
Abstract
Decompressional boiling of ascending hydrothermal waters and separation into a vapor (gas) and a liquid phase drive extensive variation in the geochemical composition of hot spring waters. Yet little is known of how the process of phase separation influences the distribution of microbial metabolisms in springs. Here, we determined the variation in protein coding genes in 51 metagenomes from chemosynthetic hot spring communities that span geochemical gradients in Yellowstone National Park. The 51 metagenomes could be divided into 5 distinct groups that correspond to low and high temperatures and acidic and circumneutral/alkaline springs. A fifth group primarily comprised metagenomes from springs with moderate acidity and that are influenced by elevated volcanic gas input. Protein homologs putatively involved in the oxidation of sulfur compounds, a process that leads to acidification of spring waters, in addition to those involved in the reduction of sulfur compounds were enriched in metagenomes from acidic springs sourced by vapor phase gases. Metagenomes from springs with evidence for elevated volcanic gas input were enriched in protein homologs putatively involved in oxidation of those gases, including hydrogen and methane. Finally, metagenomes from circumneutral/alkaline springs sourced by liquid phase waters were enriched in protein homologs putatively involved in heterotrophy and respiration of oxidized nitrogen compounds and oxygen. These results indicate that the geological process of phase separation shapes the ecology of thermophilic communities through its influence on the availability of nutrients in the form of gases, solutes, and minerals. Microbial acidification of hot spring waters further influences the kinetic and thermodynamic stabilities of nutrients and their bioavailability. These data therefore provide an important framework to understand how geological processes have shaped the evolutionary history of chemosynthetic thermophiles and how these organisms, in turn, have shaped their geochemical environments.
1. Introduction
Hydrothermal systems integrate geological processes at a planet's surface with those from the subsurface, yielding hot springs with an incredible array of geochemical compositions (Nordstrom et al., 2005, 2009; Shock et al., 2010; Lowenstern et al., 2015). Studies of extremophiles inhabiting such environments have informed our understanding of the origin and extent of life on Earth and the potential for life on other planets (Pace, 1997; Djokic et al., 2017; Amenabar and Boyd, 2019). As such, hydrothermal systems have been increasingly studied by astrobiologists interested in understanding the nature of early life on Earth and the possibility for life on other planetary bodies (Rothschild and Mancinelli, 2001).
Volcanic continental hydrothermal systems occur in a variety of tectonic settings, most notably at divergent and convergent plate boundaries. The Mid-Atlantic Ridge volcanic system in Iceland is an example of a system that has developed at a divergent plate (Gudmundsson et al., 1992) (Fig. 1A), whereas the Taupo Volcano Zone (TVZ) in New Zealand and the El Tatio hydrothermal field in Chile are examples of continental volcanic systems that result from convergent plate tectonics (Fig. 1B) (Desilva, 1989; Giggenbach et al., 1993). In addition, magma plumes underlying areas with either thin continental crust and/or weakened crust due to crustal extension and faulting can promote continental volcanic activity, such as in Yellowstone National Park (YNP), Wyoming (Fig. 1C) (Huang et al., 2015). These differing geological settings have significant consequences for the type of volcanism (e.g., primarily silicic or basaltic) and bedrock composition (e.g., mafic and felsic) that are associated with continental hydrothermal fields. Nevertheless, despite different tectonic regimes, similarities exist in the surface and subsurface geological processes that drive variation in the composition of hydrothermal fluids.
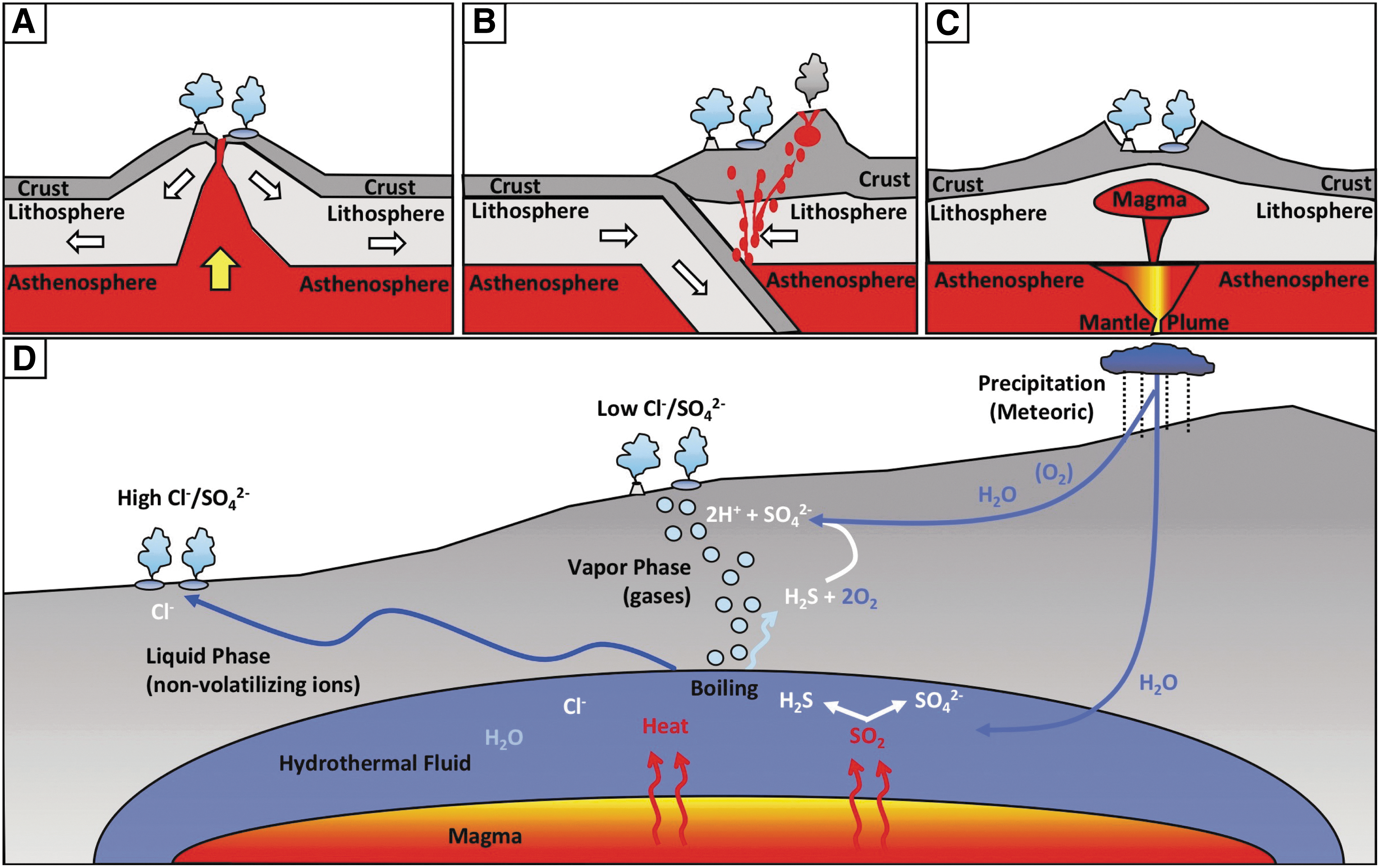
Conceptual diagram depicting different tectonic regimes
All continental hydrothermal systems comprise three common characteristics: a source of water, a source of heat, and a permeable rock stratum overlying the heat source (Heasler et al., 2009). In volcanic hydrothermal systems, that source of heat is a shallow magma body. The sources of water can be meteoric and/or marine waters that infiltrate the crust through cracks and fissures in the overlying stratum. These waters can then be heated and injected with volcanic gas (e.g., sulfur dioxide [SO2] and carbon dioxide [CO2]) that can then ascend to the surface. During its ascent to the surface, hydrothermal water can undergo the process of decompressional boiling wherein it can be separated into a vapor phase and a liquid phase (Fournier, 1989; Nordstrom et al., 2005). The vapor component of phase-separated fluids is often enriched in volatiles such as hydrogen (H2), methane (CH4), and hydrogen sulfide (H2S) (Bergfeld et al., 2014; Lindsay et al., 2019), the latter of which is generated from the disproportionation of volcanic SO2 (Nordstrom et al., 2009). The vapor can migrate toward the surface through fissures or vents resulting in fumaroles (Fournier, 1989; Lowenstern et al., 2015). Alternatively, the vapor can condense with oxygen (O2)-rich meteoric waters in the near subsurface, which can promote the oxidation of reduced sulfur species leading to the production of sulfate (SO4 2−) and acidity (Nordstrom et al., 2005, 2009) (Fig. 1D). Thus, vapor-phase-influenced hot springs tend to be gas-rich, acidic, have high SO4 2− concentrations, are limited in chloride (Cl−) since this ion behaves conservatively during boiling, and thus have elevated SO4 2− to Cl− ratios (Nordstrom et al., 2009).
