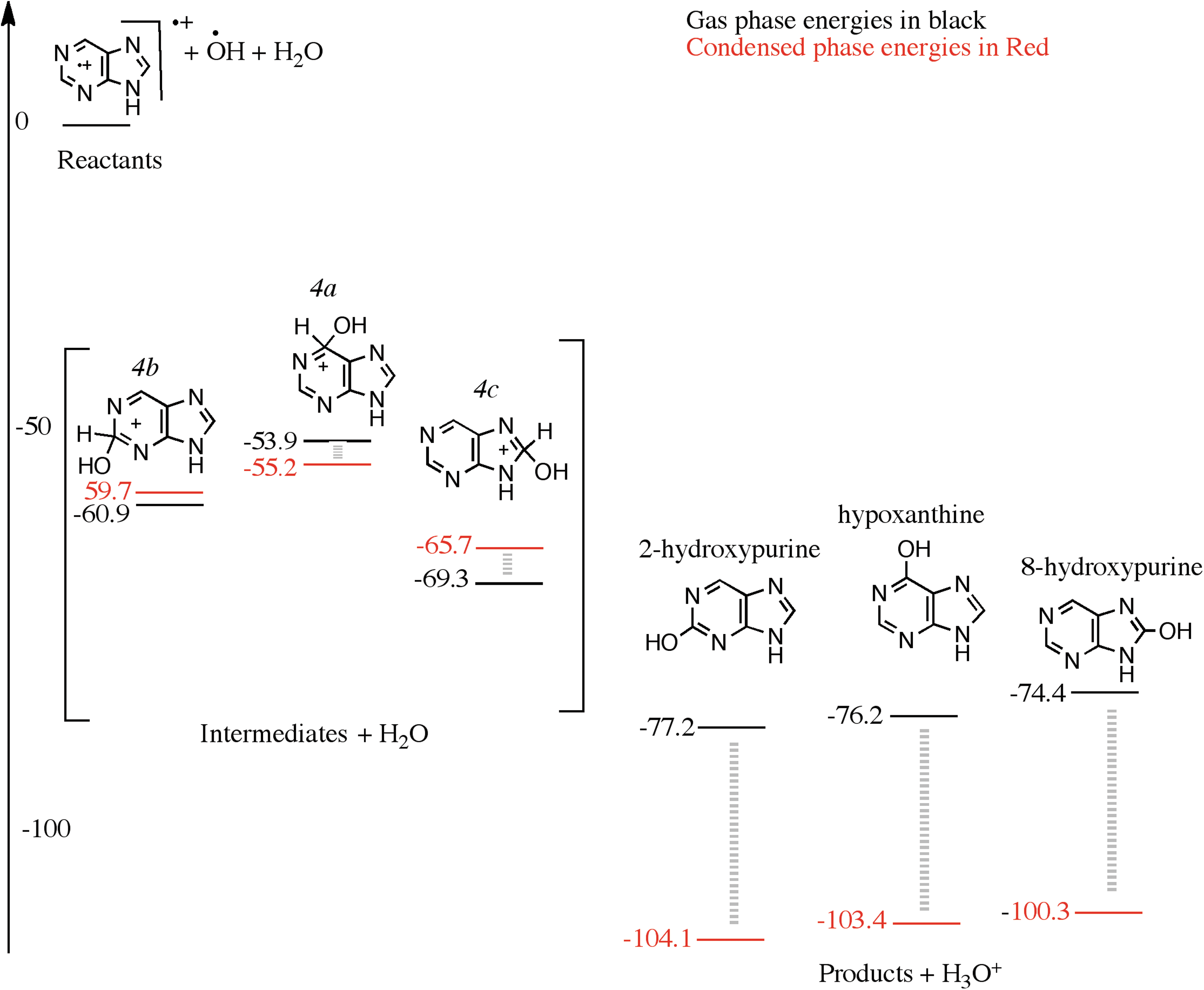
A reaction diagram for the cationic mechanism of hydroxyl group substitution to purine starting from purine cation, OH and H2O as reactants on the left, arbitrarily set to zero kcal/mol. The structures in the square brackets are those of the intermediates and, on the right, are those of the products. The black numbers were calculated using ωB97M-V/aug-cc-pVQZ in the gas phase, and the red numbers were calculated assuming a condensed phase environment, using a PCM.
