
Abstract
In this work, we provide an answer to the question formulated by Albert Eschenmoser: “How would you envisage the bridge between potentially primordial geochemistry that had been disordered and one that gradually became self-organizing?” Analysis of the free-energy profiles of some of the key reactions leading to formation of nucleotides and their oligomers shows that, whereas the first part of the pathway, up to nucleotides, is energy-driven, in the second low-energy part entropic control in the form of structural compatibility becomes more important. We suggest that the birth of modern metabolism requires structural compatibility, which is enabled by the commensurability of the thermodynamics of the synthetic steps with the stabilizing effect of those intermolecular interactions that play a key role in dictating entropic control of these reactions.
1. Introduction
A
Chemomimesis is a widely accepted concept which originates modern biosynthetic pathways in primitive chemical processes likely to occur on early Earth (Eschenmoser and Loewenthal, 1992; Pereto, 2012; Menor-Salván and Marín-Yaseli, 2013). Since NTP's origin may be as old as that of other polyphosphorylated nucleosides (Kim et al., 2016; Akouche et al., 2017), it is reasonable to raise the question: what kind of molecules could serve as energy reservoirs at the earliest beginnings of chemical evolution? To find a plausible answer to this question led us to scrutinize the free-energy profiles of some well-known prebiotic pathways from an energetic point of view.
Of course, the analogy between modern and ancient metabolic pathways cannot be complete, because modern biochemical systems are optimized to overcome high kinetic barriers, which was made possible due to the emergence of shape selectivity. This however presumes the existence of molecules which are already relatively complex. Thus, in prebiotic chemistry in addition to the thermodynamics we have to deal also with the kinetics of the reactions. Importantly, kinetics and thermodynamics are not separable, and a general rule applies saying that the activation energy of an endothermic reaction is at least as high as the free-energy balance of the reaction.
Commonly, biochemical systems operate on an energy scale of a few kilocalories per mole, that is, in the range which is compatible with the thermal energy attainable in an aqueous solution. Is this the result of some adaptation, or is it possible to reconstruct a prebiotic pathway working on this energetic level from the earliest beginnings? The free-energy diagrams of some popular prebiotic synthetic routes may provide a plausible answer to this question.
2. Free-Energy Profiles Characterizing the Synthesis of Prebiotic Building Blocks
In 2011 we analyzed (Šponer et al., 2011) the free-energy profile of the multistep synthesis of cytidine nucleotides elaborated by the Sutherland group (Powner et al., 2009). The results show that the synthetic pathway is completely exergonic and the individual reaction steps involve relatively small energy changes. The activation energy of some key reaction steps, like oxazole ring closure, is relatively low, which is consistent with a low-temperature aqueous synthesis (Šponer et al., 2011).
In contrast, the formamide-based synthesis of nucleobases (Saladino et al., 2007, 2009) is an endergonic process (Šponer et al., 2012; Wang et al., 2013a, 2013b) with a considerable activation energy, which is made possible by the 100-degrees-higher boiling point of formamide. Logically, a question has arisen whether it is possible to convert this endergonic chemistry to an exergonic one by using high-energy initiators, like CN radicals created, for example, in the afterglow of an impact plasma. 1 The synthetic pathways described in the works of Ferus et al. (2014, 2015) clearly prove that high-energy initiators, that is, radicals, generated in an extraterrestrial impact event, are well-suited for this purpose because they are able to induce a highly exergonic chemistry, which partially utilizes the chemical energy conserved in the triple bond of CN radicals to energetically support the chemical transformations leading to the building blocks of nucleic acids. In addition, radicals, in contrast to closed-shell species, are able to form favorable orbital combinations with a number of other species, which leads to an unprecedented reduction of the activation energies of their reactions. This high activity is at the basis of the lower selectivity of radical reactions and also of the fact that their effectiveness is rapidly lost within a short time interval.