In contrast, the liquid component of phase-separated fluids is typically volatile poor, enriched in Cl−, and lacks abundant SO4 2−. Springs sourced by the liquid component of phase-separated fluids tend to be circumneutral to alkaline in pH and have low SO4 2− to Cl− ratios (Nordstrom et al., 2009). The prevalence of these two geochemical end member spring types results in a bimodal distribution of spring pH in hydrothermal fields, with vapor-phase-influenced springs having a pH <5.0 and liquid-phase-influenced springs having a pH >6.5 (Fournier, 1989; Nordstrom et al., 2005, 2009; Kaasalainen and Stefánsson, 2012). This bimodal distribution in the pH of hot springs is typical for those in YNP, Iceland, the TVZ, and others, with distribution peaks typically around pH approximately 2–3 and approximately 6–7 due to buffering by sulfuric acid and bicarbonate, respectively (Brock, 1971; Nordstrom et al., 2009). Numerous other factors contribute to further variation among the geochemical compositions of springs both within and between hydrothermal fields, including the source of waters infiltrating the subsurface hydrothermal reservoir (i.e., freshwater or marine), differences in the host bedrock, and differences in the sulfidation state of the system.
The extensive geochemical variation among hot springs presents an opportunity to examine its influence on the taxonomic and functional diversity of microbial communities that inhabit these springs. At temperatures above ∼50°C, macroorganisms are absent in hydrothermal systems, and ecosystem productivity is driven exclusively by microorganisms (Rothschild and Mancinelli, 2001). In addition, above temperatures of ∼60°C, all eukaryotic microbial life (i.e., fungi and algae) is absent (Tansey and Brock, 1972), and the productivity of these systems is driven by chemosynthetic/photosynthetic bacteria or chemosynthetic archaea (Rothschild and Mancinelli, 2001). However, at temperatures >73°C in alkaline springs and >54°C in acidic springs in YNP (Cox et al., 2011; Boyd et al., 2012) (Fig. 2), and at slightly lower temperatures in TVZ and Iceland for undefined reasons, photosynthetic bacteria are excluded and all life is supported by chemical energy. In these nonphotosynthetic systems, the pH of hot spring waters has been shown to explain the most variation in the taxonomic and functional potential of thermal spring communities, with temperature exerting a secondary role (Mitchell, 2009; Boyd et al., 2010, 2013; Inskeep et al., 2013a; Colman, 2015; Colman et al., 2016). However, aside from the physiological stress imparted on cells by extremes in pH, the mechanisms that underlie the influence of pH on the functional diversity of hot spring communities has not explicitly been addressed, although several studies have begun to assess this question via physiological inference from 16S ribosomal RNA (rRNA) gene data (Meyer-Dombard et al., 2005; Colman et al., 2016).
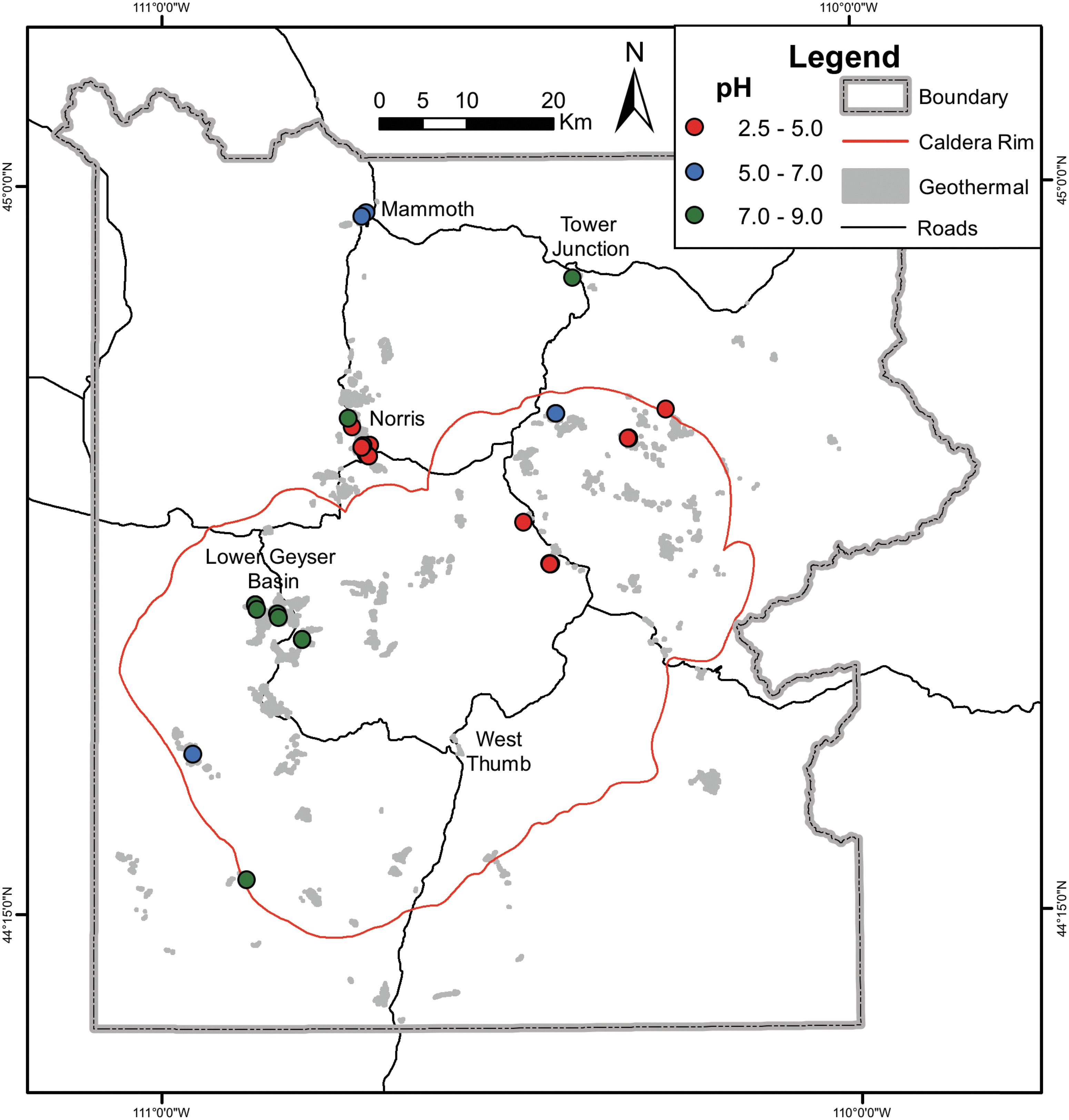
Map of YNP constructed with hydrothermal, caldera, and road reference layers. The geographic locations of hot springs with metagenomes from chemosynthetic communities are plotted and color-coded according to the pH of the hot spring. Geothermal areas are colored gray, the caldera rim is depicted by a red line, and roads are depicted by black lines. Reference layers (geothermal, caldera rim, and road layers) were obtained from the USGS GIS database (Christiansen, 2001). YNP, Yellowstone National Park.
pH can be considered an umbrella variable that influences numerous aspects of geochemistry that in turn can influence microbial metabolism, most notably the availability of nutrients. For example, pH can influence the chemical speciation and volatility of substrates [e.g., pKa = ∼7.5 at 100°C for NH4 + (aq)/NH3(g)] (Amend and Shock, 2001) and it can influence the chemical stability of substrates by influencing precipitation, hydrolysis, or disproportionation reactions. As an example, thiosulfate (S2O3 2−) is chemically unstable in aqueous solutions with pH <4.0 and rapidly disproportionates to form elemental sulfur (S°) and sulfite (SO3 2−), the latter of which is also unstable at low pH and rapidly oxidizes abiotically to form SO4 2− (Xu et al., 2000; Nordstrom et al., 2005). S° is chemically stable at temperatures of less than 100°C, and this can lead to its accumulation in acidic springs where it can serve as an electron donor or acceptor (or both during S0 disproportionation) that supports microbial metabolism (Boyd et al., 2007; Amenabar and Boyd, 2018; Amenabar et al., 2018). Moreover, acidic springs sourced by vapor phase gases are also likely to be enriched in volatiles such as H2 and CH4 that can serve as electron donors (Bergfeld et al., 2014; Lindsay et al., 2019) and generally have a higher total sulfur content (Nordstrom et al., 2009; Colman et al., 2018). Importantly, the acidity generated by sulfur oxidation in acidic springs can promote chemical weathering and the release of cations into solution, including iron that can serve as an electron donor or acceptor in the metabolism of thermoacidophiles (Kozubal et al., 2012; Amenabar et al., 2017; Amenabar and Boyd, 2018; Lindsay et al., 2018).
Based on these observations, we hypothesized that the inferred subsurface process of phase separation and the influence that this has on the pH of hot springs drive variation in the functional composition of chemosynthetic communities. Specifically, we hypothesized that the availability of metabolic substrates (e.g., sulfur and volatiles like H2 and CH4) in hot springs, which is controlled at first order by subsurface processes such as phase separation and subsequent influences on source waters, drives functional variation among chemosynthetic communities. To address this hypothesis, we compiled available metagenomes of chemosynthetic microbial communities of hot springs and investigated how broad differences in functional potential are correlated with differences in spring pH, temperature, and chemical proxies for vapor and liquid phase input. The number of metagenomes available from YNP hot spring communities is unparalleled among hydrothermal systems, and for this reason, we focus our efforts on these existing data sets. Results are discussed in the context of the role that geological processes have in influencing gas-, aqueous-, and solid-phase geochemistry and thereby in shaping the distribution of microbial functions in continental hydrothermal systems.