An interesting difference from the hydroxylation of pyrimidine molecule is that it is relatively easier to deprotonate the hydroxypurine cationic intermediates. In the case of pyrimidine hydroxylation (Bera et al., 2010), multiple H2O molecules, that is, a condensed phase–like environment, were necessary for hydroxylation to proceed. In the case of purine, we see that only one H2O molecule is enough for the reaction to proceed. As shown below, however, the reaction still proceeds more readily in the ice matrix rather than in the gas phase via a purely bimolecular process.
In the condensed phase, the energies of the intermediates 8(8H)-hydroxypurine, 2(2H)-hydroxypurine, and 6(6H)-hydroxypurine cations (4c, 4b, and 4a, respectively, in Fig. 4) are 65.7, 59.7, and 55.2 kcal/mol below the reactants, respectively. The three products 2-hydroxypurine, 6-hydroxypurine, and 8-hydroxypurine, along with the H3O+, are 104.1, 103.4, and 100.3 kcal/mol below the reactants, respectively. The products are stabilized by about 30 kcal/mol in the condensed phase due mainly to charge delocalization in the water matrix. In the gas phase, the charge is more localized on the purine cation. The condensed phase calculations prove that the H2O matrix helps abstract the proton from the intermediate and facilitate the reaction. The relative energy ordering of the three products stays the same in both the gas phase and condensed phase calculations.
In the second step below, we will examine the amine and hydroxyl group addition to the preferred products after the first step.
3.2. Step 2: Second substitution
Following the amine and hydroxyl group additions to the purine ring, the process may continue, and further substitution of amine or hydroxyl groups may take place. In the following section we address the addition of NH2 to 2- and 6-hydroxypurine and OH to 2- and 6-aminopurine. We subsequently consider amine group addition to monoaminopurine and hydroxyl group addition to hydroxypurine.
3.2.1. Amino group addition to 2- and 6-hydroxypurine(s)
Following mechanisms similar to those we investigated in the previous steps, we investigate the reactions of the 6-hydroxypurine and 2-hydroxypurine cations with the NH2 radical in Fig. 5. In Fig. 5, the combined energy of the 6-hydroxypurine radical cation, NH2 radical, and H2O is set to be zero. All the other species' energies are plotted with respect to this zero energy. The 2-hydroxypurine cation is 6.3 kcal/mol below the energy of the 6-hydroxypurine cation. The energies and the structures of the intermediates and products that originate from the 2-hydroxypurine cation are depicted in blue in Fig. 5. The cationic intermediates, produced after nucleophilic attack of the NH2 radical on 6-hydroxypurine cation, are 46.9 and 65.8 kcal/mol below the reactants (colored black in Fig. 5). Upon proton loss to a nearby water molecule, the two products, guanine and 8-amino-6-hydroxypurine, are 63.9 and 58.3 kcal/mol below the reactants, respectively. Guanine (along with H3O+) is below the reactants and the proton-attached intermediate (5a) in terms of energy; the other intermediate (5b) is below the deprotonated product by 7.5 kcal/mol.
