
Abstract
Phosphorus ester hydrolysis is one of the key chemical processes in biological systems, including signaling, free-energy transaction, protein synthesis, and maintaining the integrity of genetic material. Hydrolysis of this otherwise kinetically stable phosphoester and/or phosphoanhydride bond is induced by enzymes such as purple acid phosphatase. Here, I report that, as in previously reported aged inorganic iron ion solutions, the iron oxide nanoparticles in the solution, which are trapped in a dialysis membrane tube filled with the various iron oxides, significantly promote the hydrolysis of the various phosphate esters, including the inorganic polyphosphates, with enzyme-like kinetics. This observation, along with those of recent studies of iron oxide, vanadium pentoxide, and molybdenum trioxide nanoparticles that behave as mimics of peroxidase, bromoperoxidase, and sulfite oxidase, respectively, indicates that the oxo-metal bond in the oxide nanoparticles is critical for the function of these corresponding natural metalloproteins. These inorganic biocatalysts challenge the traditional concept of replicator-first scenarios and support the metabolism-first hypothesis. As biocatalysts, these inorganic nanoparticles with enzyme-like activity may work in natural terrestrial environments and likely were at work in early Earth environments as well. They may have played an important role in the C, H, O, S, and P metabolic pathway with regard to the emergence and early evolution of life. Key Words: Enzyme—Hydrolysis—Iron oxide—Nanoparticles—Origin of life—Phosphate ester. Astrobiology 18, 294–310.
1. Introduction
T

2. Materials and Method
Deionized water (DIW) that was used for preparing standards and reagents was first purified by a distilling unit and then processed by a Millipore Super-Q Plus Water System that produces water with 18 MΩ cm resistance. All samples and reagents were stored in polypropylene bottles or Teflon or polyethylene terephthalate glycol containers that had been immersed in a 10% HCl solution overnight. This was followed by rinsing three times with DIW and then drying at 60°C in an oven for 5–10 h prior to use.
2.1. Preparation of IO and the DMT-IO solution
Iron oxide (IO) was synthesized following the basic protocol of Atkinson et al. (Atkinson, 1967). A 5 M NaOH solution was added dropwise (5 mL min−1) to a stirred 0.1 M Fe(NO3)3·9H2O or 0.1 M FeCl3 aqueous solution until the pH reached 7.5 ± 0.5. Nitrogen (N2) was continuously bubbled in the reaction vessel during the addition of NaOH to reduce carbon dioxide (CO2) contamination. The resulting suspension was aged at different temperatures (4°C, 20°C, 50°C, and 80°C) for 2 weeks in a capped Teflon container in order to produce IO nanocrystals. The synthesized iron oxide and its specific surface area depend upon the source of ferric ion (III), the temperature, the time of aging, and uncontrolled laboratory conditions (van Geen et al., 1994; Schwertmann et al., 1999; Colombo et al., 2015). It is assumed that ferrihydrite precipitates first but “matures” to hematite (Schwertmann et al., 1999; Jolivet et al., 2006; Fu et al., 2011; Colombo et al., 2015). These IO suspensions were then introduced into a dialysis membrane tube (DMT; Spectra/Por 1 Membrane, molecular weight cut-off [MWCO] of 6000–8000 Da) and washed repeatedly with fresh DIW until its electrical resistance exceeded 15 MΩ cm. It should be noted that some IO nanoparticles can be as small as 1 nm (Rose et al., 1997; Baumgartner and Faivre, 2015) and penetrate the dialysis membrane. The prepared iron oxides, based on the ferric sources and aging temperature, are listed in Table 2.
In this method, a 1 L sized Teflon container was filled with 800 mL fresh DIW. A 10 cm DMT, previously filled with the cleaned IO at room temperature (20–22°C), was then placed within the container. After soaking in the DIW for 2 or 3 weeks, the solution in the Teflon container was ready for study. The effect of the soaking duration of DMT-IO in DIW on the catalytic activity of hydrolysis phosphate ester was then explored. In the early stages of these experiments, I employed either azide (NaN3) or chloroform (CHCl3) to test for potential microbial contamination. No significant difference was observed between inhibitor-added and the inhibitor-absent experiments. Thus, in subsequent experiments reported here, microbial inhibitors were not used.
2.2. Measurement of hydrolysis rate of G6P, ATP, and PPi
In these experiments, the concentrated organic phosphate esters or the condensed inorganic phosphates (e.g., 100 μL) were added to 50 mL of the solution bearing the iron oxide nanoparticles or 50 mL of the fresh DIW (as blank control) and mixed together, and the period of time taken for the hydrolysis was measured. The increase in concentration of inorganic orthophosphate (Pi) resulting from the hydrolysis of a phosphate ester, for example, G6P, was measured at suitable intervals with a modified Murphy-Riley colorimetric method after G6P was added to the DMT-IO solution (Huang and Zhang, 2012). The concentration of G6P at a given time, t, was calculated from [G6P]
t
= [G6P]0 − [Pi]
t
, where [G6P]0 is the initial concentration of G6P and [G6P]
t
and [Pi]
t
are the time, t. A small amount of Pi initially present in the G6P as an impurity was subtracted as blanks. The first-order hydrolysis rate constant, k (s−1), was determined from the slope (K) of the linear portion of a ln [G6P]
t
-time plot (Huang and Zhang, 2012). The typical correlation coefficient of the linear fitting was 0.99*; the duration of the hydrolysis experiments ran up to 24 or 48 h. The half-life (t
0.5) of the first-order reaction is given by
2.3. pH adjustment and buffer solution
The pH of DMT-IO solution is generally mildly acidic, around 5.5–6.5. Sodium bicarbonate (NaHCO3) was employed for neutralization or pH adjustment. After the iron oxide nanoparticle solution was separated from the DMT-IO, up to 10% of the total volume of solution was added at a range of NaHCO3 concentrations (0.2–40 mM). The solutions were mixed with a magnetic stir bar for 2 h in the container and then brought to equilibrium within another 48 h. The pH was checked prior to the addition of 100 μL of the organic phosphate ester or condensed phosphate to 50 mL of each equilibrated nanoparticle solution, and the length of time taken for hydrolysis was recorded. It was noted that the buffer capacity of these nanoparticle solutions, after pH adjustment, was also changed.
To approach the large range of pH, four different buffer solutions (as 10% of the total volume) were further tested in this work. They included 0.1 M citrate buffer (pH 4.0–6.2), 1.0 M acetate buffer (pH 4.0–5.6), 0.2 M dimethyl glutaric acid buffer (pH 4.2–6.8), and 0.01 M bis-Tris buffer (pH 5.8–7.2). The difference of equilibrium pH, after 48 h, of nanoparticle solution and added buffer solution was less than 0.05 units. However, it was observed that the catalytic activity of the nanoparticle solution became negligible within 2 h after 0.1 M citrate buffer or 0.01 M bis-Tris buffer was added.