2. Materials and Methods
Assembled metagenomic data representing YNP hot spring community metagenomes as of September 17, 2018, were retrieved from the Integrated Microbial Genomes (IMG) database (Markowitz et al., 2012). Metadata associated with the metagenomes were used to screen for those that were from YNP hot springs and that represented samples from natural environments (Supplementary Table S1). Two additional metagenomes from moderately acidic spring sediment communities (“MV2” and “SJ3”), which were generated in our laboratory as components of other ongoing studies (Colman et al., 2019), were also included. Putative replicate metagenome entries were removed by choosing (1) the assembly that was released later, (2) choosing the metagenome with the latest version number, or (3) the metagenome with the larger assembled protein coding gene (PCG) count, depending on available data.
Metagenomes were also screened for evidence that they were not derived from enrichment experiments or consortia that were not clearly identifiable as belonging to environmental samples. Finally, only metagenomes from chemosynthetic communities (i.e., nonphotosynthetic) were selected from the data set based on published information for the metagenomes, IMG metadata, or otherwise by previous publications for the springs indicating the lack of photosynthetic activity. This screening resulted in 51 metagenomes of chemosynthetic communities from 27 springs across YNP with several metagenomes sampled from different locations within a given spring or at different times. The majority of these metagenomes were also used for analyses in the work of Colman et al. (2019).
Metadata (GPS coordinates and spring geochemistry) associated with the springs were collected from the IMG entries, or otherwise from published reports (Supplementary Table S1). In some instances, temperature, pH, chemical data, or GPS coordinates were not available through IMG or published reports and were instead collected from the publicly available survey of thermal features through the YNP Research Coordination Network (
To approximate the functional content within each metagenome, the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) assignments for each assembled metagenome were collected from IMG-processed annotations on the IMG server. The KO data were used to construct an abundance-weighted table for each metagenome using custom scripts in MATLAB. To account for unequal sequencing depths among metagenome assemblies, the KO abundances were normalized to the total abundance of KOs for that metagenome using the normalize.rows function of the vegetarian R software package v.3.4.1 (Charney and Record, 2012). A Bray–Curtis dissimilarity matrix was then constructed from the normalized data set by using the “vegdist” function within the vegan package (v.2.4–4) for R (Charney and Record, 2012). Due to the lack of available read-mapping and/or sequencing coverage data for the contigs of most publicly available metagenomes, the KO counts within each metagenome do not represent abundance-weighted estimates but are rather a richness-like estimate of each KO within each metagenome. Of the entire KO data set, only those database annotations associated within the “Metabolism” category were used in further analyses to limit the use to data that were most pertinent in discerning the relationship between spring geochemistry and microbial metabolism.
To identify patterns in the functional composition among metagenomes, the dissimilarity matrix was subjected to complete linkage clustering analysis using the “hclust” function in the base R package. The dissimilarity matrix was also subjected to principal coordinates analysis (PCO) by using the “pco” function in the labdsv v.1.8-0 package (Roberts, 2016). The percent variation that was explained by each axis of the ordination was calculated from the relative contributions of PCO eigenvalues.
To identify metabolic pathways and functional genes that were associated with overall differences in community metabolism among metagenomes, groups of metagenomes were defined based on clustering analyses, and the enrichment of KOs in each group, relative to the other defined groups, was investigated. Metagenome groups were defined based on broad geochemical differences that were consistent with the higher order branching of metagenomes within the cluster dendrogram. The “Metabolism” KEGG category includes protein encoding genes involved in pathways of carbohydrate, lipid, nucleotide, and amino acid metabolism in addition to other various categories including glycan biosynthesis, cofactor and vitamin metabolism, and secondary metabolite biosynthesis, among others. To limit the data being considered to only that which was most relevant to our hypothesis, only KOs within the “energy metabolism” subset (n = 818 non-zero KOs in the data set) of the broader “Metabolism” KEGG category (n = 6891 non-zero KOs) were used. We specifically focused on pathways of CH4, sulfur, and nitrogen metabolism in addition to oxidative phosphorylation, and carbon fixation pathways, among others. KOs enriched in each group were then investigated based on the distributions and relative abundances of each KO normalized within each sample, and within each metagenome group. Specifically, the relative enrichment of KOs was statistically analyzed by using indicator values that take into account both the “relative abundance” within individual samples of the group and their frequency among metagenomes within the group, as implemented in the “ind.val” function in the lab.dsv R package. KOs that were significantly (p < 0.05) enriched in a metagenome group were manually organized into broader metabolic pathways based on annotation information and inferred functionalities.
3. Results
3.1. Overview of metagenomes
Forty-nine publicly available metagenomes from chemosynthetic communities of YNP thermal springs were recovered from the IMG database (Supplementary Table S1). Two additional unpublished metagenomes from moderately acidic springs were also included (“SJ3” and “MV2”) (Colman et al., 2019). The compiled metagenomes represented spring communities from 12 geyser basins across YNP (Fig. 2) and spanned a range of pH (2.5–9.0) and temperature (54.0–93.5°C) regimes. A plot of the pH, temperature, and HS−/H2S concentrations (Fig. 3) for these spring waters (for those with available data) revealed that the communities were largely from springs with geochemical conditions previously shown to exclusively host chemosynthetic microorganisms (Boyd et al., 2010, 2012; Cox et al., 2011; Hamilton et al., 2012). Importantly, temperature, pH, and sulfide were only previously shown to account for 67% of the variance in the distribution of phototrophs in YNP springs (Boyd et al., 2012), suggesting that other unaccounted factors further constrain the distribution of this metabolism. Perhaps of little surprise, in several instances, metagenomes from springs analyzed in this study exhibited temperature and pH combinations that plotted within parameter space that can support photosynthetic metabolisms (Fig. 3A). However, these springs either exhibited elevated HS−/H2S that apparently restricts phototrophs (Castenholz, 1977; Oren et al., 1979; Miller and Bebout, 2004) or otherwise lacked evidence for photosynthetic populations in the corresponding metagenomic data. Of the seven springs with temperature and pH values that could theoretically allow for photosynthetic communities (Fig. 3A), “YNP10.Mammoth” exhibited sulfide concentrations above the apparent photosynthetic threshold (Inskeep et al., 2013b), whereas “SJ3,” “YNP17.OPP,” and “Bison3” did not exhibit similarly high levels of sulfide, but nonetheless lacked evidence for photosynthetic populations (Swingley et al., 2012; Inskeep et al., 2013b; Colman et al., 2019). Furthermore, sulfide measurements for the Mound and Bijah spring samples were not available, but the populations present in these metagenomes did not represent canonical photosynthetic primary producers (Yu et al., 2017). Finally, while sulfide concentration data were not available for the “Lib.Cap” sample, the metadata (IMG ID: 3300000343) indicated that it was retrieved from “streamers” at a temperature (72°C) consistent with Aquificales-dominated chemosynthetic streamer communities (Takacs-Vesbach et al., 2013). Thus, the metagenomic data set from chemosynthetic communities spanned broad geochemical and geographical ranges within the YNP geothermal ecosystem.