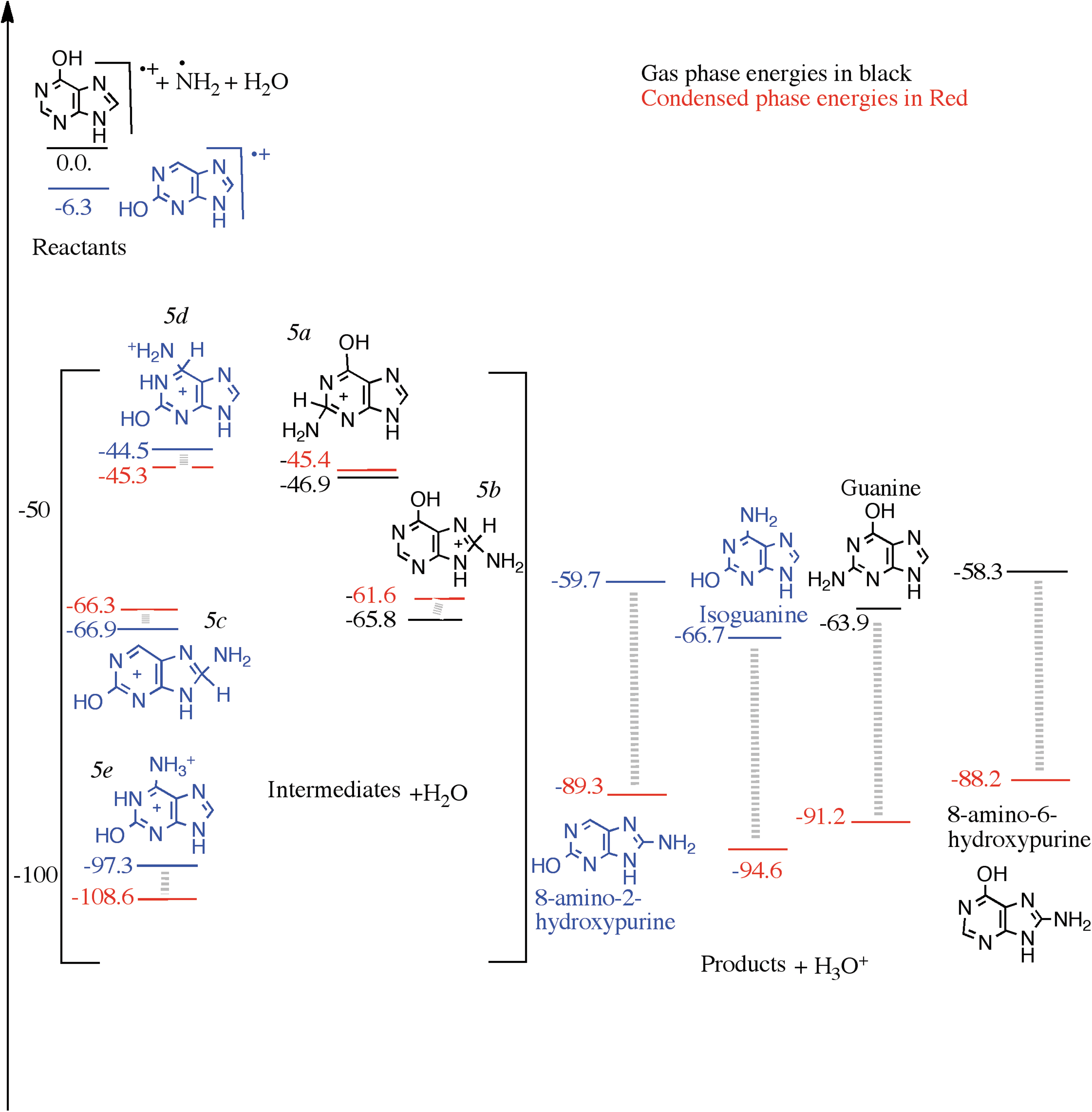
A reaction diagram for the cationic mechanism of amino group substitution to 2-hydroxypurine and 6-hydroxypurine starting from their cations, NH2 and H2O as reactants on the left, arbitrarily set to zero kcal/mol. The structures in the square brackets are those of the intermediates and, on the right, are those of the products. The black numbers were calculated in the gas phase at the ωB97M-V/aug-cc-pVQZ level of theory, and the red numbers were calculated assuming a condensed phase environment, using a PCM.