2.4. Environmental temperature
All the iron oxide nanoparticle solutions in this experiment were collected at room temperature (22°C ± 2°C). They were then placed in an environmental temperature incubator at 12°C, 22°C, 38°C, 52°C, or 72°C, respectively, for the temperature tests. After 4 h at the incubator, 100 μL of the phosphate ester or condensed phosphate was added in to 50 mL of each equilibrated nanoparticle solution, and the length of time taken for hydrolysis was recorded. It was noted that catalysis activity ceased as the solutions reached equilibrium after 16 h at 72°C. The hydrolysis rate constant (
where
For the storage experiment, the iron oxide nanoparticle solutions were first separated from the DMT-IO and then stored at the different target temperatures (−18°C [as F], 4°C [as L], 22°C [as R], and 50°C [as H]) in Teflon or polyethylene terephthalate glycol containers. The treatments included seven combinations of different temperature and period of time (total 11 days). For the 11R and 11L treatments, the solution was kept at 22°C or 4°C for 11 days, respectively. For the 5L6R treatment, the solution was initially stored at 4°C for 5 days and then kept at 22°C for an additional 6 days. For 5H6L, 5H6R, and 9H2R treatments, the solution was initially stored at 50°C for 5 days or 9 days and then kept at 4°C for an additional 6 days or 22°C for an additional 6 days or 2 days, separately. For the 5F6L treatment, the solution was initially stored at −18°C for 5 days then melted and kept at 4°C for an additional 6 days.
3. Results
As shown in Fig. 2a, the concentration of phosphate esters and condensed inorganic phosphate decreased, and inorganic orthophosphate (Pi) increased in the solution as a result of the hydrolysis reaction catalyzed by the iron oxide nanoparticles in the solution, which consists of a DMT (e.g., Spectra/Por 1 Membrane, MWCO 6000–8000 Da) filled with iron oxide (DMT-IO). At a fixed concentration of organic phosphate ester or condensed phosphate, the change of state of phosphorus can be described by a pseudo-first-order reaction (Huang and Zhang, 2012). For example, the 25 μM phosphorus concentration of the three different organic phosphate esters and two inorganic condensed phosphates at room temperature in an IO nanoparticle solution (pH 6.4 in DIW soaked with DMT-IO for 20 days prepared from Fe(NO3)3·9H2O and NaOH aged at 80°C for 2 weeks) can be expressed as a function of hydrolysis time, t, as
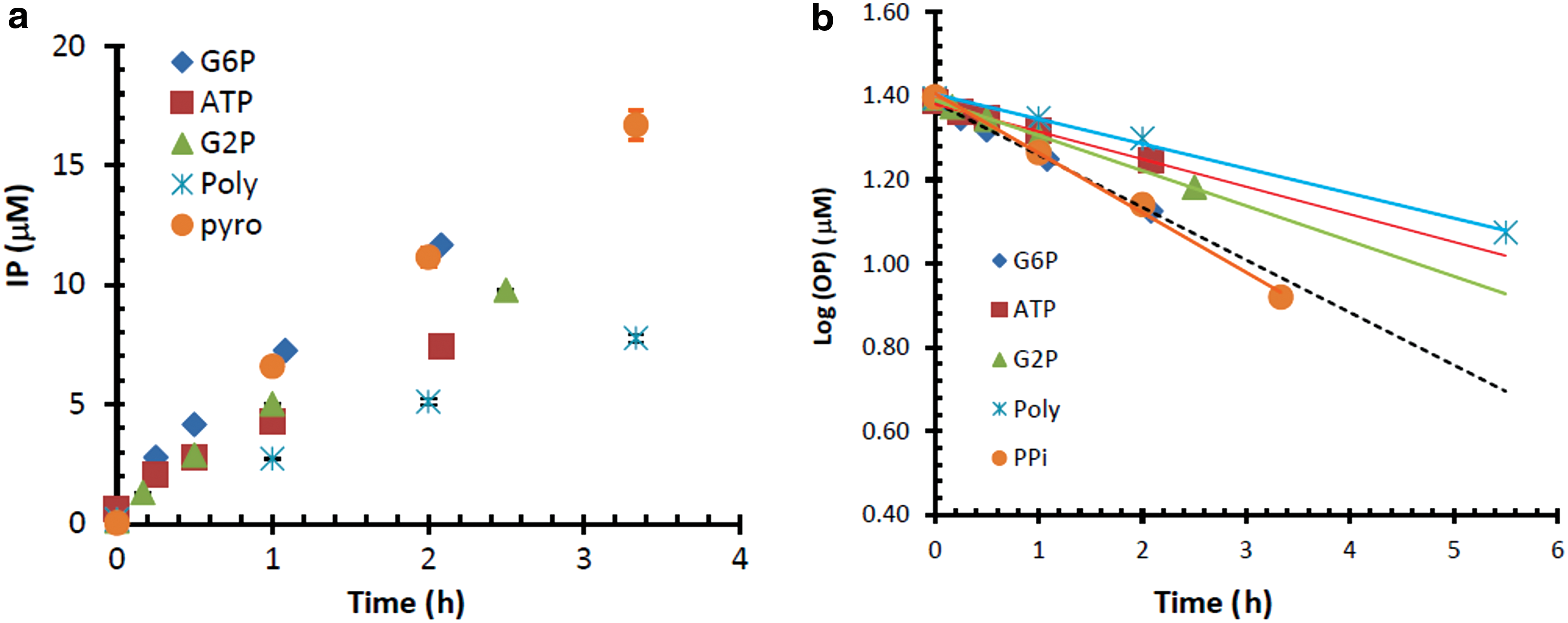
Hydrolysis kinetics of different phosphate esters and condensed phosphate in a DMT-IO (IO-D) solution. (
where the unit of organic phosphate esters or condensed phosphate is in μM and t is in h (Fig. 2b). The corresponding hydrolysis rate constant (k) for 25 μM G6P, G2P, ATP, poly-Pi, and PPi was 8.03 × 10−5, 5.37 × 10−5, 4.22 × 10−5, 3.78 × 10−5, and 9.11 × 10−5 s−1, respectively. The corresponding half-life (t 0.5) of phosphorus was 2.4, 3.6, 4.6, 5.1, and 2.1 h, respectively.
Similar to the natural PAPs, the t 0.5 of various organic phosphate esters or condensed phosphates at different concentrations was not constant. As shown in Fig. 3, the catalytic activity of the different concentrations of phosphorus can be described by the typical Michaelis-Menten equations. Based on the Lineweaver-Burk linear equation (1/V is a linear function of 1/[S]), the Michaelis-Menten constant (K m) and maximum velocity (V m), as well as the range of concentration of phosphorous among these compounds, were determined (Table 1). Usually, monophosphate has a large range with higher hydrolysis velocities. In contrast, the bi- or triphosphates have low concentrations and relatively low velocities, which may relate to the released orthophosphate and one of the inhibiting factors for its catalysis (Huang and Zhang, 2012). The same inhibiting patterns were also observed in these IO nanoparticles (data not shown). Meanwhile, the catalysis activity was still observed even when the phosphorus exceeded the range of the Michaelis-Menten equations, as with many of the natural enzymes, including the PAP (Bardsley et al., 1980).