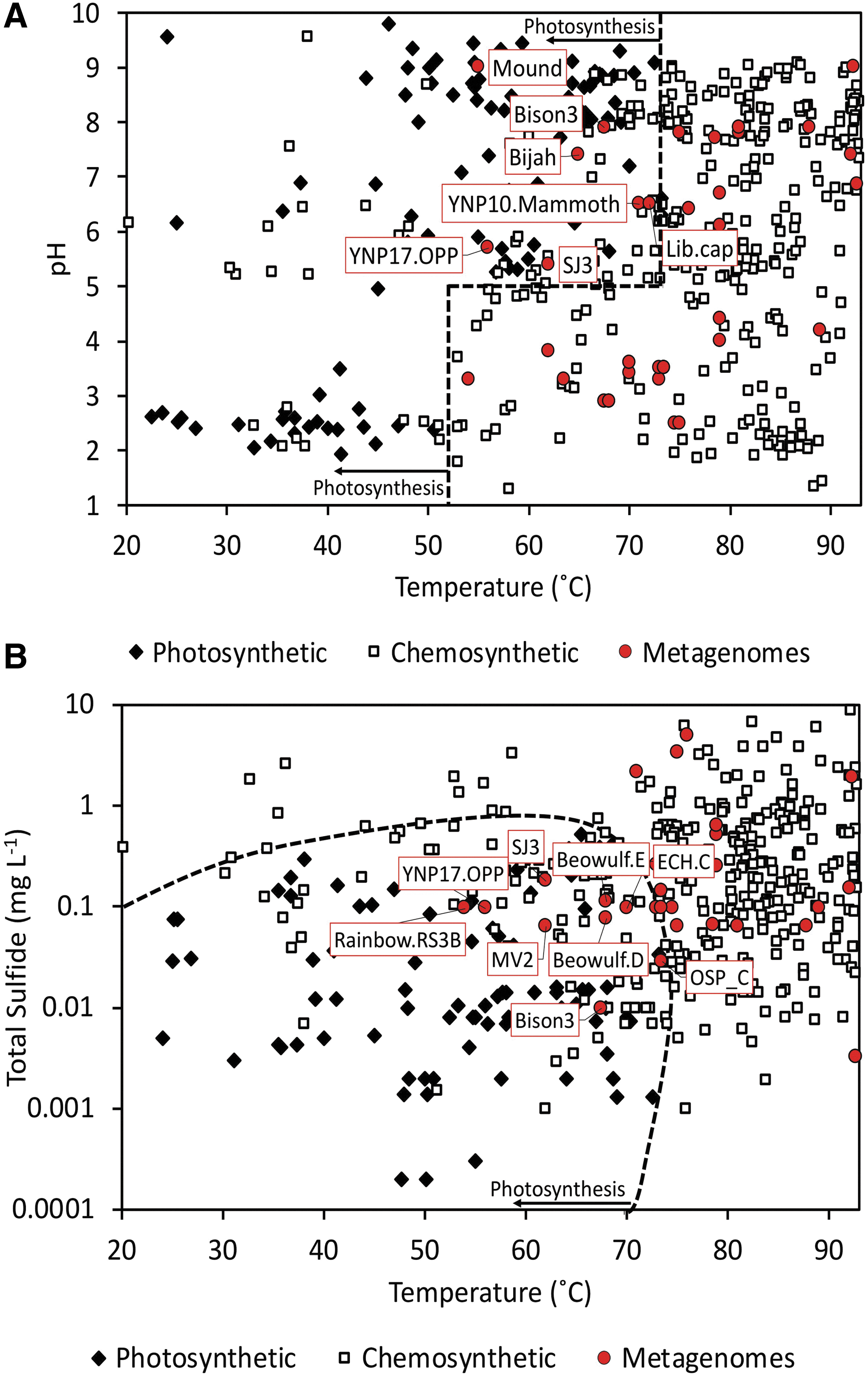
The presence (closed diamonds) or absence (open squares) of phototrophs in 439 springs plotted as a function of spring pH and temperature
3.2. Variation in metagenomic functional content
Variation in the PCG functional content among metagenomes was evaluated based on differences in the composition of KEGG-annotated PCGs in the “Metabolism” category for entire metagenomes. PCO of the relative abundances of KOs among metagenomic assemblies revealed discrete clustering of metagenomes by both pH (Fig. 4A) and temperature (Fig. 4B). Accordingly, regression of PCO axis 1 (which explained 38% of the variation in the ordination) against corresponding pH values for springs resulted in a highly significant correlation (adjusted R 2 = 0.695, p < 1 × 10−13, n = 50) (Supplementary Fig. S1). Furthermore, regression of PCO axis 2 (13% of variation) against temperature resulted in a highly significant relationship, although weaker than that of pH (R 2 = 0.432, p < 1 × 10−6, n = 50) (Supplementary Fig. S1). Nevertheless, the major dichotomy among spring metabolic PCG profiles corresponded to spring pH. It should be noted, however, that this distinction was not discrete, with moderately acidic springs (pH approximately 5–7) variously positioned as intermediates between lower and higher pH metagenomes, or otherwise clustered with the lower and higher pH groups (Fig. 4A).

Ordination of the dissimilarity in protein coding genes among 51 metagenomes from chemosynthetic communities in YNP and relationship to primary spring geochemical parameters.
To further evaluate the relationships among spring metagenome functional profiles and their correspondence to geochemical profiles, the metagenome KO distribution dissimilarities were clustered using complete linkage hierarchical clustering (Fig. 5). As in the PCO analyses, the 51 metagenomes clustered primarily by the pH of spring waters and secondarily by spring temperature (Fig. 5A–C). Five higher level groups (I–V) were defined based on metagenome clustering and geochemical attributes of the springs (Fig. 5A). Metagenomes that formed groups I and II were demarcated from other metagenomes by deriving from springs with acidic pH (generally pH <5) (Fig. 5B). However, these two groups could be further demarcated based on being from high (group I) or low (group II) temperature springs (Fig. 5C).
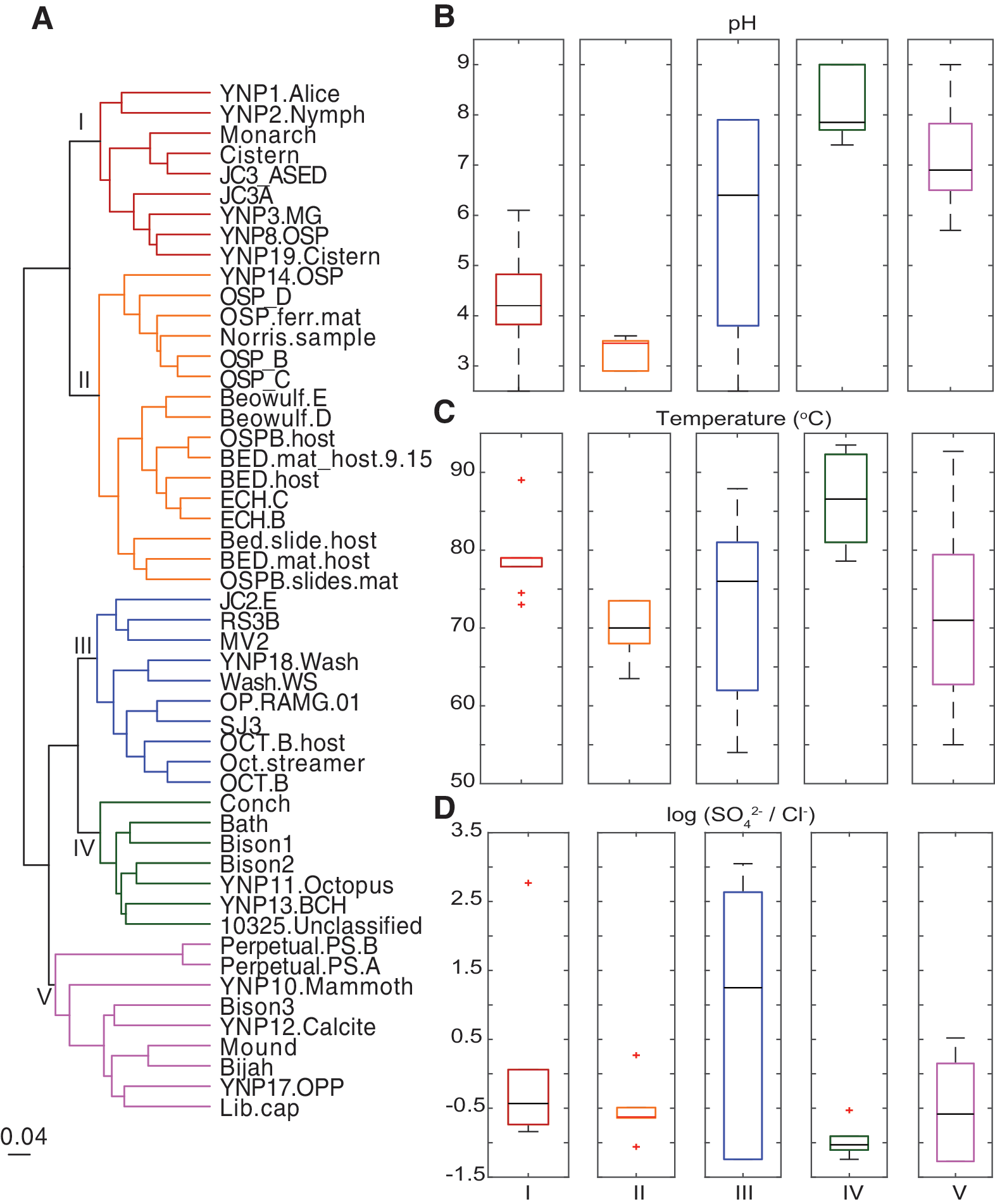
Dissimilarity in protein coding gene profiles among metagenomes from 51 chemosynthetic YNP springs and their corresponding geochemical attributes.
Metagenomes that formed group III were from springs that exhibited a wide range in pH (2.5–7.9) but that were, on average, moderately acidic (Fig. 5B). The metagenomes within this group were from springs that exhibited a large range in temperatures (54–88°C) but that were, on average, from lower temperature environments (Fig. 5C). With the exception of metagenomes from Octopus Spring, a defining feature of metagenomes that formed group III were that they were from springs that host waters with elevated SO4 2−/Cl− ratios (Fig. 5D). These geochemical signatures can be interpreted to indicate a higher input of reduced volcanic gas, including sulfide, that when oxidized contributes sulfate to the waters (Nordstrom et al., 2009). The low amounts of Cl− indicate relatively little input of deeper sourced hydrothermal waters (Fournier, 1989). Consequently, these springs are likely sourced by volcanic gas that condensed in the presence of meteoric water (Nordstrom et al., 2009; Lindsay et al., 2019).