Some of the intermediates associated with NH2 attack on the 2-hydroxypurine cation include structure 5c (−66.9 kcal/mol), 5d (−44.5 kcal/mol), and a rearranged form of 5d at 97.3 kcal/mol below the reactants. The unusually low energy of the latter intermediate, which is isoguanine protonated at the amine group (5e), relative to the other intermediates stems from the proton having moved from the carbon atom (C6) to the amine group, giving the nitrogen atom a formal positive charge. This is also the structure that results from direct attack of NH3 at the C6 position of 2-hydroxypurine cation. Similar rearrangements of intermediates 5a, 5b, and 5c are possible and, because of the relatively high basicity of amines, should also be significantly lower in energy. Upon loss of a proton, the lowest-energy intermediate produces isoguanine with energy 66.7 kcal/mol below reactants, and the other intermediate produces the 8-amino-2-hydroxyl-purine with relative energy of −59.7 kcal/mol. The monohydroxylated purines prefer to attach an amine group on the six-membered ring. 2-Hydroxypurine, which is the preferred product at the first hydroxylation step (Fig. 5), produces isoguanine, and 6-hydroxypurine produces guanine after amine group addition.
In the condensed phase, the combined energies of the reactants, that is, 6-hydroxypurine cation (or 2-hydroxypurine cation), OH radical, and one H2O molecule calculated in the presence of the solvent, have been again set to zero. Relative energies of the intermediates (OH group attached cationic intermediates and H2O) and the products (monoamino monohydroxy purine and H3O+) are also plotted in Fig. 5. As for the gas phase, one of the intermediates (5e) is unusually stabilized due to the movement of the proton from carbon to the NH2 group. The products are lower in energy compared to the reactants as well as the intermediates except for that one case in which the proton has moved. The loss of the proton from the C6 carbon, however, is energetically feasible and likely driven by the large gas phase proton affinity of isoguanine.
3.2.2. Hydroxyl group addition to 2- and 6-aminopurine(s)
Starting from 2-aminopurine and 6-aminopurine cation, a monohydroxylation produces four cationic intermediates. The two intermediates, 2(2H)-hydroxy-6-aminopurine and 8(8H)-hydroxyl-6-aminopurine (labeled 6a and 6b in Fig. 6) produced from the 6-aminopurine and hydroxyl radical, are 41.1 and 65.3 kcal/mol below reactants (colored black). The intermediates 6c and 6e (colored blue) produced from the 2-aminopurine cation and OH radical are 71.2 and 87.1 kcal/mol below the reactants. These energies are lower than the product energies as seen in Fig. 6. The two products produced from the monohydroxy-2-aminopurine intermediates are guanine, which is 51.5 kcal/mol below the reactants, and 8-hydroxy-2-aminopurine, which is 41.5 kcal/mol below the reactants. The products associated with the 6-aminopurine are isoguanine, which is 54.3 kcal/mol below the reactants, and 8-hydroxy-6-aminopurine, which is 43.5 kcal/mol below the reactants. In this case, although well below the reactants, the products are not below the intermediates when only one water molecule is involved. A water environment is therefore necessary to deprotonate the intermediates and produce the products. Again, the monoaminated purines prefer to attach a hydroxyl group to the six-membered ring and especially either on the 2 or the 6 carbons. Isoguanine is slightly more energetically favored over guanine. Both isoguanine and guanine can be found in the experimental samples, isoguanine being more abundant than guanine.