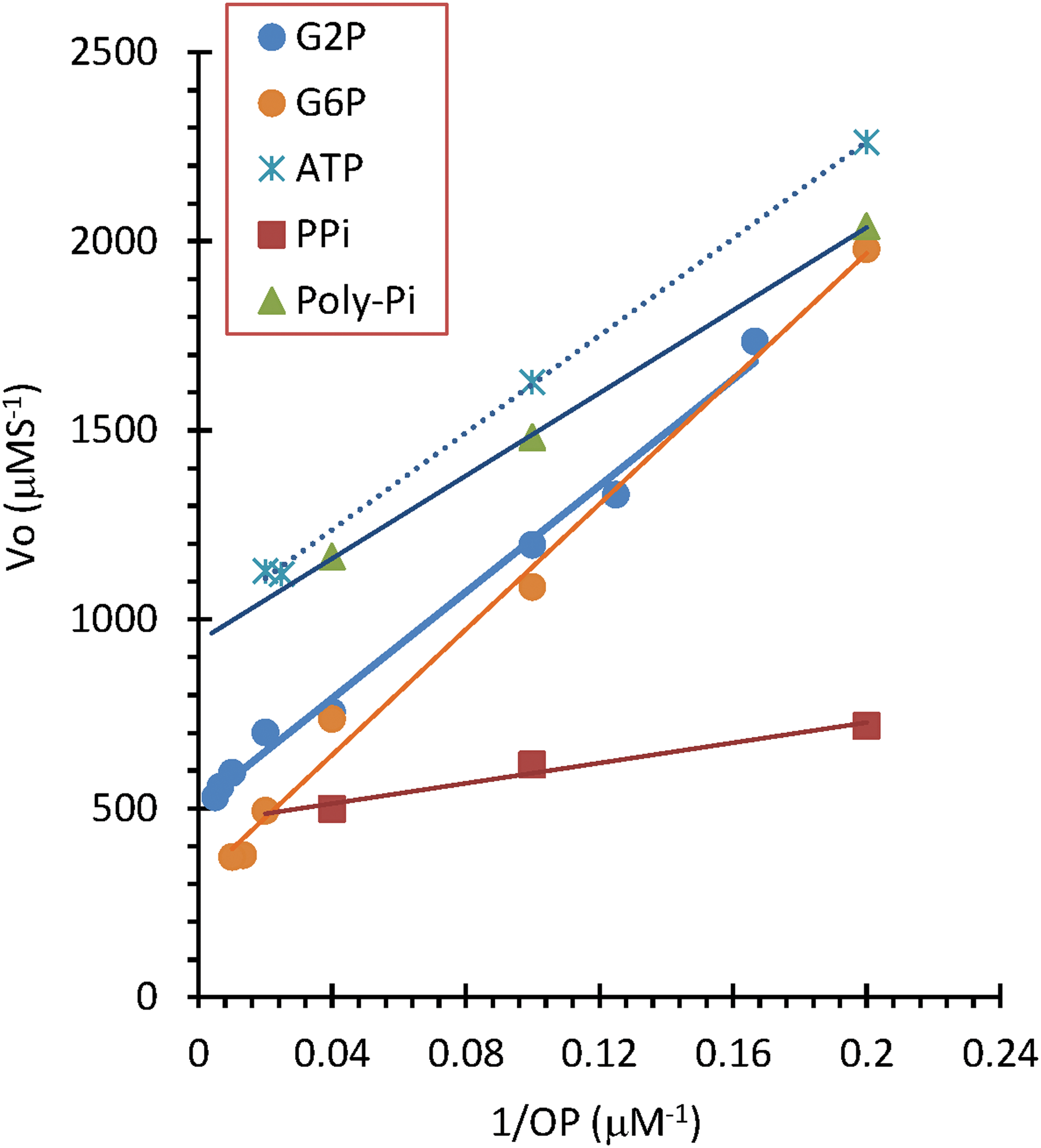
Lineweaver-Burk plot of different phosphate compounds in a DMT-IO solution (IO-D).
As expected, the catalytic activity, for example, the k, hydrolysis rate constant, is related to the soaked time of DIW with DMT-IO and the nature of IO. The kinetics of k of 100 μM G6P and ATP in three different IOs is presented in Fig. 4. These results can be explained by the changes of the nanoparticle concentration in the water. It was expected that the concentration of the IO nanoparticles in these solutions would initially increase up to 10 days and then reach equilibrium. However, the total dissolved iron concentrations in these solutions were still beneath the detection limits of iron (0.1 nM) (Zhang et al., 2001).
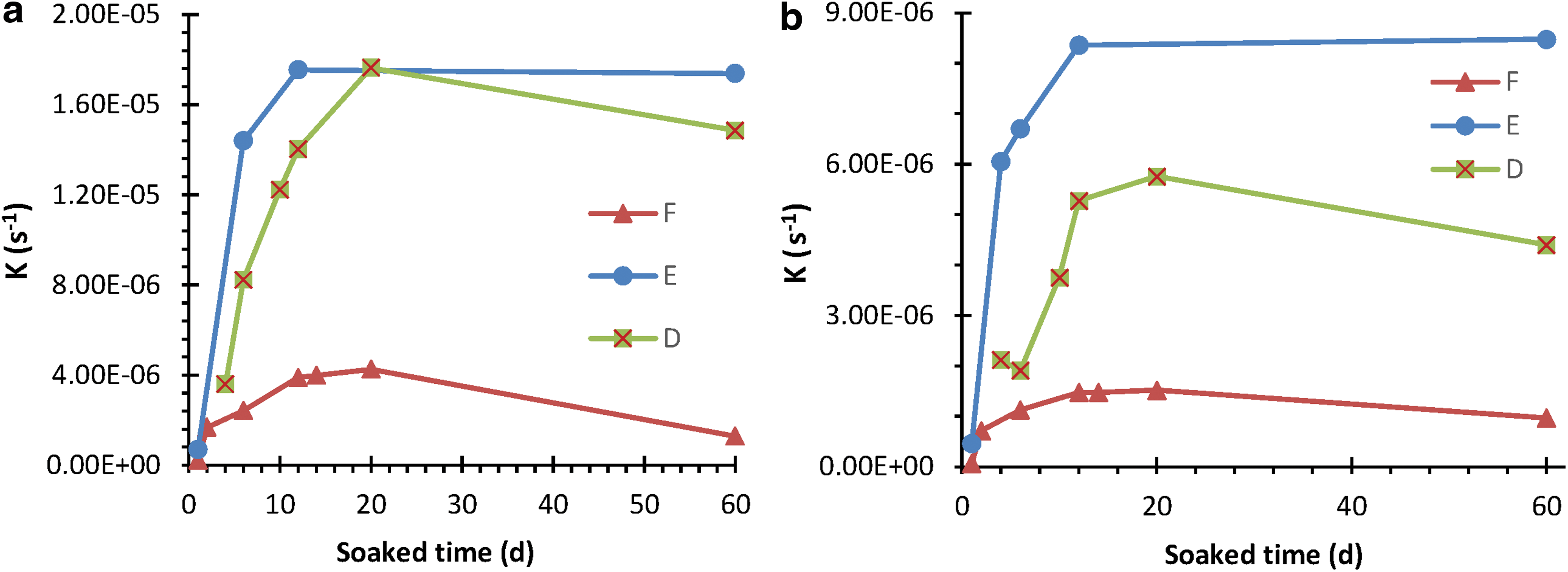
Relationship between the soaked time of IO and hydrolysis rate of phosphorus in different DMT-IO solutions. (
Table 2 further indicates, as expected, that the behavior of catalysis depends on the sources of iron, whether FeCl3 or Fe(NO3)3, and even on the different manufacturers, as well as with the different aging temperatures (5–80°C) (Schwertmann et al., 1999; Guo and Barnard, 2013; Baumgartner and Faivre, 2015). No clear relationships between ferric ion (III) sources, age processing, and catalytic activity, with the hydrolysis rate constant (k), were observed.