Metagenomes that formed groups IV and V were from springs with circumneutral to alkaline pH, despite that they did not cluster into the same dendrogram group (Fig. 5B). Group IV metagenomes were from springs with elevated temperature when compared with those representing metagenomes that formed group V (Fig. 5C). Metagenomes in these groups also tended to be from springs that host waters with low SO4 2−/Cl− ratios (Fig. 5D), an observation that likely reflects predominant sourcing of these springs by deeply sourced, circumneutral, Cl−-rich waters (Nordstrom et al., 2009).
It should be noted, however, that variation was observed for both metagenomes from the same spring and in the geochemical attributes of the springs that typified each of the five metagenome cluster groups. As an example, metagenomes from Octopus Spring clustered in both groups III and IV, despite this spring exhibiting geochemical signatures associated with being sourced by deep, Cl−-rich, high temperature waters typical of springs that host group IV metagenomes. Likewise, the two metagenomes from Obsidian Pool clustered with both groups III and V, despite this spring featuring characteristics typical of group III springs, including moderate-to-high SO4 2−/Cl− ratios and moderately acidic pH (Meyer-Dombard et al., 2005; Shock et al., 2005; Colman et al., 2016). Nevertheless, the majority of metagenome assemblies from the same spring clustered exclusively or otherwise within the same metagenome group, including those from Perpetual Spouter, the higher temperature Bison Pool samples, three of the Octopus Spring samples, “Washburn Spring,” “Beowulf Spring,” Echinus Geyser, “OSP Spring,” Monarch Geyser, Cistern Pool, and “JC3A Spring.”
3.3. Enrichment of metabolic functions among chemosynthetic community groups
Statistical analysis of KO enrichment among the five metagenome groups revealed metabolic functions that differentiated them. The results of these analyses are described below for three groups of metagenomes from low pH (groups I and II), mid pH (group III), and high pH (groups IV and V) springs. Groups were defined not only by higher order clustering but also by shared geochemical characteristics. Consequently, although groups IV and V spring metagenomes did not form a cohesive cluster in the cluster analyses (and group IV clustered with group III), they are nevertheless discussed together because they shared similar geochemical properties (i.e., high pH and low SO4 2−/Cl−) and broadly similar enriched functional gene profiles. It should be noted that the significant enrichment of KOs within a particular group does not preclude their presence in other metagenomes or groups, but that their “relative abundances” and distribution discriminated a particular group from others. A subset (n = 114) of the total (n = 298) significantly enriched KOs comprising sulfur, CH4, 1-carbon, H2, and nitrogen metabolism is shown in Fig. 6, whereas the remainder (respiratory-associated, ATP synthesis, carbon metabolism, unidentified functions, others, and photosynthesis-related) are shown in Supplementary Fig. S2.
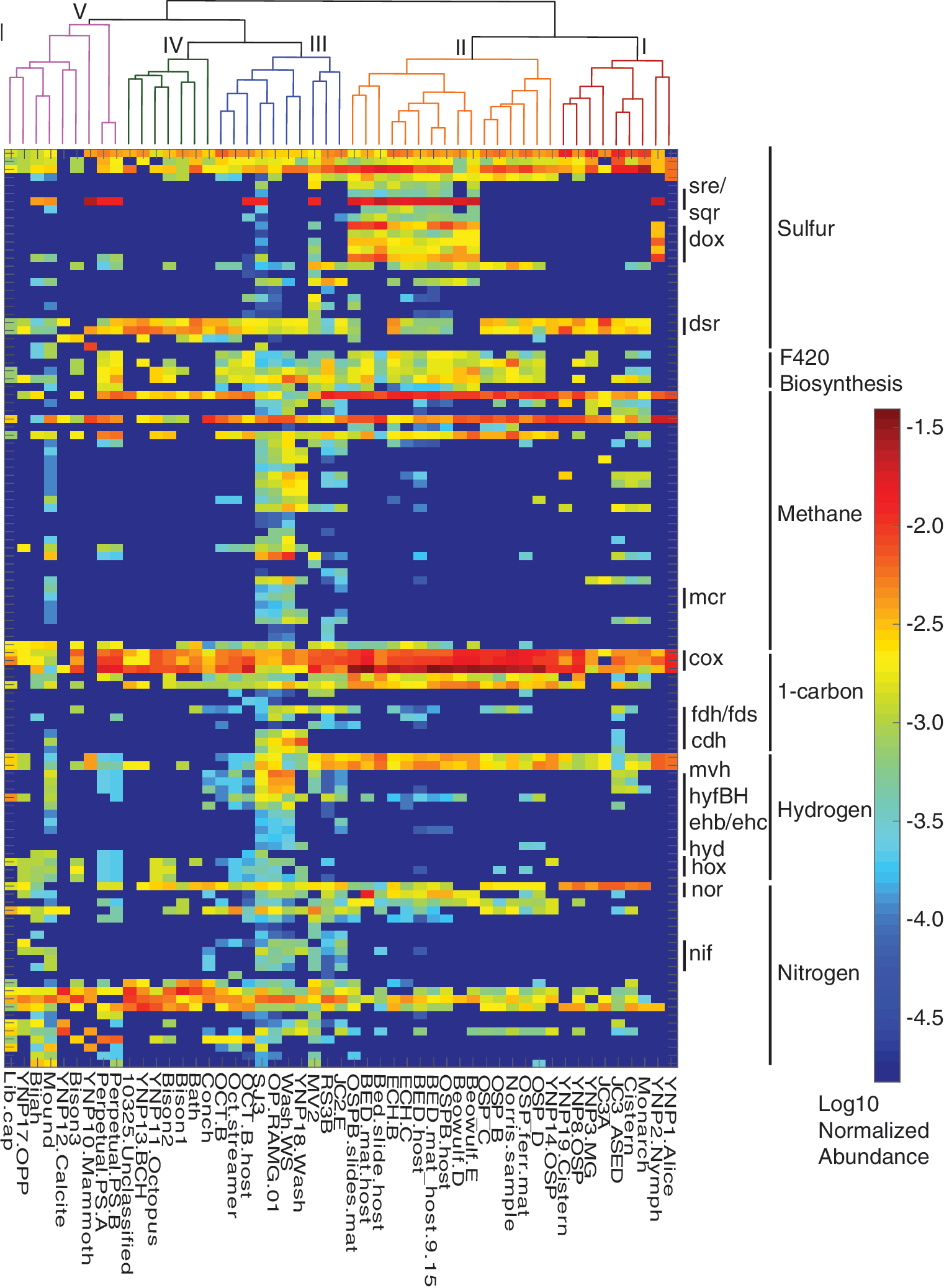
Enrichment of KO metabolic functions among metagenome groups. Heatmap shows the log-transformed normalized abundances of KO groups that were significantly (p < 0.05) enriched among the five metagenome groups. The color scale on the right indicates normalized abundances. Statistical significance of KO enrichment was assessed using indicator value analyses that accounts for “relative abundance” within groups (e.g., groups I–V), and the relative frequency among metagenomes within a group (i.e., the fidelity of the KO for metagenomes within a group). KOs were manually grouped according to higher levels of metabolic category classification. KOs that were specifically highlighted in the text are shown on the right of the heatmap, and additional information about the KOs including full names of functional homologs are provided in Supplementary Table S3 in the same order shown as the heatmap. The dendrogram above the heatmap is the same as in Fig. 5, and individual metagenome names are provided below the heatmap. Scale bar at the top left of the figure shows the scale (0.04) for community distances. KO, KEGG Orthology.
3.3.1. Metabolic functions enriched in groups I and II metagenomes
The predominant KO category that distinguished metagenomes from low pH springs (groups I and II) from others was sulfur metabolism (Supplementary Table S2). Several KOs with predicted functions in the oxidation of reduced sulfur compounds or reduction of oxidized sulfur compounds were enriched in metagenomes of groups I and II. In particular, KOs annotated as molybdopterin sulfur reductase (Sre) subunits involved in sulfur reduction (Laska et al., 2003) and sulfide:quinone oxidoreductases (Sqr) involved in sulfide oxidation (Shahak et al., 1992) were enriched in group II metagenomes. In addition, both the thiosulfate-quinone oxidoreductase subunits (DoxAD) implicated in the oxidation of sulfur compounds (i.e., S2O3 −) by thermoacidophilic archaea and terminal oxidases associated with sulfur oxidation (DoxBCE) (Quatrini et al., 2009; Auernik and Kelly, 2010) were enriched in group II metagenomes. In addition, sulfur oxygenase reductases (Sor) that are used in sulfur oxidation within thermoacidophilic archaea (Kletzin, 1989) were enriched in metagenomes of group II, but the relationship was slightly above the significance cutoff (p = 0.067) for inclusion in Fig. 6.