Proton flux delivered to Earth by solar flares in the form of proton irradiation is another plausible source (Airapetian et al., 2016) of radicals and was demonstrated to contribute to a versatile systems chemistry (Saladino et al., 2015, 2017). This goes beyond nucleic acid building blocks and could induce formation of short oligonucleotides as well (Costanzo et al., 2017). As quantum chemical calculations demonstrate (Saladino et al., 2017), in the course of the proton irradiation–mediated nucleosidation between adenine and ribose, OH radicals formed upon radiolysis induce an exergonic chemistry, which proceeds on a slightly lower energy scale than the CN-radical-based impact chemistry (Ferus et al., 2014, 2015). This enables higher selectivity and yields. In line with this, the reaction can be used to trigger formation of short oligonucleotides via oligomerization of cyclic nucleotides (Costanzo et al., 2017).
3. Self-Assembly and Oligomerizations
There is increasing evidence that prebiotic oligomerization reactions are preceded by the formation of a supramolecular architecture that enables the transphosphorylation reactions leading to phosphodiester linkages (Usher and Yee, 1979; Costanzo et al., 2009, 2017; Da Silva et al., 2015; DeGuzman et al., 2014; Šponer et al., 2015a). The energy balance of phosphodiester bond formation is low (a few kcal/mol) and is very close to that of modern biochemical processes (see Fig. 1). This is the reason why at this level of complexity structural compatibility becomes as important as in modern biochemical systems. In contrast, the energetic compatibility of the reactants dominates up to the level of the chemical synthesis of prebiotic building blocks, such as nucleobases, nucleosides, or nucleotides.
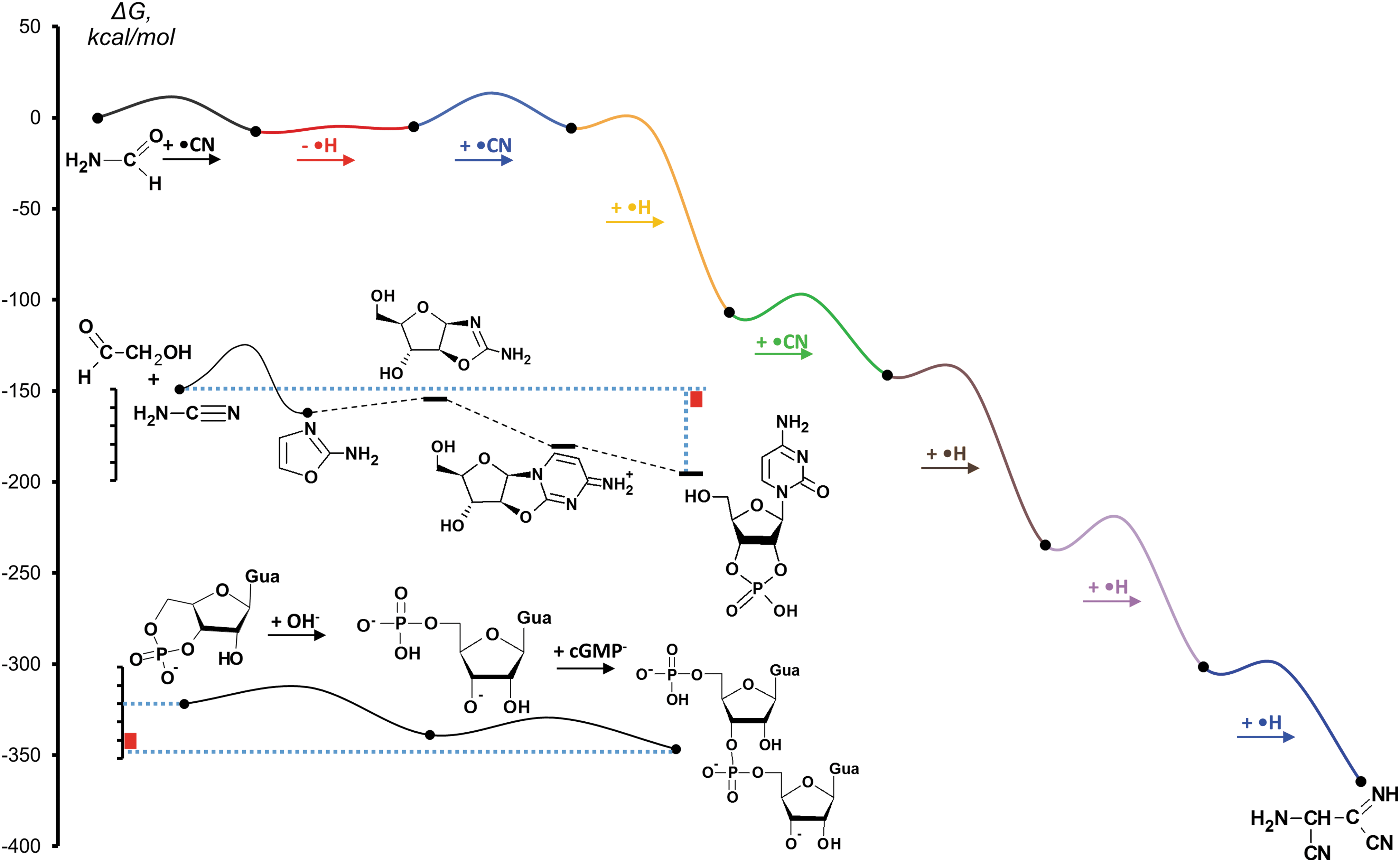
Free-energy (ΔG 0) diagrams of selected prebiotic synthetic pathways. For comparison, the red bars indicate the computed free-energy change for the reaction of ribose-1-diphosphate(2-) with guanine (-8 kcal/mol, taken from Šponer et al., 2011), that is, a simple alternative route contemporary organisms may utilize for the biosynthesis of guanosine. The uppermost pathway depicts the CN-radical assisted synthesis of diaminomaleonitrile from formamide in the course of an extraterrestrial impact (Ferus et al., 2015). The