The hydrolysis reaction rate of 20 μM G6P and ATP in DIW is 1.84 × 10−8 and 2.07 × 10−8 s−1, and the corresponding half-life is 10,450 and 9,290 h.
As with the natural enzyme, the hydrolysis rate constant of these IO nanoparticles is sensitive to temperature. The optimum temperature for the phosphate ester hydrolysis reaction was around 50°C (Fig. 5a, 5b), which is comparable to recent observations on the natural enzymes (Hobbs et al., 2013; Elias et al., 2014; Schipper et al., 2014; Arcus et al., 2016). However, catalytic activity of the IO nanoparticles in solution was lost as the temperature was raised to 90°C for an hour or to 72°C for 16 h. This behavior is similar to the thermal denaturation of the natural enzyme. Moreover, the temperature coefficient, Q 10, a measure of the hydrolysis velocity, is also decreased as a consequence of increasing the temperature by 10°C (Fig. 5d). This effect, too, is comparable to the general patterns of enzyme behavior in biological systems (Hobbs et al., 2013; Schipper et al., 2014). Taken together, these observations also carry the implication that moderate, that is, 50°C, and not high temperatures were likely favorable to the catalytic reactions from the view of efficiency and speed of the catalysis. There are some differences among the temperature responses to different forms of organic phosphate ester and condensed phosphate (Fig. 5). The Q 10 of inorganic condensed phosphate seems to be higher than that of phosphate ester at the same temperature, especially at the low temperature. In actuality, the catalytic activity of the nanoparticles remained high after removal from their source (IO) for days, even when stored at −18°C, demonstrated by the storage experiments (Table 3).
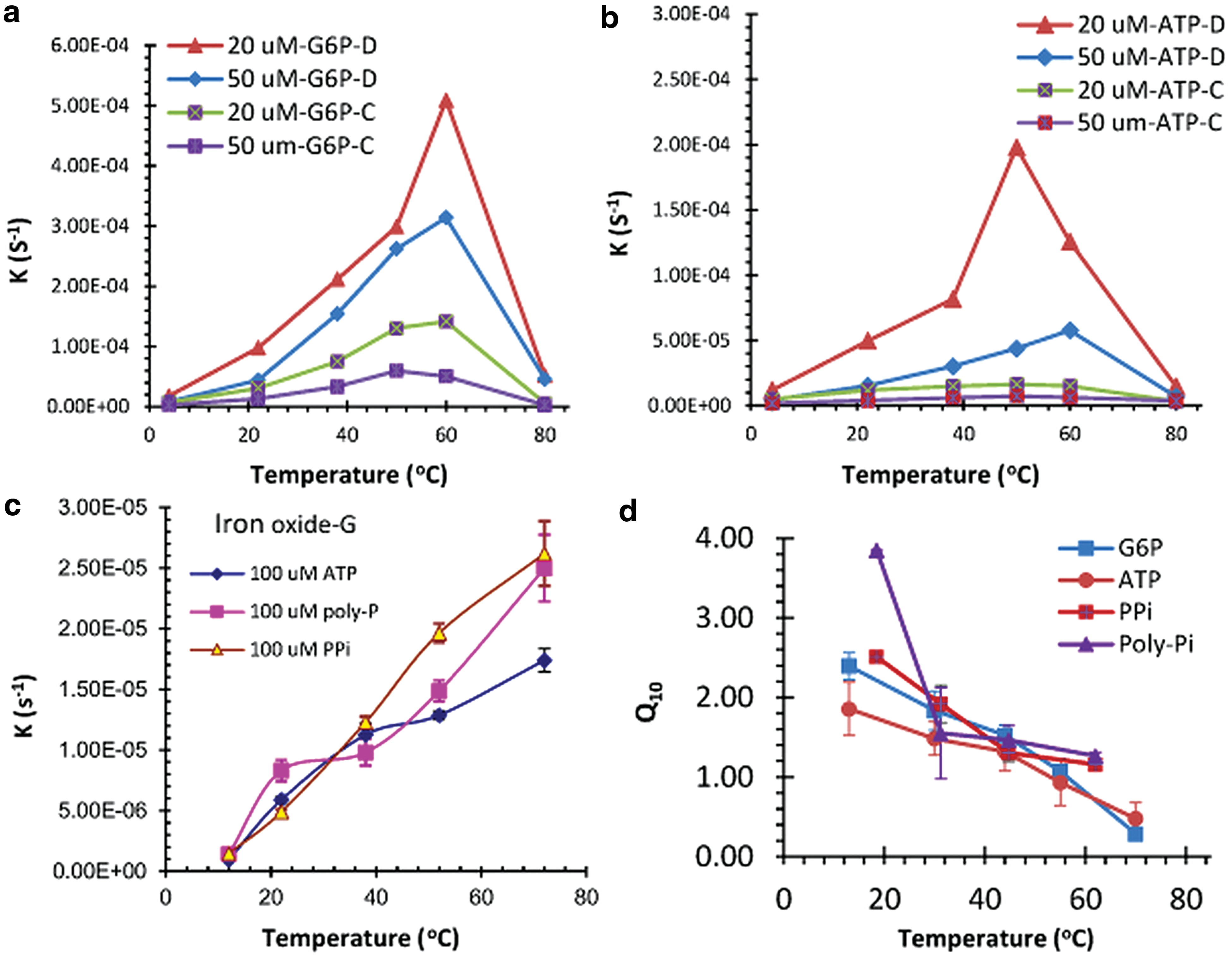
Relationship between environment temperature and the hydrolysis rate of phosphorus. (
The inorganic phosphate (IP) of the 20 μM G6P (DIW, control) after 120 h at room temperature was changed from 1.67 ± 0.008 to 1.83 ± 0.027 μM. The corresponding half-life was 10,450 h. The IP of the 20 μM ATP (DIW, control) after 120 h at room temperature was changed from 1.89 ± 0.016 to 2.06 ± 0.037 μM. The corresponding half-life was 9290 h.