A number of other PCGs involved in dissimilatory sulfur metabolism were enriched in group II metagenomes, including anaerobic sulfite reductases (Asr) and alkanesulfonate monooxygenase (Ssu). Furthermore, other sulfur-metabolism-related genes were enriched in group I (adenylylsulfate kinase, CysC; phosphoadenosine phosphosulfate reductase, CysH; and sulfate adenylyltransferase, Sat) and group II (assimilatory sulfite reductase, CysI) metagenomes. HdrB (heterodisulfide reductase subunit B) was also significantly enriched in group I metagenomes but is annotated within the CH4 metabolism pathway of the KEGG database. This is despite the prevalence of this homolog in the genomes of thermoacidophilic, non-methanogenic archaea and bacteria where it has been suggested to be involved in S° oxidation (Quatrini et al., 2009).
While a few KOs involved in methanogenesis were enriched in group I or II metagenomes (16% of the total, including HdrB), they were not core functional homologs involved in methanogenesis (Fig. 6). Intriguingly, however, KOs involved in F420 biosynthesis were relatively enriched in many of the group II metagenomes. F420 is an integral cofactor for methanogens, present in other Archaea (Lin and White 1986), and recent metagenomic analyses have indicated the presence of F420 biosynthesis pathways in thermoacidophiles of YNP springs (Jay et al., 2018), which is consistent with their enrichment in high temperature acidic spring metagenomes. While protein homologs involved in metal metabolism are particularly poorly annotated, the single enriched KO involved in metal metabolism, mercuric reductase (MerA), was significantly enriched in group II metagenomes. This is consistent with enrichment of MerA homologs among the genomes of thermoacidophiles (Boyd and Barkay, 2012) and merA polymerase chain reaction amplicons in high temperature acidic springs in YNP (Wang et al., 2011). Furthermore, of the KOs associated with 1-carbon metabolism, only aerobic carbon monoxide (CO) dehydrogenase variants (Cox) were enriched in group II metagenomes, in addition to formamidases and an assembly factor for formate dehydrogenases (FdhD). Finally, the enrichment of KOs involved in nitrogen metabolism was noticeably absent in group I or II metagenomes, with the exception of nitric oxide reductase subunits (Nor) and a KO encompassing nitrilases.
3.3.2. Metabolic functions enriched in group III metagenomes
Group III metagenomes were derived from springs that exhibited wide ranges in pH and temperature. With the exception of metagenomes from Octopus Spring, one of the defining features of group III metagenomes were that they were from springs with geochemical signatures that are consistent with being sourced by volcanic gas that has condensed with meteoric waters (e.g., “SJ3,” “Washburn,” and Obsidian Pool). Of the enriched KOs for group III metagenomes, those involved in CH4 metabolism, 1-carbon metabolism, and H2 metabolism were most prominent (Fig. 6 and Supplementary Table S3). Indeed, aside from KOs involved in F420 biosynthesis, the majority of CH4-metabolism-associated KOs were enriched in group III metagenomes (80% of the total enriched CH4-metabolism-related KOs). This included subunits from the methyl coenzyme reductase complex (Mcr), and other protein homologs involved in methanogenesis including formylmethanufaran dehydrogenase (Fwd), among others (Fig. 6). However, Mcr homologs were only identified in metagenomes from three springs: Smokejumper 3, “Washburn Spring,” and Obsidian Pool. Indeed, recent characterizations of previously unknown putative methanogens or methan-/alkan-otrophs from these three springs have highlighted the discrete enrichment of novel methane cycling organisms in them (Berghuis et al., 2019; Borrel et al., 2019; Colman et al., 2019; McKay et al., 2019; Wang et al., 2019) and likely others with similar geochemical characteristics. In addition, several other PCGs putatively involved in methylotrophic methanogenesis were enriched in this group, including trimethylamine-corrinoid protein Co-methyltransferases, [methyl-Co(III) methanol-specific corrinoid protein]:coenzyme-M methyltransferase, methanol-5-hydroxybenzimidazolylcobamide Co-methyltransferase, and [methyl-Co(III) methylamine-specific corrinoid protein]:coenzyme M methyltransferase (Mtt, MtaA, MtaB, and Mtb, respectively). While heterodisulfide reductases (Hdr) are integral to many methanogens via their participation in the electron bifurcating [NiFe]-hydrogenase-Hdr complex (Kaster et al., 2011), HdrB homologs were also highly prevalent in a number of other metagenomes where evidence for methanogenesis was rare or absent. This may reflect their widespread involvement in a number of hypothesized or inferred physiological processes primarily associated with sulfur metabolism (e.g., sulfur oxidation and SO4 2− reduction, among others; Mander et al., 2004; Quatrini et al., 2009; Pereira et al., 2011).
In addition to KOs associated with CH4 metabolism, KOs putatively involved in 1-carbon compound metabolism were particularly enriched in group III metagenomes. Indeed, of the thirteen KOs involved in 1-carbon metabolism, eight were enriched in group III (the primary exception being subunits of aerobic CO dehydrogenases that were enriched in group II metagenomes). The eight KOs comprised subunits for the anaerobic metabolism of CO (Cdh) and the metabolism of formate (Fdh, Fds, Hyc), in addition to a KO associated with S-(hydroxymethyl)mycothiol dehydrogenase. Homologs of carbonic anhydrase, which interconverts HCO3 − and CO2 and is integral in the acquisition of inorganic carbon for fixation in Archaea and Bacteria (Zimmerman and Ferry, 2008), were also enriched in group III metagenomes. KOs involved in H2 metabolism were also particularly highly enriched in metagenomes of this group (63% of the total enriched hydrogen metabolism KOs). These KOs comprised components of diverse hydrogenase isoforms including bacterial [FeFe]-hydrogenases (Hyd) and several [NiFe]-hydrogenases including the group 4 type energy-converting hydrogenase (Ech/Eha/Ehb) and formate hydrogenlyases (Hyf), in addition to group 3 isoforms typically associated with hydrogenotrophic methanogenesis (methyl-viologen reducing hydrogenase, Mvh) (Vignais and Billoud, 2007; Peters et al., 2015; Greening et al., 2016; Poudel et al., 2016). Importantly, the catalytic subunits were among the enriched KOs for several of these isoforms, including HydA, EchE, and MvhA.
Finally, several KOs involved in sulfur metabolism (24% of the total) and nitrogen metabolism (39% of the total) were enriched in group III metagenomes. The KOs enriched in group III and involved in sulfur metabolism were primarily involved in transport or assimilation of sulfur compounds (Fig. 6). In contrast, KOs representing subunits involved in the fixation of nitrogen (nitrogenase, NifH, NifB, and NifE), nitrification (hydroxylamine dehydrogenase, Hao), and denitrification (nitrate reductase, NapB and NapC; nitrite reductase, NirB and NirD), in addition to a KO annotated as a formate-dependent nitrite reductase (NrfF), were enriched in group III metagenomes.
3.3.3. Metabolic functions enriched in groups IV and V metagenomes
Group IV and V metagenomes were derived from springs that were generally circumneutral to alkaline in pH and that exhibited geochemical characteristics consistent with sourcing from deep Cl−-rich hydrothermal waters (Nordstrom et al., 2009). In contrast to groups I–III, metagenomes in groups IV and V lacked any enriched KOs associated with CH4 metabolism or 1-carbon metabolism. Likewise, only three KOs associated with sulfur metabolism (12% of the enriched KOs involved in sulfur metabolism) were enriched in group IV metagenomes and these included key genes involved in dissimilatory SO4 2−/SO3 2− reduction (DsrAB), in addition to a SO3 2− dehydrogenase involved in SO3 2− oxidation to SO4 2− (Kappler et al., 2000). The clustering of group III and IV metagenomes to the exclusion of group V metagenomes could have been potentially due, in part, to some protein functionalities that were nearly universally present in the former groups (although at varying “relative abundances”), but less universally present in group V metagenomes (Supplementary Table S3). While these protein annotations included several KO functions involved in core carbon metabolism, some KO functions involved in dissimilatory metabolism, including DsrAB, were also universally present in group III metagenomes (Supplementary Table S3). In addition to the above, a single KO annotated as QmoA, which is a component of the energy conservation pathway of SO4 2− reducers (Pereira et al., 2011), was enriched in group V metagenomes. Unlike groups I and II metagenomes, those from groups IV and V exhibited greater numbers of KOs involved in nitrogen metabolism (17% and 30% of the enriched KOs, respectively). These KOs corresponded to homologs that catalyze several nitrogen cycling processes including denitrification (ferredoxin-nitrite reductase: NirA, enriched in group IV; NO-forming nitrite reductase: NirS, group V; nitrate reductase: NapA and NapH, group V; nitrous oxide reductase: NosZ, group V), ammonification (hydroxylamine reductase: Hcp, group V), and the oxidation of nitronates (nitronate monooxygenase: Ncd, group IV). In addition, other nitrogen assimilation or transporter-associated functions were identified in the KOs enriched in groups IV and V metagenomes (Fig. 6).