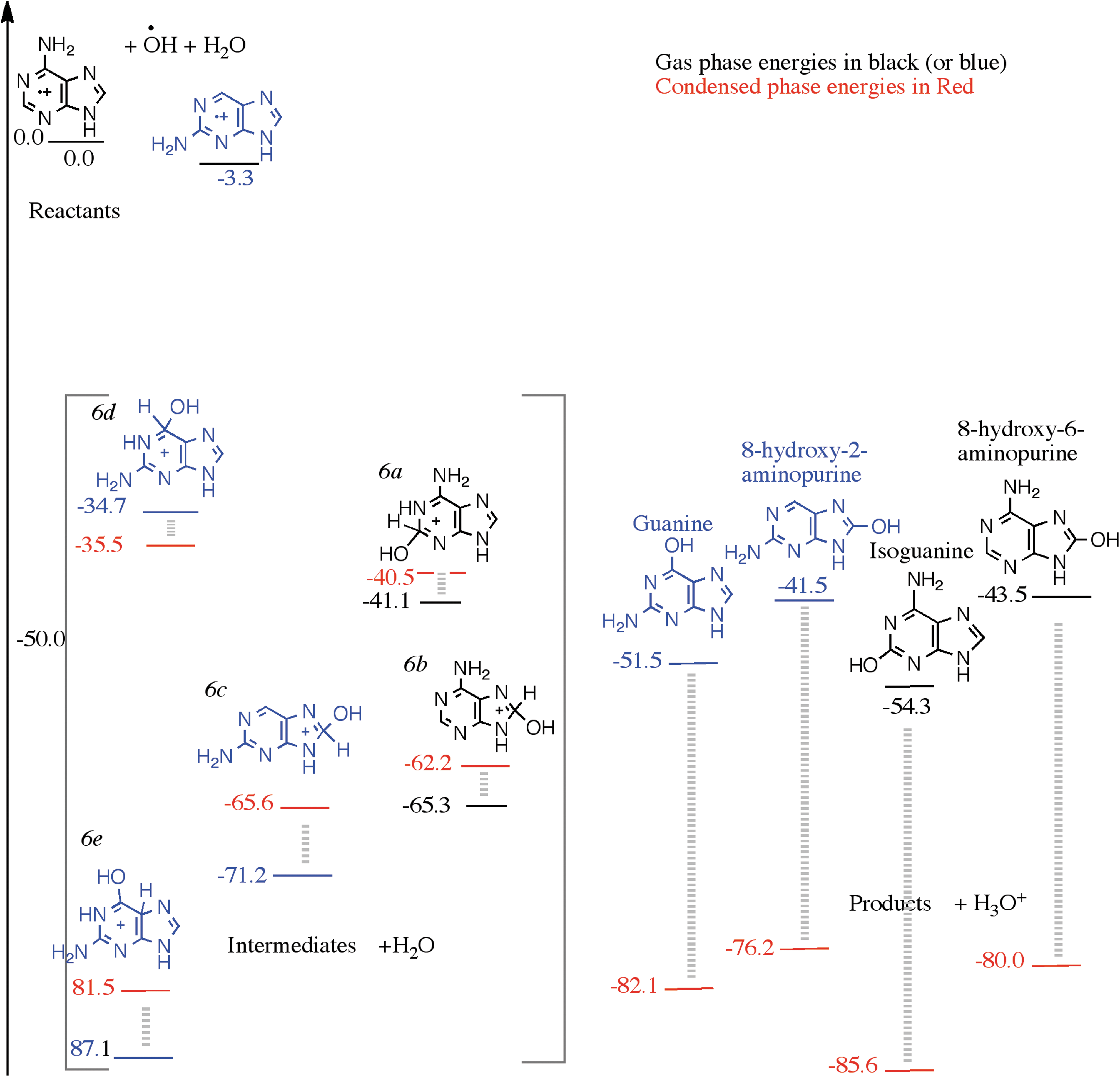
A reaction diagram for the cationic mechanism of amino group substitution to 2-aminopurine and 6-aminopurine starting from their cations, OH and H2O as reactants on the left, arbitrarily set to zero kcal/mol. The structures in the square brackets are those of the intermediates and, on the right, are those of the products. The black numbers were calculated using the ωB97M-V/aug-cc-pVQZ level of theory, in the gas phase, and the red numbers were calculated assuming a condensed phase environment, using a PCM.
When examining the reaction by the condensed phase calculations, we again keep the reactants 2-aminopurine and 6-aminopurine cations, OH radical, and H2O as the starting point. The monohydroxy-monoaminopurines are products, along with H3O+. The products are again stabilized by about 30 kcal/mol compared to the gas phase due to the H2O solvent effect. The intermediates remain more or less at the same level as in the gas phase. Three out of the four products are below the energies of the intermediates shown in Fig. 6 in the condensed phase.