As with the natural enzyme, pH is another key factor in enzyme-like activity of IO nanoparticles. Various concentrations of bicarbonate were introduced in the DMT-IO system to simulate the early ocean environment, but in all cases enzyme-like activity for phosphate ester hydrolysis remained quite high (Fig. 6a). In general, the most favorable pH of the enzyme-like activity was found to be between 6 and 7, though the phosphorus source, the concentrations of bicarbonate, and the type of DMT-IO also influenced its activity (Fig. 6b, 6c). When pH was raised beyond 7 (e.g., pH 7, 7.2, and 8), the catalysis coefficient, k, decreased as the concentration of HCO3 increased, especially for the DMT-IO-D. When pH in solution was less than 7 (e.g., pH 6.2, 6.4, and 6.8), however, there were no clear patterns of k with respect to the concentration of HCO3 and both the ATP and G6P in these two nanoparticle-bearing solutions. At the same time, k at weak acidic conditions (pH 6.2–6.8) was much higher than at weak base conditions (pH 7–8). This conclusion was further supported by an additional experiment involving the hydrolysis of ATP, whereby the pH values of DMT-IO solution were extended from 4 to 9.3 units by employing four different buffer systems (1.0 M acetate buffer [pH 4.0–5.6], 0.2 M dimethyl glutaric acid buffer [pH 4.2–6.8], 20 mM NaHCO3 [pH 7.6–9.3], and 40 mM NaHCO3 [pH 5.8–9.3]) (Fig. 6d).
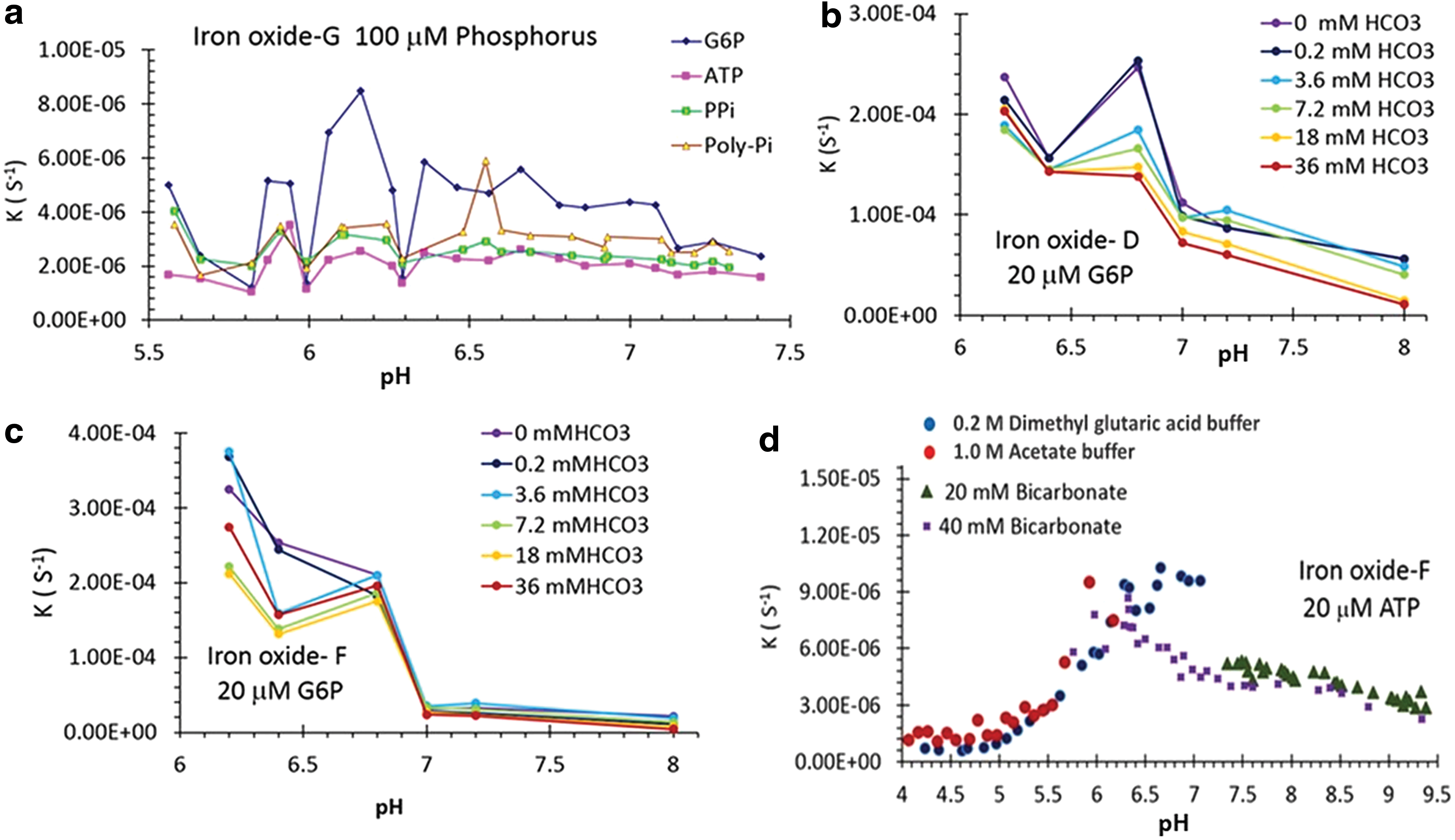
Relationship between pH and the hydrolysis rate of phosphorus. (
4. Discussion
Although ferrous ion (Fe II) is presumed to be the dominant form of iron in the earliest ocean (Holland, 2003; Raiswell and Canfield, 2012), low levels of ferric ion (Fe III) would have been produced at the ocean's surface as a result of photooxidation, even in the absence of oxygen in the atmosphere (Braterman et al., 1983; Bekker et al., 2013). Also, ferric ion, precipitated for example in green rust in the Archean banded iron formations, was also likely to have been present in the Hadean or the early Archean (Arrhenius et al., 1993, 1997; Holland, 2003; Bekker et al., 2013; Nitschke et al., 2013; Génin et al., 2014; Posth et al., 2014; Shibuya et al., 2016; Halevy et al., 2017). IO nanoparticles with 3.5 Å oxo-Fe bindings (e.g., doubly shared iron octahedra) such as ferrihydrite, goethite, hematite, magnetite, and even green rust (fougerite) can be formed from solution depending on aging and present in the natural environment, including the early Earth (Fig. 1b, adapted from the iron oxides figure from Dr. Jean Pierre Jolivet [personal communication]) (Rose et al., 1997; Schwertmann et al., 1999; Benali et al., 2001; Jolivet et al., 2006; Michel et al., 2007, 2010; Trolard et al., 2007; Navrotsky et al., 2008; Zegeye et al., 2012; Guo and Barnard, 2013; Génin et al., 2014; Baumgartner and Faivre, 2015) (SI-Fig. 5). The solubility of IOs indicates that the critical ferrihydrite nucleus with an equivalent diameter of ∼15 Å and containing only ∼30 Fe atoms is stable in aqueous solution (Hiemstra, 2015). The 10 Å discrete iron-oxo cluster (known as the Keggin ion, Fe13) is also soluble as an aqueous ion (Sadeghi et al., 2015) (SI-Fig. 6). Although the μ-oxo iron ion was only identified in situ in the high concentrated inorganic iron solution (e.g., 0.1 M Fe(NO3)3 [Zhu et al., 2013] [SI-Fig. 7] and 0.1 M FeCl3 [Hellman et al., 2006]), density function theory calculations and molecular dynamics simulation support the presence of aqueous di-iron or poly-irons, in which the Fe-Fe distance is 3.0–3.5 Å, with bonds by oxo bridge or hydroxo bridge (Panina et al., 2010; H. Zhang et al., 2015) (SI-Fig. 8). Experiments and chemical models have also demonstrated that temperature impacts the stability of the aqueous poly-iron formation (Milburn and Vosburgh, 1955) and the nanostructure of IO in the solution (Navrotsky et al., 2008; Guo and Barnard, 2013). The dissolved CO2 in the IO nanoparticle solution was also observed to result in nanosize-induced phase transformation and changes in the nature of the Fe–Fe bond in the surface structure (Chernyshova et al., 2013). Therefore, it is reasonable to suggest that the oxo-bridged Fe–Fe structure in the aqueous IO nanoparticles contributes to the catalysis of phosphate ester hydrolysis. This compares to the activity of the aged nanomolar inorganic iron ion solutions (Huang and Zhang, 2007, 2012) and mimics of artificial phosphatases (Than et al., 1999; Belle and Pierre, 2003; Schenk et al., 2013). The common feature between these IO nanoparticles, either from the DMT-IO or the aged inorganic iron ion solutions, as well as the natural or synthesized biomimetic phosphoesterase constitutes a kind of acceleration of electron transfer rate in the structure of the μ-(hydr)oxo ligand between the metals, particularly iron (Sträter and Lipscomb, 1996; Mitić et al., 2006; Lassila et al., 2011; Huang and Zhang, 2012; Duval et al., 2013; Schenk et al., 2013). In other words, the hydrolysis of phosphate ester is entirely dependent on its catalysis on this special Fe-oxo-Fe structure. It should be pointed out that the K m, which denotes the affinities of the phosphate ester to catalysis, are significantly (up to 3 orders of magnitude, Table 1) lower than are the IO nanoparticles, whether from DMT-IO or from the aged inorganic iron ion solution, as compared to the natural PAPs (Vincent et al., 1991; LeBansky et al., 1992; Cashikar et al., 1997; Bozzo et al., 2004; Huang and Zhang, 2012). This finding indicates that the IO nanoparticles are either much more sensitive to the low concentration of phosphate ester in the environment or they have a much higher affinity for phosphate ester compared to the natural enzyme, although the maximum velocity of the hydrolysis was relatively low with these IO nanoparticles, especially for high phosphate ester concentrations.