middle curve shows the free-energy profile (Šponer et al., 2011) computed for the synthesis of 2',3' cCMP (Powner et al., 2009). The bottom pathway depicts the free-energy profile of the transphosphorylation reactions between 3',5' cGMPs, as described in Šponer et al. (2015a). Solid connecting lines indicate the approximate kinetic barrier heights inferred from computations, while dashed lines are used to connect those intermediate states where only thermodynamic data are available. Note that all free-energy changes are given according to the physical chemistry definition of the standard state (which is different from that used in biochemistry; for details see, e.g., Voet and Voet, 2011).
Another similarity to modern functional biomolecules is that in supramolecular architectures formed by the self-assembly of nucleotide monomers, structural compatibility is mediated by noncovalent interactions, like H-bonding and stacking interactions. In order for them to play a decisive role, the free-energy balance of the synthetic processes and that of the formation of supramolecular complexes must be commensurable. Early supramolecular assemblies were by evolution replaced by the astonishing variability of biomolecular systems, whose structural dynamics still relies on the very same noncovalent interactions.
An obvious manifestation of this principle is the proton irradiation–induced oligomerization of cyclic nucleotides. Whereas the synthetic steps induced by impact chemistry reported in works of Ferus et al. (2014, 2015) proceed on an energy scale of ca. 50–100 kcal/mol, the OH-radical chemistry induced by proton irradiation involves a slightly smaller energy range, in the order of several tens of kilocalories per mole (Costanzo et al., 2017; Saladino et al., 2017). The latter energy scale is roughly the same as that of mild heat treatment in an aqueous environment. This is why irradiation with slow protons at room temperature, in contrast to impact chemistry, may induce oligomerization of nucleotides without the disruption of relatively unstable supramolecular architectures (Costanzo et al., 2017). Radiation chemistry might be capable to balance the conditions for the simultaneous synthesis of the monomers (Saladino et al., 2015, 2017) with their subsequent polymerization (Costanzo et al., 2017).