4. Discussion
4.1. Subsurface geological processes influence the availability of substrates that support chemosynthetic microbial metabolism
Metabolic functionalities that support chemosynthetic organisms in high temperature hot spring communities in YNP are nonrandomly distributed and were collectively assigned to one of the five groups in our analyses. These five groups of metagenomes correspond largely to differences in the geochemistry of springs, which at first order is controlled by the source of waters and gases to those springs. The primary geological process thought to demarcate the different sources of hydrothermal fluids that supply hot springs is decompressional boiling of ascending hydrothermal waters and the separation of fluids into liquid and vapor phases. As mentioned above and expounded upon below, condensation of vapor with oxidized meteoric waters can drive the oxidation of HS−/H2S and promote the generation of acid and the acidification of hot springs. Thus, hot spring waters influenced by vapor tend to be moderately acidic to hyperacidic, whereas liquid-phase-influenced hot spring waters tend to be circumneutral to alkaline.
The dichotomy in the functional composition of hot spring communities is evident in the high correlation between chemosynthetic community functional profiles and spring pH, with temperature exerting a secondary influence (Fig. 4A). Indeed, broad clusters of metagenomes are apparent that correspond to whether they were sampled from acidic or alkaline/circumneutral springs; a third metagenomic group was additionally observed that comprised metagenomes from springs with intermediate-type geochemistry (Figs. 4A and 5). This hierarchy of environmental factors that influence YNP thermal spring communities has been previously described for taxonomic compositions (Mitchell, 2009; Boyd et al., 2013; Colman, 2015; Colman et al., 2016), functional genes (Boyd et al., 2010), and smaller subsets of these metagenomes (Inskeep et al., 2013b; Alsop et al., 2014). Moreover, the primary and secondary roles of pH and temperature, respectively, in controlling 16S rRNA gene taxonomic compositions across continental geothermal fields have been observed for hot springs in China (Xie et al., 2014) and those of the Taupo Volcanic Zone in New Zealand (Power et al., 2018). Despite the widespread recognition of the integral role of pH in influencing thermal spring community ecology, little effort has been directed toward understanding why this relationship exists. The present investigation represents the largest analysis of thermal spring functional profiles from metagenomes. The results reported here provide important context for understanding why and how subsurface geological processes are likely to influence microbial communities across continental hydrothermal springs at a mechanistic functional level (Fig. 6).
4.2. Enrichment of sulfur-dependent metabolisms in communities inhabiting acidic springs
Metagenomes from acidic YNP hot springs were enriched in metabolic functions that allow for the use of sulfur compounds in their energy metabolism. This includes metabolic functionalities associated with the oxidation of HS−/H2S (Sqr) or those involved in the oxidation/reduction of intermediates generated during the oxidation of H2S/HS− (e.g., oxidation of S2O3 2− via Dox, reduction of S° via Sre). These observations are consistent with other studies that have documented the prevalence of organisms in acidic springs with the ability to utilize sulfur compounds (Brock et al., 1972; Boyd et al., 2007, 2009; Inskeep et al., 2013b; Amenabar et al., 2017, 2018; Colman et al., 2018).
Enrichment of KOs involved in the use of sulfur compounds in the energy metabolism of thermoacidophiles is consistent with current models for the generation of acidity in hot springs and with the expected bioavailability of sulfur compounds, which is controlled in large part by kinetic and thermodynamic instabilities of several key intermediate sulfur compounds. When volcanic SO2 is injected into subsurface hydrothermal aquifers, it can disproportionate to SO4 2− and H2S (Nordstrom et al., 2009), the former remaining in hydrothermal waters as a nonvolatile solute, and the latter apportioning into the vapor phase upon phase separation. Condensation of H2S-enriched vapor with oxidized meteoric waters results in the oxidation of H2S/HS−, primarily by O2 (Nordstrom et al., 2005, 2009). This reaction occurs abiotically and rapidly in aqueous solutions with circumneutral pH (pH >6.0) (Chen and Morris, 1972; Zhang and Millero, 1993) and at high temperature (Nordstrom et al., 2005). At lower pH and temperatures, abiotic oxidation of H2S with O2 is kinetically inhibited (Chen and Morris, 1972; D'imperio et al. 2008), providing an opportunity for microorganisms to utilize this substrate as an electron donor.
Oxidation of H2S, either abiotically or biotically regardless of pH, can result in the formation of S2O3 2−, which is stable at circumneutral pH, but disproportionates to form S° and SO3 2− at pH <4.0 (Xu et al., 2000; Nordstrom et al., 2005), the latter of which is also unstable at pH <4.0 and oxidizes abiotically under oxic conditions to form SO4 2− (Nordstrom et al., 2005). In contrast, S° is stable at temperatures <100°C and can accumulate in acidic springs where it can serve as an electron donor/acceptor, as described above. To this end, we suggest that enrichment in encoded functions involved in the metabolism of reduced sulfur compounds (e.g., H2S/HS−, S°) in microbial populations at acidic pH is consistent with the kinetic and thermodynamic stabilities of these compounds under these conditions, which therefore lead to higher bioavailabilities.
Seemingly inconsistent with this hypothesis, the high concentrations of SO4 2− in acidic springs (up to 5 mM) (Nordstrom et al., 2009) would otherwise be expected to lead to enrichment of encoded proteins involved in the use of SO4 2− as an electron acceptor. However, hyperacidic environments are among the most oxidizing environments on the planet (Boyd et al., 2019) and such conditions are thought to select against strict anaerobes (Colman et al., 2018) since anaerobic electron transfer pathways and electron carriers are considered to be tuned to operate at much lower reduction potentials (Moore et al., 2017; Poudel et al., 2018). Furthermore, it is possible that SO4 2− reducers are not competitive in acidic environments due to competition for electron donors and carbon sources among organisms operating higher energy yielding metabolisms, such as aerobic respiration (Shock et al., 2005). Nonetheless, SO4 2− reducing bacteria have been enriched from mining impacted environments in aqueous medium with a pH as low as 4.0 (Sanchez-Andrea et al., 2013), and isotopic tracer studies suggest that SO4 2− can be reduced by microorganisms associated with sediments collected from hot springs with temperatures as high as 88°C and pH as low as 2.3 in the Norris Geyser Basin of YNP (Fishbain et al., 2003). However, SO4 2− rates were sporadic among replicate cores in the latter study and were considerably lower than those sampled from circumneutral to alkaline springs, which could be due to a low abundance of populations capable of reducing SO4 2− in these systems. This interpretation is consistent with genes for SO4 2− reduction not being especially enriched in metagenomes from acidic YNP springs.
4.3. Enrichment of gas-supported metabolisms in communities inhabiting moderately acidic springs
Group III comprised metagenomes that were from springs that spanned a wide range of temperature and pH. However, an attribute that was generally consistent among these springs was the relatively high SO4 2−/Cl− ratios of their waters. As discussed above, high SO4 2−/Cl− ratios have been interpreted to reflect springs that are sourced by oxidized meteoric waters enriched with volcanic gas (i.e., vapor) without significant input of hydrothermal water (Nordstrom et al., 2009). Concentrations of dissolved H2 and CH4 in “SJ3” are the highest measured in YNP springs, and this spring exhibits a high SO4 2−/Cl− ratio (Colman et al., 2019; Lindsay et al., 2019). H2 and CH4 are also enriched in total gases sampled from springs in the Washburn area, Mud Volcano area (i.e., Obsidian Pool and “MV2”), and Hot Springs Basin (e.g., “RS3” and “JC2”) (Bergfeld et al., 2014), and these springs host waters with the highest SO4 2−/Cl− ratios measured in YNP (Bergfeld et al., 2014; Lindsay et al., 2019). Consequently, the enrichment of gas-dependent metabolisms in this metagenome group is consistent with current models for fluid sourcing to these springs (Nordstrom et al., 2009) and suggests that resident populations are adapted to take advantage of these gases as electron donors and/or carbon sources. Indeed, ∼80% of the dominant population-level genome bins assembled from SJ3 encode for hydrogenase homologs that are biased toward H2 oxidation (Lindsay et al., 2019). Moreover, a number of population-level genome bins from SJ3 were shown to be enriched in functionalities that allow for CH4 metabolism and metabolism of other volatiles such as CO (Colman et al., 2019).
Methanogens have been documented in photosynthetic mats in YNP hot springs (e.g., Ward et al., 1998). However, with the exception of a few individual sites (e.g., Imperial Geyser, 81.5°C, pH 4.1) (Boyd et al., 2013), investigations of the microbial diversity in chemosynthetic communities in YNP springs have generally highlighted the conspicuous absence of methanogens (Inskeep et al., 2013a; Colman, 2015). Recent studies, however, suggest that putative methanogens are present, if not widespread in YNP thermal springs, but belong to uncultured archaeal taxonomic lineages (Berghuis et al., 2019; Borrel et al., 2019; Colman et al., 2019; McKay et al., 2019; Wang et al., 2019) that are phylogenetically distinct from the canonical euryarchaeal lineages that host methanogens (Bapteste et al., 2005). For example, putative hydrogenotrophic methanogens affiliated with the Archaeoglobales order have been identified in “SJ3” spring (Colman et al., 2019), and these are evolutionarily and metabolically distinct from canonical members of the Archaeoglobales that metabolize SO4 2−, SO3 2−, and S2O3 − or reduce Fe(III) (Garrity and Holt, 2001). Moreover, putative methanogens or methanotrophs within the candidate division ‘Verstraetearchaeota’ which is related to the Crenarchaeota (Vanwonterghem et al., 2016), were identified in “SJ3” (Colman et al., 2019). Likewise, these taxa, in addition to putative methanogens or methan-/alkane-otrophs, were recently described via metagenomic data from Obsidian Pool and “Washburn Spring” that were associated with Korarchaeota and “Hadesarchaeota” (Berghuis et al., 2019; Borrel et al., 2019; McKay et al., 2019; Wang et al., 2019). Importantly, the key enzyme complex for CH4 activation, McrABG, is required for Archaea catalyzing both methanogenesis and methanotrophy. McrABG are also apparently involved in the anaerobic oxidation of short-chain hydrocarbons such as butane (Laso-Perez et al., 2016) and ethane (Chen et al., 2019), making it difficult to definitively associate directionality (e.g., CH4 formation or oxidation) or substrate utilization (e.g., CH4 or butane) with these taxa. Nonetheless, enrichment of functionalities such as hydrogenases, Hdr, and Mcr that are associated with methanogenesis, methanotrophy, or short-chain hydrocarbon cycling is consistent with an elevated input of volcanic gases that commonly contain H2, CH4, and short-chain hydrocarbons into these springs (Bergfeld et al., 2014).
