
Abstract
The search for, and characterization of, organic matter on Mars is central to efforts in identifying habitable environments and detecting evidence of life in the martian surface and near surface. Iron oxides are ubiquitous in the martian regolith and are known to be associated with the deposition and preservation of organic matter in certain terrestrial environments, thus iron oxide-rich sediments are potential targets for life-detection missions. The most frequently used protocol for martian organic matter characterization (also planned for use on ExoMars) has been thermal extraction for the transfer of organic matter to gas chromatography-mass spectrometry (GC-MS) detectors. For the effective use of thermal extraction for martian samples, it is necessary to explore how potential biomarker organic molecules evolve during this process in the presence of iron oxides. We have thermally decomposed iron oxides simultaneously with (z)-octadec-9-enoic and n-octadecanoic acids and analyzed the products through pyrolysis-GC-MS. We found that the thermally driven dehydration, reduction, and recrystallization of iron oxides transformed fatty acids. Overall detectability of products greatly reduced, molecular diversity decreased, unsaturated products decreased, and aromatization increased. The severity of this effect increased as reduction potential of the iron oxide and inferred free radical formation increased. Of the iron oxides tested hematite showed the least transformative effects, followed by magnetite, goethite, then ferrihydrite. It was possible to identify the saturation state of the parent carboxylic acid at high (0.5 wt %) concentrations by the distribution of n-alkylbenzenes in the pyrolysis products. When selecting life-detection targets on Mars, localities where hematite is the dominant iron oxide could be targeted preferentially, otherwise thermal analysis of carboxylic acids, or similar biomarker molecules, will lead to enhanced polymerization, aromatization, and breakdown, which will in turn reduce the fidelity of the original biomarker, similar to changes normally observed during thermal maturation.
1. Introduction
1.1. The search for organic matter on Mars
Organic molecules may be relatively widespread on Mars. They will at least have been delivered to the planetary surface from exogenous sources such as meteorites, comets, or interplanetary dust particles (Gibson, 1992; Flynn, 1996; Sephton, 2012). They may also have formed from indigenous martian sources due to igneous, hydrothermal, and atmospheric processes (Chyba and Sagan, 1992; ten Kate, 2010; Steele et al., 2012, 2018; Konn et al., 2015).
Habitable conditions are believed to have existed on ancient Mars (Grotzinger et al., 2014) and may transiently occur up to the present day (Jones, 2018). Potential martian habitability raises the exciting possibility that organic matter has been generated from the biological processes of extinct or extant life, and may, under certain conditions, be preserved in near-surface sediments, sedimentary rocks, or regolith (Summons et al., 2011; Vago et al., 2017). Thus, alongside other potential biosignatures such as stable isotopes, atmospheric gases, biosedimentary structures and macrotextures (Summons et al., 2011), organic biomarker compounds are a target for both past habitability (e.g., as sought by Mars Science Laboratory [MSL]) and life-detection (e.g., as sought by ExoMars 2022) efforts (Des Marais et al., 2008; Vago et al., 2017).
The majority of attempts to detect martian organic molecules have, however, only found simple organochlorine molecules (Biemann et al., 1977; Glavin et al., 2013; Leshin et al., 2013; Ming et al., 2014; Freissinet et al., 2015). So far, the lack of complex organic compound detection has been partly attributed to the deleterious effects of oxidizing (and chlorinating) minerals on the martian surface where perchlorates and potentially sulfates cause the oxidation of organic matter during thermal extraction experiments (Blake et al., 2013; Glavin et al., 2013; Leshin et al., 2013; Ming et al., 2014; Freissinet et al., 2015; Lewis et al., 2015; Miller et al., 2016). The organochlorine molecules are, therefore, suggested to be the reaction products of more complex martian organic molecules, such as polycyclic aromatic hydrocarbons (PAHs) or organic acids, being transformed in the presence of oxidizing minerals.
Recent detections of nonchlorinated organic molecules on Mars have identified relatively simple species such as alkanes, thiophenes, alkylbenzenes, and naphthalene (Eigenbrode et al., 2018; Freissinet et al., 2019). The aromatic compounds have been attributed to be the pyrolysis products of a macromolecular source (Eigenbrode et al., 2018), while it has been suggested that the alkanes may be obtained from the decarboxylation of carboxylic acids (Freissinet et al., 2019), suggesting that a variety of sources and types of organic matter do exist on Mars.