It is noted that the temperature of IO formation, for example, even >200°C (Mottl and McConachy, 1990; McCollom and Shock, 1997; Russell and Hall, 1997; Kilias et al., 2013; Nitschke et al., 2013; White et al., 2015) might be different from the temperature where the IO nanoparticles act as biocatalysts, as demonstrated by the storage experiments. IO nanoparticles can be displaced a considerable distance from their source and maintain catalytic activities for a considerable time. Meanwhile, low temperatures, even frozen conditions, also favor the persistence of catalytic activity from these IO nanoparticles. If prebiotic reactions are rapid, as is suspected, at a temperature of 50°C or even a little higher, the catalyzed reactions still take place. Such moderate temperatures for their catalytic activity support the suggestion that life might start at submarine hydrothermal vents on wet and icy worlds (Russell et al., 2014), and this is consistent with the low ocean temperature assumed for early Earth (Kasting et al., 1984; Hren et al., 2009; van den Boorn et al., 2010).
Solutions of DMT-IO are generally mildly acidic (pH 5.5–6.5), that is, comparable to the early ocean (Canfield, 2005; Russell et al., 2014), which would have been rendered somewhat acidic by a high concentration of carbon oxide in the early atmosphere (Kasting et al., 1984; Macleod et al., 1994). The responses of catalytic activity of IO on hydrolysis of phosphate ester in different pH experiments imply that the significant change was not due to the pH itself but to the interactions between nanoparticles and chemicals in the environment. It was observed that the catalytic activity dropped precipitously after a small amount of citrate buffer (pH 4.0–6.2) or when tris(hydroxymethyl)aminomethane (Tris; pH 5.8–7.2) was introduced into DMT-IO solution. Both citrate (Spiro et al., 1967; Silva et al., 2009) and Tris (Gupta et al., 2013) can react with Fe (III) in solution to form the aqueous Fe-complex, particularly at high ratios of citrate or Tris to Fe (>>10000:1 molar ratio). It has been reported that some buffer systems can significantly inhibit the activity of natural enzyme, for example, citrate on special PAPs (Nuttleman and Roberts, 1990) and alkaline phosphatase (Evered and Steenson, 1964) and Tris on aminopeptidase and RimO methylthiotransferase (Desmarais et al., 2002; Molle et al., 2016), due to structure changes and metal-complex formation (Palmer and Drummond, 1988; Silva et al., 2009; Gupta et al., 2013). However, enzyme-like activity of DMT-IO is not inhibited by high concentrations of acetate (e.g., 0.1–1 M), which is significant in view of the proposed synthesis of acetate in the alkaline hydrothermal mound (Nitschke and Russell, 2009; Russell et al., 2014). Indeed, an acetate buffer is commonly used in the purification of natural enzymes, including PAPs (Herrala et al., 2013). The differences of buffer response might also relate to the stability of metal structure in the active center. As expected, the stability of μ-(hydr)oxo ligand between the irons in the DMT-IO or aged inorganic iron solution is probably weak compared to that in the metal center in natural PAPs, which would explain loss of activity in the Tris and citrate buffer system. In contrast, most PAPs are still active, even when these same buffers are used in purifications (Herrala et al., 2013). All of these results further indicate that the catalysis mechanisms of DMT-IO as phosphatase are very complex, which, as with the natural phosphatase and its mimic, cannot be simplified to either intra-intermolecular or acid-base catalysis (Cassano et al., 2004; Mitić et al., 2006; Kamerlin et al., 2008; Lassila et al., 2011; Schenk et al., 2012; Zhang et al., 2014; Daver et al., 2016). Both intra-intermolecular and acid-base catalysis mechanisms were reported in the natural phosphatase as well as its mimics (Kady and Tan, 1995; Nakamura and Tajima, 1997; Molenveld et al., 1998; Miclet et al., 2001; Piatek et al., 2004; Kirby et al., 2006, 2009, 2011, 2013; Orth et al., 2008; Gerasimova et al., 2010).
Furthermore, these IO nanoparticles, acting as phosphatases, have the potential to release “free energy” from the phosphoanhydride bond in ATP and PPi; it is believed that these compounds are far from thermodynamic equilibrium considering their hydrolysis products (Baltscheffsky et al., 1999; Kornberg et al., 1999; Achbergerová and Nahálka, 2011; Branscomb and Russell, 2013). It is assumed here that the free energy released on the hydrolysis processing has the potential to drive endergonic reactions such as the synthesis of serine, threonine, and tyrosine and the formation of the backbones of RNA and DNA, all essential requirements for early life and its evolution (Deamer, 1997; Eschenmoser, 2007; Ruiz-Mirazo et al., 2014), though the mechanisms of free energy conversions are disputed and are in need of further investigation and experimentation (Hill, 2004; Branscomb et al., 2017). These IO nanoparticles may also be involved in the reversible phosphorylation of three hydroxylamine acids—serine, threonine, and tyrosine residues—in proteins as a regulatory mechanism (Hunter, 2012).