With the oligomerization reactions, molecular evolution arrives at an energy level which is very similar to that of modern biochemical systems (see Fig. 1); yet the extent of self-organization of the system (i.e., entropic effects) is much smaller, and the system has to cope with relatively high activation barriers. A possible way to overcome high activation energies is to activate the phosphate with positively charged cations (see Fig. 2). This strategy may choose among (at least) two possible options: (i) in weakly basic environments, diamines or amino-alcohols work (Verlander et al., 1973), whereas (ii) under strongly acidic conditions (Da Silva et al., 2015), relevant to geothermal fields, coordination of positively charged cations may activate the phosphate for transphosphorylations. In both cases, this happens because the positive partial charge transferred to the phosphate makes the phosphorus a better target for a nucleophilic attack (Šponer et al., 2015b). Finally, the anionic ring-opening oligomerization of cyclic GMPs is achieved via a slightly different pathway, with essential contribution from the activation of the nucleophile by deprotonation (Šponer et al., 2015a). In this case, formation of a special stacked supramolecular architecture, well-known from X-ray crystallography studies (Chwang and Sundaralingam, 1974), precedes the oligomerization. The supramolecular arrangement provides favorable steric conditions for the attack of O3'-deprotonated nucleotides at the next phosphate and subsequent propagation of the reaction. In addition, in the absence of highly mobile cations the stacked architecture protects the anionic center from deactivation. Thus, here the principle is the same as in modern replication: in addition to providing favorable conditions for the transphosphorylation reactions, the nucleophile is protected by its chemical environment from being deactivated. This enables a self-sustaining chemistry in the form of a living polymerization (Šponer et al., 2017). Let us note that up to now this mechanism is the only reported living polymerization among the plausible prebiotic template-free oligomerization scenarios.
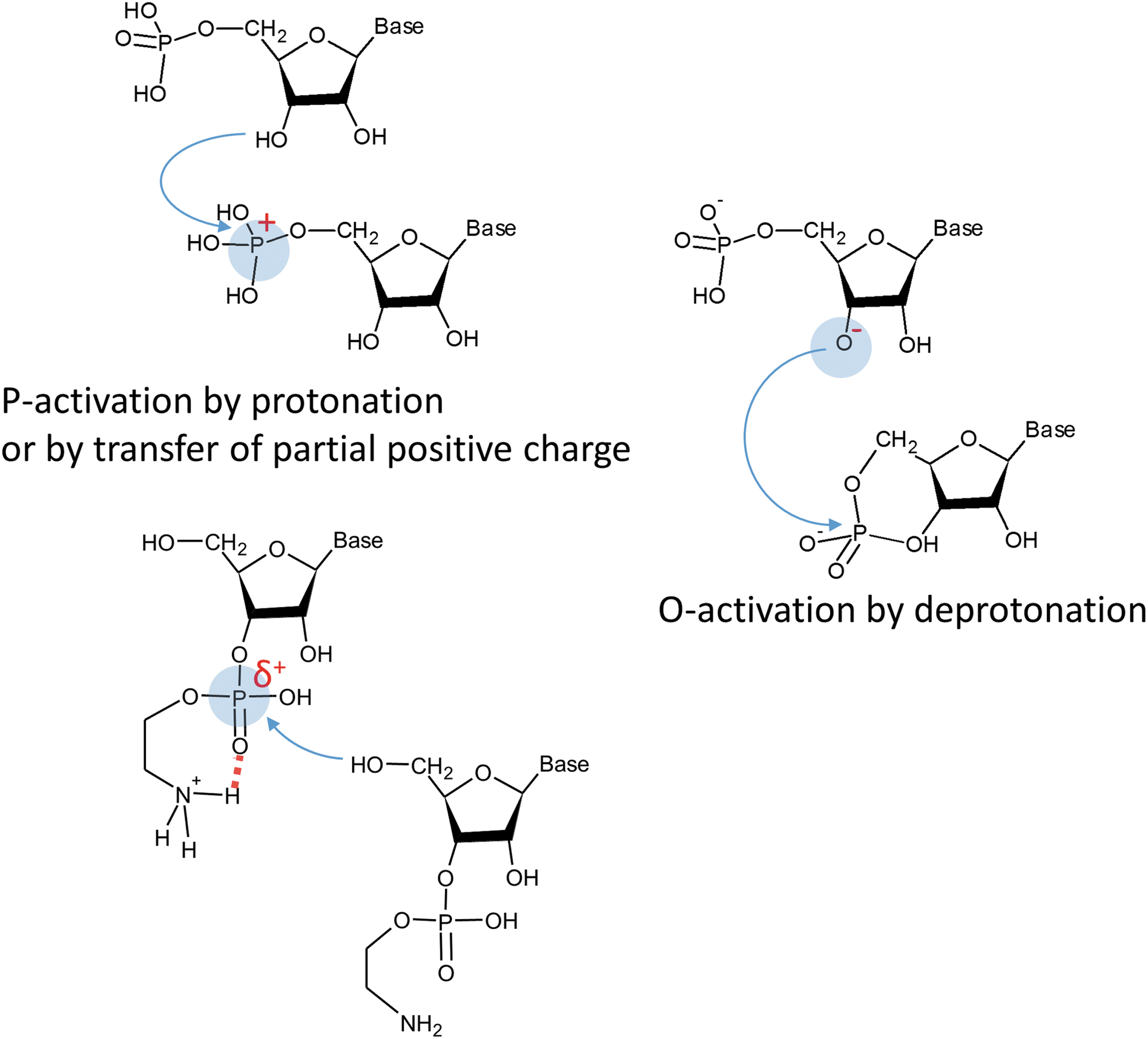
Cationic (Verlander et al., 1973; Da Silva et al., 2015) and anionic (Šponer et al., 2015a) strategies of phosphate activation used to trigger the template-free non-enzymatic oligomerization of nucleotides. The blue arrow indicates the direction of the nucleophilic attack. Activated centers are highlighted with a light blue background. The red dotted line highlights the H bond between the ammonium moiety and the phosphate oxygen responsible for the transfer of partial positive charge (δ+) to the phosphorus.