One intermediate [derived from 6d by proton migration from C6 to C5 but not shown in Fig. 6 (6e)] shows larger stabilization and is 83.5 kcal/mol below the reactants. This is due to allowing the structure to pucker slightly in the gas phase. This intermediate (plus a H2O) has energy comparable to the product guanine and H3O+. Other lower-energy protonated intermediates are possible if solvent-assisted proton migration is feasible from the site of hydroxyl attack to an amine site, which is more basic. This yields amine-protonated guanine or isoguanine, and so on. The icy environment could facilitate such proton shuttling. Finally, both in the gas phase and in the condensed phase calculations, we find that isoguanine has a lower energy relative to guanine. The relative energy ordering of the products also remains the same.
3.2.3. Amine group non-addition in pure ammonia ice
When the experiments were performed in a pure ammonia ice, no products were observed, except for perhaps a trace amount of adenine (Materese et al., 2017). These results are similar to the pyrimidine irradiation in pure ammonia ices. This lack of product formation is in sharp contrast to the amounts of products produced in the mixed H2O and NH3 molecular ices and pure H2O ices. The UV source used for the irradiation (10.2 eV) is enough to ionize NH3 to its cation (gas phase threshold is 10.1 eV), as well as to photolyze it to yield NH2 radicals (NH2) as discussed earlier. A reaction mechanism could be imagined in which a purine radical cation reacts with the amine (NH2) radical to produce initial intermediates, which lose a proton to the neighboring NH3 molecules to produce the products and NH4 + ion. This reaction sequence is plausible since the proton affinity of NH3 is large, 211 kcal/mol (Czakó et al., 2008), and there are 10 NH3 molecules to each purine molecule in the pure NH3 ices. Lack of experimentally detected products in the pure ammonia ice, however, suggests that some characteristic of the ammonia ice precludes this pathway.
Indeed, in a similar ice experiment, Cuylle et al. (2012) showed that, in Lyman α irradiated ammonia ice seeded with pyrene, its anions are exclusively produced as a result of free electrons generated by ionization of NH3, and the fact that the ratio of NH3 to pyrene was large (5000:1). On the other hand, in water ices (also 5000:1 ratio), pyrene cations are produced presumably by direct ionization, as Lyman α does not ionize water molecules; therefore, stray free electrons are not available. In mixed H2O and NH3 ices, when keeping the ice:pyrene ratio at 5000:1, the results depended on the relative abundances of H2O and NH3. Under conditions similar to the experiments we are modeling (H2O:NH3 ratio is 10:1), anion signal was quenched, and only pyrene cations were produced.
Purine has a larger electron affinity than pyrene and, hence, would exclusively yield purine anions in Lyman α irradiated NH3 ice experiments. A reaction of the purine anion with NH2, or other radicals, or formation of a coordination complex with NH3, would be unfavorable from an electrostatic point of view. This is presumably the reason that that product formation was not observed in experimental studies of purine in NH3 ice (Materese et al., 2017). In a mixed ice with H2O:NH3 ratio of 10:1, we expect the cations of purine to be present, and the cationic reaction mechanism would yield all the products that have been predicted in this work or seen by the experiments.
4. Discussion
Interestingly, we find that the amine group preferentially binds at the 6 position on the purine ring, while the hydroxyl group prefers the 2 position. As a consequence, adenine is the thermodynamically preferred monoaminated product, and 2-hydroxypurine is the monohydroxylated preferred product. In Fig. 1, the upward or the left paths seem to be the way forward after the first step.
Both NH2 and OH radicals are very effective nucleophiles. From our calculations, we do not observe much difference in reaction energies for the reaction of purine with either NH2 or OH. Both the set of monoaminated and monohydroxylated products are expected to form after the first set of substitutions. Based on thermodynamics, the first NH2 substitution leads to adenine, while the first OH substitution leads to hypoxanthine. Therefore, after the first step, adenine and 2-hydroxypurine are expected to be the most favored products, although 2-aminopurine and 6-hydroxypurine, that is, hypoxanthine, are also expected to form as they are very close in energy, and there is no barrier to these reactions. Hypoxanthine and 2-hydroxypurine are more likely to form, as H2O is 10 times more abundant in the reaction mixture, and therefore hydroxylation has a higher chance of occurring. Experimentally, hypoxanthine was found to be the most abundant product followed by adenine; 2-hydroxypurine could not be tested, as a standard was not available. Nevertheless, the most favored products give us a path forward for further reactions.