Similar to phosphatases, the active metal centers of most peroxidases and catalases in nature also comprise the transition metals, for example Fe (horseradish peroxidase, HRP [Gajhede et al., 1997], heme catalases [Bravo et al., 1995], rubrerythrin [LeGall et al., 1988]), Mn (manganese peroxidase [Sundaramoorthy et al., 1994], manganese catalases [Dismukes, 1996; Pace et al., 2012]), or V (haloperoxidases [Renirie et al., 2000]), all of which exhibit the oxo-ligand structure. Different metal oxide nanoparticles, for example, Fe3O4 (Gao et al., 2007), α-Fe2O3 (Chaudhari et al., 2012), γ-Fe2O3 (Chen et al., 2012), γ-FeOOH (Peng et al., 2011), Co3O4 (Mu et al., 2014), MnFe2O4 (Peng et al., 2015; Vernekar et al., 2016), MFe2O4 (M = Mg, Ni, Cu) (Su et al., 2015), ZnFe2O4 (Su et al., 2012), NiO (Ray et al., 2014), VO2 (Nie et al., 2014), and MnO2 (Yang et al., 2015) were recently observed to have peroxidase-like or catalase-like activity. Also, even some biogenic magnetic nanoparticles exhibit peroxidase-like activity (Guo et al., 2012; Pan et al., 2014). And, as with these observations on inorganic phosphatases, almost all the reported K m of these inorganic nanoparticles evince some activity, although significantly lower than HRP, the corresponding natural enzyme (Table 4, 3,3′,5,5′-tetramethylbenzidine, TMB). The catalytic activities depend to some degree on the surface area of these nanoparticles, and not merely on particle size (Liu et al., 2011; Chaudhari et al., 2012; Wei and Wang, 2013; Mu et al., 2014; Wu et al., 2014; Peng et al., 2015; Tian et al., 2015; K. Zhang et al., 2015). Various polymers or other organic compounds, for example, porphyrin rings, the backbones of short peptides, amino acids, and even DNA, have been employed in the stabilization of the oxo-bridged Fe–metal center in different iron oxides, and this has been confirmed to have the same or higher catalytic activity as peroxidase (Yu et al., 2009; Singh et al., 2010; Liu and Yu, 2011; Wang and Huang, 2011; Wang et al., 2012; Wang et al., 2013; Liu et al., 2014; Liu and Liu, 2015, 2017). It has also been demonstrated that introducing histidine on the surface of the Fe3O4 nanozyme significantly improves the affinity for H2O2 and enhances its catalytic activity (Fan et al., 2017) (SI-Fig. 11). It has been discerned that two histidine residues, His42 and His170, are located distally and proximally to iron in the active site of HRP (Berglund et al., 2002), and the distal imidazole, His42, assists the location of H2O2 into the active site of HRP through H-bond interaction and the formation of an initial complex for its catalysis (Derat and Shaik, 2006). In these cases, the catalysis of hydrolysis of hydrogen peroxide is a consequence of the high-valent-metal oxygen, for example, the Fe(IV) = O structure (Penner-Hahn et al., 1986; Dawson, 1988; Berglund et al., 2002; Groves, 2006; Hersleth et al., 2006; Coggins et al., 2013).
The similarly elevated CO2 levels that likely prevailed in the early atmosphere could have resulted in a neutral or mildly acidic ocean (Kasting et al., 1984; Macleod et al., 1994; Canfield, 2005; Russell et al., 2014; Halevy and Bachan, 2017); the relatively high concentrations of the hydrogen peroxide in the anaerobic conditions are also suggested in the early ocean for emergent life (Kasting et al., 1984; McKay and Hartman, 1991; Borda et al., 2001; Foustoukos et al., 2011; Liang et al., 2006; Pecoits et al., 2015). Some metal oxide nanoparticles with the high-valent-metal oxygen, for example, the Fe(IV) = O structure, which act as peroxidase, promote the oxidation of various organic compounds by using naturally occurring hydrogen peroxide or organic hydroperoxides (R-OOH) as electron acceptors, breaking down toxic organic compounds to harmless substances. Some of these metal oxide nanoparticles, acting as catalases, generate free oxygen, which protects the cell from oxidative damage by reactive oxygen species (ROS) due to the decomposition of hydrogen peroxide. It has also been suggested that the decomposition of hydrogen peroxide might drive the formation of amino acids and the replication of primordial RNA (Slesak et al., 2012; Ball and Brindley, 2014). These processes exploit the supply of free energy, that is, the disequilibrium, for all life (e.g., PPi and ATP with respect to their hydrolysis products). They are also essential in the building of the backbones of RNA, DNA, and protein, as well as for much of the rest of metabolic processing. The potential for IO nanoparticles (and also manganese oxides) to have acted as the catalases at the emergence of life should also be pointed out, and it should be said that thereafter they could have contributed directly as active centers to the first biotic enzymes (Blankenship and Hartman, 1998; Kirschvink and Kopp, 2008; Slesak et al., 2012).
Some mammalian PAPs are bifunctional, catalyzing both phosphate ester hydrolytic reactions and peroxidations (Bozzo et al., 2002; Kaija et al., 2002). The latter are facilitated by the redox-active iron in the binuclear active site (Schenk et al., 2013), so it is not surprising that both peroxidase and phosphoesterase-like activities are also a feature of iron oxide nanoparticles. As mentioned, catalysis is due to the metal center comprising the nanoparticles, and not simply to their surface (ab intra and Huang and Zhang, 2007, 2012). Similar conclusions may be drawn from leaching experiments (Su et al., 2015; K. Zhang et al., 2015); the duration of experiments are, of course, a factor that may explain why no catalytic activity has been reported for CoFe2O3 and Fe3O4 in experiments lasting only 6 h (see Supplementary Information—Fig. 5, K. Zhang et al. [2015], and Gao et al. [2007]). Overall, it is reasonable to assume that “primitive” forms of peroxidase comprised mineral nanoparticles with comparable oxo-ligand structures.
Because a similar architecture of the active site of PAPs with respect to the amino acid residues in the Fe-oxo-Fe center was also observed in the V-oxo-V center of the haloperoxidases (Messerschmidt and Wever, 1996; Hemrika et al., 1997), it was expected that the nanomaterials of V2O5 should have similar enzyme-like behavior for hydrogen peroxidase, as indeed has been demonstrated in the ocean environment (André et al., 2011; Natalio et al., 2012) (SI-Fig. 9). In this regard, it is notable that a vanadium pentoxide nanowire was also observed to have high antioxidant enzyme-like (glutathione peroxidase) activity on the introduction of cellular glutathione at physiologically relevant conditions (Vernekar et al., 2014) (SI-Fig. 10). Comparable to the response of IO nanoparticles, the efficiency of vanadium pentoxide catalysis changed significantly depending on which buffer system was used, even at the same pH, thus emphasizing the stability of the structure (André et al., 2011; Natalio et al., 2012). The catalytic activity of peroxidase is due to the common metal center that has the same V-oxo-V bond in both the nanoparticles and the haloperoxidases (Messerschmidt et al., 1997; Zampella et al., 2005; Natalio et al., 2012). For the mechanistic comparison with glutathione peroxidase, the intermediate products of reaction, for example, the peroxido species, were detected by Raman spectroscopy in the case of the reaction of nanowire of V2O5 with hydrogen peroxide (Vernekar et al., 2014). This comparable outcome is assumed to be due to the surface-exposed 010 planes of V2O5 (André et al., 2011).