Diverse hydrogenase isoforms and protein homologs associated with 1-carbon compound (e.g., CO and formate) metabolism were particularly enriched in metagenomes within group III (Fig. 6). Intriguingly, the diverse array of hydrogenase isoforms that were enriched in group III metagenomes (Hyd, Ech, Ehb, Hyf, and Mvh) differed from the single isoform enriched in group V metagenomes (Hox) that are from lower temperature circumneutral springs. This suggests potentially different physiological roles of hydrogenases associated with these functional groups in different hot spring settings. This is consistent with previous work showing that hydrogenase isoforms are distinctly associated with specific physiologies (Vignais and Billoud, 2007; Schut et al., 2013; Boyd et al. 2014; Peters et al., 2015; Greening et al., 2016; Poudel et al., 2016). It is possible, if not likely, that the enrichment of metabolic functionalities to utilize gaseous substrates such as H2, CO, CH4, or short-chain hydrocarbons in springs featuring elevated supplies of these gases results from selection for their inclusion during the assembly of resident communities. Springs sourced by liquid-phase input (i.e., circumneutral springs) tend to have less H2 (Bergfeld et al., 2014; Lindsay et al., 2019), and thus, the inclusion of populations adapted to utilizing H2 as an electron donor would be expected to be lower in these systems.
4.4. Enrichment in nitrogen and respiratory functions in circumneutral and alkaline spring communities
In contrast to the metagenomes associated with low pH and moderately acidic springs, the circumneutral and alkaline YNP spring community metagenomes lacked enrichment in many of the metabolic functionalities that defined the other groups, including sulfur metabolism, CH4 metabolism, H2 metabolism, and 1-carbon compound metabolism. Higher pH springs are thought to be sourced by the liquid-phase component from the phase separation process, as described above. As such, these springs would generally not be expected to exhibit high levels of gas and are likely to have lower total sulfur content than groups I–III spring types. Thus, the lack of enrichment in KOs associated with metabolism of volatile gases and sulfur compounds in the communities of these higher pH springs is consistent with proposed models for the sourcing of these springs with volatile and sulfur poor fluids (Fournier, 1989; Nordstrom et al., 2005, 2009). It is important to note that HS−/H2S can be enriched in circumneutral/alkaline springs (Zinder and Brock, 1977; Cox et al., 2011). However, the kinetics of abiotic oxidation of HS−/H2S are faster at alkaline pH (Chen and Morris, 1972; D'imperio et al 2008), which might preclude involvement of microbial metabolism in its oxidation for the purposes of energy generation (Shock and Boyd, 2015).
Nevertheless, the capacity for dissimilatory SO3 2−/SO4 2− reduction (via Dsr) was one of the few sulfur-based metabolic functionalities that was uniquely enriched in either group IV or group V metagenomes. It is unclear if the SO4 2− that would support these organisms is sourced from the disproportionation of volcanic SO2 in the deeper part of the hydrothermal system, which is estimated to contribute a baseline of approximately 73–98 mg/L SO4 2− to the liquid-phase component of phase-separated fluids (Nordstrom et al., 2009), or if it is derived from abiotic/biotic sulfide oxidation. Regardless, unlike in highly oxidizing acidic springs that may select against anaerobes, in particular those that operate lower energy yielding reactions such as SO4 2− reducers or methanogens (Shock et al., 2005), alkaline environments are more reduced (due to predominant sourcing by circumneutral/alkaline hydrothermal waters) (Boyd et al 2019). Thus, these waters may be expected to be more compatible with anaerobic organisms and their metabolisms, such as SO4 2− reduction.
Of the few KOs that were enriched in the higher pH metagenomes, those involved in the reduction of oxidized nitrogen species were particularly prevalent (Fig. 6). In addition, prevalent protein homologs involved in heterotrophy and respiratory pathways were enriched in groups IV and V metagenomes from circumneutral to alkaline springs (Supplementary Fig. S2). At the same time, protein coding functions involved in lithotrophic metabolisms were not enriched in metagenomes from these spring types. These observations could point to an increased dependence on respiratory functions involving oxidized nitrogen compounds (e.g., NO3 −) and organic carbon in circumneutral to alkaline hot springs. Many of the metagenomes from higher pH spring communities were generated from filamentous biofilm communities that have previously been shown to depend on respiratory functions and particularly aerobic respiration (Takacs-Vesbach et al., 2013; Beam et al., 2015), and heterotrophic metabolism (Schubotz et al., 2015). Paradoxically, the lack of meteoric water input into these predominantly hydrothermally sourced springs should lead to a lack of available oxidants (e.g., O2, NO3 −) within the spring source waters. However, the biofilm communities described above are generally located at the surface of spring runoff channels, which feature locally higher levels of dissolved O2 due to atmospheric in-gassing during water movement and turbulence (Inskeep et al., 2005; Fouke, 2011, Takacs-Vesbach et al., 2013). Moreover, Schubotz et al. (2015) demonstrated that the carbon that supports heterotrophic metabolism in filamentous streamer communities of several circumneutral to alkaline springs is likely to be exogenous to the spring and perhaps derived from soil runoff or dust deposited by wind.
5. Conclusions
The results described herein suggest a link between subsurface geological processes, geochemistry, and the metabolic processes that support microbial life in high temperature hot springs. In particular, data suggest that distinct geochemical provinces are generated in hot spring environments, and the generation of these distinct environments is driven primarily by the process of phase separation. These include three broad groups of spring types: (1) acidic sulfur-rich springs whose geochemical composition is influenced by input of the vapor component of phase-separated waters, (2) circumneutral to alkaline springs that are influenced by input of the liquid-phase component of phase-separated waters, and (3) moderately acidic gas-rich hot springs that are sourced by meteoric water and condensed vapor.
Our data suggest that pH, corresponding with the different pH provinces defined above, explained the most variation in the functional composition of communities, which is likely due in part to the physiological stress that extremes of pH (e.g., hyperacidity) impose on cells. However, these data also suggest that the relationship between community functional composition and pH is due to the sources of waters that supply these springs with nutrients that support chemotrophic microbial metabolisms. In particular, chemosynthetic organisms inhabiting acidic springs are generally adapted to integrate sulfur into their energy metabolisms, whereas microorganisms inhabiting moderately acidic gas-rich springs are adapted to utilize those gases as sources of carbon and/or reductant to fuel biosynthesis and metabolism. Microorganisms that inhabit circumneutral to alkaline springs appear to be adapted to heterotrophic metabolism involving oxidized nitrogen compounds, oxygen, or sulfate as electron acceptors.
Variation in the predominant metabolisms that support hot spring populations is likely to result from the effects of phase separation on the sourcing of hot springs with volatiles, which can explain differences in the metabolisms supporting organisms inhabiting moderately acidic springs (group III) versus circumneutral to alkaline springs (groups IV and V). In turn, microbial acidification of hot spring waters influences the kinetic and thermodynamic stabilities of nutrients and their bioavailability to support microbial metabolism (groups I and II). Thus, these data provide an important geobiological framework to understand how geological processes have shaped the evolutionary history of thermophiles, in particular those that are dependent on mineral sources of energy, and how thermophiles in turn have shaped the habitability of their geochemical environments.
Footnotes
Acknowledgments
We thank Christie Hendrix, Stacey Gunther, and Annie Carlson at YNP for research permitting.
Author Disclosure Statement
The authors declare no commercial associations or conflicts of interest with the present investigation.
Funding Information
This work was supported by a NASA EPSCoR grant (MT-19-EPSCoR-0020) to E.S.B. and D.R.C, a grant from the Montana Space Grant Consortium grant to D.R.C. and E.S.B, a grant from the National Science Foundation to E.S.B. and D.R.C. (EAR-1820658), and a grant from the NASA Astrobiology Institute (NNA13AA94A) to E.S.B. M.R.L. acknowledges support from the NASA Earth and Space Science Fellowship program (NNX16AP51H).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.