If the NH2 group addition is preferred over OH group, then adenine would be the energetically most-favored product after the first NH2 group addition, and it would be a logical place to start for further NH2 or OH group addition. An OH group preferentially attaches with adenine at the 2 position leading to isoguanine, which again is one of the main products. If we started from 2-aminopurine, then guanine is the most favored product after OH group addition. Isoguanine is lower in energy compared to guanine by 3.5 kcal/mol in the condensed phase calculations and thus should be an energetically preferred product. It can of course add another NH2 group to produce 2,6-diaminopurine, and it does form in the experiments. Therefore, there is no direct path to make guanine from adenine, but there exists a path to form isoguanine, which is why isoguanine is a major product in the experiment.
If, however, OH group addition is preferred over NH2 group addition in the first step (following the horizontal paths in Fig. 1), then we expect to see either 2-OH-purine or hypoxanthine after the first step. Since 2-hydroxypurine is the most favored product, it can act as a starting point for further functionalization. An NH2 group addition to 2-hydroxypurine would preferentially make isoguanine over the other products as seen in Fig. 5. If, however, we started from hypoxanthine, which is a less favored product after the first hydroxylation, then we get guanine.
Thus, starting from either adenine or 2-hydroxypurine—the major products of the first step—we see that isoguanine is the most favored product. To produce guanine, we need to start OH or NH2 group addition to the less favored products after the first step. This indicates that isoguanine would probably be significantly in higher abundance than guanine, because of the difficulty in formation of the latter. Experimentally, Materese et al. (2017) found that the singly substituted products hypoxanthine, adenine, and 2-aminopurine are the most abundant products. Doubly substituted products are an order (or orders) of magnitude less abundant than the singly substituted ones. Xanthine, isoguanine, guanine, and 2,6-diaminopurine are the most abundant doubly substituted products, in order of their abundance.
4.1. Validation of cationic mechanisms for hydroxylation and amination
Lack of detection of products in the pure NH3 samples validates the cationic reaction mechanism over the other types of mechanisms. If other mechanisms were responsible, or dominant over the cationic mechanism, for hydroxylation and amino group substitution, then we would expect to see products formed in irradiated pure NH3 ices as well. Since products are not formed in the pure NH3 ices, we can conclude that anionic purine does not lead to hydroxyl or amine substitution, at least in an efficient manner. On the other hand, positive detection of products in the 10:1 H2O:NH3 and pure H2O irradiated ices, where cations of purine would be produced as shown by Cuylle et al. (2012), strongly indicates that the reaction proceeds through the cationic mechanism. Ions are reported to play an important role in hydroxyl group addition to polycyclic aromatic hydrocarbons as well (Guennoun et al., 2011a, 2011b; Cook et al., 2015).
4.2. Astrobiological significance
Our results indicate that the nucleobases adenine and guanine can be photochemically synthesized in the condensed phase in a mixture of purine, water, and ammonia under UV irradiation. The reaction proceeds via an ion-radical-type mechanism and does not require going over any barriers and therefore could perhaps happen in cold temperatures, such as on the icy mantles of meteorites. Adenine and hypoxanthine are the most likely mono-substituted products, whereas isoguanine, followed by guanine, are the most likely bi-substituted products. We show a sequential nature of functional group substitution in purine. Of course, nothing in our calculations indicates that other processes leading to the formation of less energetically favorable products are prohibitive. Other possibilities abound, but further exploration is beyond the present scope.
Hypoxanthine (also known as d-Inosine) has been demonstrated to preferentially base pair with cytosine, indicating its guanine surrogate capability (Budke and Kuzminov, 2006). Further, a type of RNA editing in cells converts adenosine to inosine (hypoxanthine) in order to exploit its base-pairing abilities with cytosine (Nishikura, 2010). These facts indicate that hypoxanthine, despite not being one of the biological bases present in DNA and RNA, could have played an important role in prebiotic chemistry. Our results show the mechanism by which hypoxanthine, as well as the nucleobases adenine and guanine, can be synthesized easily in the irradiated ices if purine is present.