4. Crystals and Structural Compatibility
A means of decreasing the system's entropy is crystallization. Indeed, all the so-far documented non-enzymatic template-free oligomerization protocols involve a solid phase synthesis. One of the reasons for this is the obvious fragility of phophodiester bonds in water. The second reason is that the solid phase provides better steric conditions for the oligonucleotide formation. Of course, crystals may impair the oligomerization as well, if the nucleotides adopt an orientation inconsistent with the inline attack. A remarkable correlation can be noticed between the crystal geometry and the length of oligomers formed upon the oligomerization of 3',5' cyclic nucleotides. While oligomerization of 3',5' cGMP yields up to 20-mers (Costanzo et al., 2009, 2012; Morasch et al., 2014; Šponer et al., 2015a), only 3- or 4-mers are observed when oligomerizing 3',5' cAMP under the same conditions (Costanzo et al., 2016). cUMP does not oligomerize at all (Šponer et al., 2017). Analysis of crystal geometries clearly explains this behavior. Whereas the crystal structure of 3',5' cGMPs provides optimum conditions to support the oligomerization reaction, crystals of 3',5' cAMP, and especially so that of cUMP, are less appropriate for the purpose (Šponer et al., 2017). Thus, 3',5' cGMP has an intrinsic propensity to form oligonucleotides without the need of involvement of any other external factors. This is in contrast to 2',3' cyclic nucleotides, which do not oligomerize unless diamines or amino-alcohols are present in the reaction mixture (Verlander et al., 1973).
5. Continuity of the Transition from Energetic to Structural Compatibility
In the formamide-based scenario of the origin of life (Saladino et al., 2012; Šponer et al., 2016), the continuity between the energetically and structurally controlled parts of the pathway is represented by 3',5' cGMP, because this molecule accumulates sufficient energy to push through the transphosphorylation reactions required to form oligomers, yet enabling the existence of a stable supramolecular architecture under the operating conditions of the oligomerization reaction. As it is demonstrated by the free energies of hydrolysis, the energy stored inside the 3',5' cyclic phosphodiester linkage is slightly higher than that in 2',3' cyclic nucleotides (Rudolph et al., 1971; Sturtevan et al., 1973; Gerlt et al., 1980). This 1–2 kcal/mol energy difference in combination with the favorable steric conditions might be responsible for the fact that 3',5' cGMP spontaneously oligomerizes in the biologically relevant temperature range (above 30°C) without any phosphate-activating agent.