In addition, vanadium would also have been dissolved to a limited degree in the early ocean (Emerson and Huested, 1991; Wanty and Goldhaber, 1992; Calvert and Pedersen, 1993; Huang et al., 2015), where it could have acted as a peroxidase (Hager et al., 1966). Although the solubility of molybdenum would have been lower than vanadium in the anaerobic Hadean ocean (Emerson and Huested, 1991; Calvert and Pedersen, 1993; Erickson and Helz, 2000; Anbar et al., 2007), it would have been available in alkaline springs (Helz et al., 2014). In these conditions, molybdenum would have served as a proto-enzyme and subsequently as the mononuclear metal center for many enzymes that would, thereby, be rendered capable of supplying either one electron or two electrons at any one time (Feng et al., 2007; Schwarz et al., 2009; Schoepp-Cothenet et al., 2012). In this regard, it is not surprising that molybdenum trioxide (MoO3) nanoparticles have been shown to have intrinsic biomimetic sulfite oxidase activity (Ragg et al., 2014) (SI-Fig. 12). Similar to the nanoparticles of IO and vanadium pentoxide, the catalysis of molybdenum trioxide is due to the oxo ligand, as revealed in the metal center of sulfite oxidase (Feng et al., 2007; Schwarz et al., 2009).
There are many different enzymes to scavenge hydrogen peroxide in nature (Mishra and Imlay, 2012), including glutathione peroxidase with selenocysteine as the active catalytic center (Epp et al., 1983) and alkyl hydroperoxide reductase with a disulfide bond structure (Poole and Ellis, 1996; Hall et al., 2011). Similar to the metal oxides, it was also observed that the transition metal selenide and sulfide nanoparticles (FeSe [Dutta et al., 2012a], α-MnSe [Wang et al., 2015], FeS [Dutta et al., 2012b], Fe3S4 [Ding et al., 2016], Fe7S8 [Yao et al., 2013], CuS [Dutta et al., 2013], CuZnFeS [Dalui et al., 2015], CdS [Maji et al., 2012], MoS2 [Lin et al., 2014a], and WS2 [Lin et al., 2014b]) also exhibit catalytic function for hydrogen peroxide. Meanwhile, these sulfide and selenide nanoparticles are more sensitive to the lower concentrations of H2O2 compared to the oxide nanoparticles and even the natural enzyme (due to the low concentration of K m, Table 4), which might have served as a primitive peroxidase in the more anoxic ocean. Actually, these selenides and sulfides still can be detected in hydrothermal systems (Nitschke and Russell, 2009; Russell et al., 2014; Pogge von Strandmann et al., 2015).
Given the similarity of the architecture between the oxo-bridged Fe–Fe in the green rust with methane monoxygenase, it has even been suggested that green rust may have catalyzed the oxidation of methane to a methyl group as a step toward one tributary to the acetyl coenzyme-A pathway (SI-Fig. 13) (Yoshizawa et al., 2000; Noodleman and Han, 2006; Han and Noodleman, 2008; Nitschke et al., 2013; Shibuya et al., 2016).
5. Conclusions and Implications
Laboratory experiments on the hydrolysis of phosphate ester in water demonstrated that inorganic phosphoesterase-like activity, using various inorganic iron oxide nanoparticles, significantly promotes the hydrolysis of phosphate ester, including G6P, PPi, and ATP. These findings and the fact that this and other inorganic nanoparticles can act effectively as enzymes—for example, iron oxide as peroxidase (Gao et al., 2007; Peng et al., 2011; Chaudhari et al., 2012; Chen et al., 2012), vanadium pentoxide as bromoperoxidase (André et al., 2011), and molybdenum trioxide nanoparticles as sulfite oxidase (Ragg et al., 2014)—further support the concept of inorganic enzymes, as the biocatalysts prior to the onset of the RNA world (Huang and Zhang, 2007, 2012). The catalytic propensity of these nanoparticles is likely due to the structure of the metal oxides or metal bonds in the oxides and not merely to nanoparticle surface. Furthermore, all the enzyme-like oxide nanoparticles are composed from the transition metals, with the oxo bond acting either as the terminal ligand or as a bridge. These active nanoparticles may be considered as inorganic enzymes, that is, as “primitive enzymes” (Fry, 2000) or biocatalysts, since they were likely present in the early ocean or at least in hydrothermal precipitates at life's emergence (Holland, 2003; Nitschke and Russell, 2009; Bekker et al., 2013; Nisbet and Fowler, 2013; Nitschke et al., 2013; Shibuya et al., 2016). Therefore, all of these observations (this experiment and the previous experiments on aged inorganic iron ion solution [Huang and Zhang, 2007, 2012]), as well as those of many other studies into nanozymes, support the metabolism-first hypothesis rather than the replicator-first scenario (Gilbert, 1986; Nitschke et al., 2013). They further demonstrate that “The chain of life is of necessity a continuous one, from the mineral at one end to the most complicated organism at the other,” as proposed by Leduc (1911). These experiments provide solid evidence that biocatalysts could have been formed and functioning before the protein and RNA world occurred and thereby offer a solution to the “chicken or egg” conundrum at life's beginnings (Cairns-Smith, 1985, 2008).
Footnotes
Acknowledgments
This work is dedicated to my beloved parents (Dr. Shi-Xiong Huang and Dr. Jing-Xiong Ji) and my family (Wei Sun and Jack Jixiang Huang) for their love, endless support, encouragement, and sacrifices. X.L.H. greatly appreciates one of the referee's thoughtful suggestions and the precious comments from the late Dr. R.J.P. Williams in the past years, the kindly permissions to use the structure of iron oxide (![]() ) from Dr. Jean Pierre Jolivet, as well as the personal encouragements from Drs. Robert Atlas, Jia-Zhong Zhang, and Peter B. Ortner.
) from Dr. Jean Pierre Jolivet, as well as the personal encouragements from Drs. Robert Atlas, Jia-Zhong Zhang, and Peter B. Ortner.
Author Contributions
X.L.H. initiated, performed the experiments at Atlantic Oceanographic and Meteorological Laboratory (AOML), National Oceanic and Atmospheric Administration (NOAA), and wrote the manuscript after 9 years as an independent researcher.
Author Disclosure Statement
The author declares no competing financial interest. The experiment part of this work was initially conducted by X.L.H. from 2007 to 2008 at the AOML, NOAA, supported by the National Oceanic and Atmospheric Administration's (NOAA) Coastal Ocean Program and Climate and Global Change Program. The research was carried out, in part, under the auspices of the Cooperative Institute of Marine and Atmospheric Studies (CIMAS), a joint institute of the University of Miami and NOAA, cooperative agreement #NA67RJ0149. The statements, findings, conclusions, and recommendations are those of the author and do not necessarily reflect the views of CIMAS, NOAA, or the U.S. Department of Commerce.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.