5. Conclusions
We explored the range of possible products resulting from hydroxyl and amino group substitutions in purine present as a dopant in mixed 10:1 water-ammonia molecular ices. Based on quantum chemical calculations, we find that both amino and the hydroxyl group substitutions are energetically favorable processes and that the 2 and 6 positions in the six-membered cyclic ring are the most favored positions for these functional group substitutions. The energies of amine group substitutions were slightly lower than the energies of hydroxyl group substitutions, indicating that amino group–substituted products may be dominant over hydroxyl group products. Nevertheless, the higher abundance of hydroxyl should produce higher yield for 2-hydroxypurine and hypoxanthine over adenine, and xanthine over 2,6-diaminopurine. Starting from 2-hydroxypurine, an amine group substitution should lead to the preferential formation of isoguanine over guanine as seen from the bottom right corner of Fig. 1. Starting from adenine should also produce isoguanine following the upper-left corner seen in Fig. 1. Starting from hypoxanthine, an amino group substitution would lead to guanine. This was supported by the experimental observation that the monoamino-substituted product adenine and the monohydroxylated product hypoxanthine were found to be in 1:4 ratios despite NH3 and H2O being present in a 1:10 ratio. Mono-substituted products were also almost an order of magnitude more abundant in experimental samples compared to the bi-substituted products. The product abundance is carried forward in the biamino and bihydroxy purine ratios as well. This is due mainly to the sequential nature of the substitutions and further establishes our sequential reaction mechanisms.
H2O plays multiple critical roles in the photoprocessing of purine in ices: as a reactant, as a catalyst, and as a solvent. H2O gives birth to hydroxyl radicals that take part in the hydroxylation reactions. H2O plays a central role as a catalyst in the substitution reactions by the cationic mechanism by abstracting a proton to make products. Further, the H2O matrix is essential not only to capture protons that are by-products of the reaction but also to permit survival of the reactant cations. In the absence of H2O, that is, in pure NH3 ices, anions (rather than cations) are produced, which impedes the reactions. These crucial roles of H2O are also evident from the experimental observation of no products in pure ammonia ices.
The role of the water matrix in the amination and hydroxylation is proven by the condensed phase calculations. In some cases (Figs. 2 and 3), only one H2O molecule was enough to make the reaction proceed; but in most cases, more than one H2O molecule, that is, an H2O matrix, is necessary. The C-PCM calculations indicated that the reactants, intermediates, and products all become solvated, but to different extents. The products are solvated better than either the reactants or intermediates, which helps the reactions go forward. This is in agreement with the previous and current gas phase calculations, which shows that the reaction is incomplete in the gas phase.
From our quantum chemical calculations, we predicted that adenine and 2-hydroxypurine are the most energetically favorable mono-substituted products, and isoguanine the most favored bi-substituted product. Experimentally, adenine and hypoxanthine (2-hydroxypurine standards were not available) are the most abundant products, followed by the bi-substituted products. We showed how, in the presence of the H2O matrix, the reaction proceeds to the products by the assistance of H2O. In a pure H2O ice, the reaction could proceed via the cationic mechanism. However, in the pure NH3 medium, where the cations are not produced but anions are created, no products are identified. The experimental nondetection of the products in pure NH3 ice supports this hypothesis. Together with the catalytic and solvation effects of H2O the matrix, all evidence indicates that that the cationic mechanism is operational in pure H2O and mixed molecular ices.
Footnotes
Acknowledgments
This material is based upon work supported by the National Aeronautics and Space Administration through the NASA Astrobiology Institute under Cooperative Agreement Notice NNH13ZDA017C issued through the Science Mission Directorate. This paper has greatly benefited from constructive discussions with Dr. Christopher Materese, Dr. Michel Nuevo, and Dr. Scott Sandford, who are simultaneously publishing a paper in Astrobiology (Materese et al., ![]() ) on the experimental investigations of the photoprocessing of purine in NH3:H2O ices.
) on the experimental investigations of the photoprocessing of purine in NH3:H2O ices.