Eschenmoser addresses the problem of energetic versus structural compatibility by formulating the question: “How would you envisage the bridge between potentially primordial geochemistry that had been disordered and one that gradually became self-organizing?” (Eschenmoser, 2007). The formamide-based models described in the works of Saladino et al. (2012) and Šponer et al. (2016, 2017) provide a plausible answer to this question. We have mentioned several plausible (Sleep, 2018) formamide-based geochemical scenarios, utilizing heat, proton irradiation, and extraterrestrial impacts (all mutually compatible) which enable various ways to generate 3',5' cyclic nucleotides. The ability of 3',5' cGMPs to self-assemble and spontaneously oligomerize ensures the smooth transition toward a structure-driven chemistry; and, in addition, this polymerization process may serve as a prototype of metabolic cycles. As discussed in the work of Šponer et al. (2017), due to the symmetry of the ladder-like stacked architecture that provides optimum stereochemical conditions for the formation of a selectively 3',5'-linked oligonucleotide sequence, oligomerization of 3',5' cGMPs can be considered as the simplest form of replication. As such, the process obviously has an autocatalytic potential as well. Basically it can be considered as a two-member autocatalytic cycle (Vasas et al., 2012), in which the product, the oligoG sequence, serves as a circuit and reaction catalyst for the oligomerization of 3',5' cGMPs. OligoG templates could then anchor another key step into the catalytic circuit, that is, the formation of oligoC sequences from CMPs via Watson-Crick compatibility. Thus, this model suggests that the very first autocatalytic circuits could emerge simultaneously with the synthesis of the first informational biopolymers, oligomerization of nucleotides being the prototype of autocatalytic cores. This is enabled by the fact that self-assembly of 3',5' cGMPs proceeds on a similar energy scale as the transphosphorylation chemistry leading to oligonucleotides. A further expansion of the autocatalytic network toward even higher complexity, that is, mixed oligomers, is possible by “rare events” (see Vasas et al., 2012) mediated by the complex network of RNA-tertiary interactions. As discussed in the works of Pino et al. (2013) and Stadlbauer et al. (2015), for example, tetraloop-like overhang formation may catalyze the transfer of a nucleotide from the 5'-end of an oligonucleotide to the 3'-end of a complementary strand. Such chemical reactions may thus proceed through rarely sampled highly reactive conformations rather than the thermodynamically dominant ground state; note that a similar principle has been recently suggested also for the chemistry occurring in catalytic centers of small ribozymes, to reconcile the persistent inconsistencies between structural and mechanistic data (Sripathi et al., 2014).
In the above-referenced work, Eschenmoser says that “the larger the diversity of catalytically accessible alternative pathways and products between alternative starting materials and final products the greater the opportunities for contingent growth and complexification of chemical changes” (Eschenmoser, 2007). This principle is clearly reflected by the formamide chemistry, in which several mutually compatible segments of the geochemical pathways contribute to the emergence of the key compound, 3',5' cGMP.
6. Summary
In seeking for analogies between the free-energy profiles of modern metabolic pathways and those of prebiotic chemical models we have illustrated that a formamide-based thermal chemistry combined with involvement of free radicals may serve as the best example. Similar to ATP, in a prebiotic chemical frame radicals could provide material and energy to trigger chemical transformations otherwise inaccessible via a thermal route. Although radicals could be a reasonable solution to overcome thermodynamic and kinetic barriers, they could not control the whole process of abiogenesis due to their high energy content and, consequently, to their limited lifetime. It is plausible to assume that heat from the environment determined the main directions of molecular evolution, which was undoubtedly flavored with occasional radical chemistry, induced by a variety of environmental effects, like extraterrestrial impacts, cosmic radiation, or UV light.
Footnotes
Acknowledgments
Financial support from the grant GAČR 17-05076S is greatly acknowledged. This work was supported by the Italian Space Agency (ASI) project “Esobiologia e Ambienti Estremi” number 2014-026-R.O (CUP: F 92I14000030005).
Author Disclosure Statement
No competing financial interests exist.
Associate Editor: David Deamer