The crust of Mars has abundant iron (Taylor and McLennan, 2009), and its oxides are ubiquitous on the martian surface. The nanophase, hydrated iron oxide species ferrihydrite (5Fe2O3 · 9H2O) is believed to be widespread on the martian surface as a major component of the iron-rich amorphous soil (Dehouck et al., 2017), along with the more crystalline oxyhydroxide goethite [FeO(OH)] and the iron (II, III) and iron (III) oxides magnetite (Fe3O4) and hematite (Fe2O3). These iron species have been detected in martian meteorites (Steele et al., 2007; Muttik et al., 2014) by infrared spectroscopy (Evans and Adams, 1980; Bell et al., 1993); by the Mössbauer spectrometers on both of the Mars Exploration Rovers (MER) (Klingelhöfer et al., 2005; Morris et al., 2006a, 2006b) and by X-ray diffraction (XRD) by the Chemistry and Minerology (CheMin) instrument on board the MSL Curiosity rover (Bish et al., 2013; Blake et al., 2013; Treiman et al., 2016).
Iron oxides can preserve biosignatures through a variety of mechanisms. These include the entombment of bacteria by iron oxide crystals (Ferris et al., 1989; Peng et al., 2013), the formation of textural biosignatures by iron oxide encrustation (Potter-McIntyre et al., 2014; Williams et al., 2015, 2017), the formation of organometallic complexes (Lalonde et al., 2012), and the preservation of organic molecular biosignatures through adsorption onto the surface of iron oxides and oxyhydroxides (Parenteau et al., 2014; Tan et al., 2018). The end results of these preservative mechanisms can be clearly seen in a variety of aqueous environments on Earth. Almost a quarter of organic carbon in sedimentary deposits has been found to be directly bound to reactive iron phases with this association promoting its preservation over geological timescales; this is known as the “rusty sink” effect (Lalonde et al., 2012). Many of these terrestrial, iron-rich settings are analogous to environments present on ancient Mars (Allen et al., 2000, 2004; Schelble et al., 2004; Fernández-Remolar et al., 2005); hence, iron-oxide-rich sediments are potential targets for future life-detection missions.
Searches for life and abiotic organic molecules on Mars have so far been almost exclusively carried out by thermal extraction experiments, by the Viking Landers (Klein et al., 1976; Biemann et al., 1977), the Phoenix mission (Hecht et al., 2009; Cull et al., 2010; Kounaves et al., 2014), and the Sample Analysis at Mars instrument suite on the MSL (Mahaffy et al., 2012; Blake et al., 2013; Glavin et al., 2013; Leshin et al., 2013; Ming et al., 2014; Freissinet et al., 2015; Miller et al., 2016), and this technique will also be applied to samples from up to 2 m below the martian surface on the forthcoming ExoMars mission (Barnes et al., 2006).
Despite their potential for preserving code-posited martian organic matter, iron oxides are known to promote the oxidative breakdown of organic molecules on heating (Minicò et al., 2000), and are likely to have an effect on the detectability of any organic molecules present in an iron-oxide-rich sediment analyzed by thermal decomposition techniques. We need to know, therefore, if and how Mars-relevant iron oxides catalyze the breakdown of organic biosignature molecules during thermal decomposition and how this may lead to false-negative life detections.
1.2. Carboxylic acids as biosignatures
In this study we have focused on carboxylic (fatty) acids as unlike most other biomolecules, they are relatively chemically recalcitrant even under harsh environmental conditions and so may persist over geological timescales (Summons et al., 2008), and it has been suggested that alkanes detected at Mars may be the degradation products of carboxylic acids (Freissinet et al., 2019). Carboxylic acids are useful biosignatures with their alkyl chain length and saturation state suggestive of their source. In the context of this article, we define a short alkyl chain as ≤C5, medium as C6–C12, long as C13–C21, and very long ≥C22.
Abiotic processes yield primarily shorter length molecules and decreasing amounts of carboxylic acids of longer chain lengths in an Anderson–Schulz–Flory type distribution, typical of polymerization processes, where an alkyl chain grows probabilistically by bonding with additional methylene (CH2) groups verses chain terminating moieties (McCollom and Seewald, 2007; Smith et al., 2013). This discriminates against longer chain lengths to create a unimodal distribution and does not give any preference to either odd or even carbon numbers (Summons et al., 2008), as may be observed in experimental Fischer-Tropsch-type reactions (Mißbach et al., 2018) and abiotic settings such as meteorites (Cronin and Pizzarello, 1990; Sephton, 2002). Any deviations from this probability distribution, usually in the form of increased medium- or long-chain carboxylic acids, are indicative of formation processes that preferentially select for longer chain lengths. In addition, these formation processes must dominate over random addition reactions to be expressed in carboxylic acid profiles.
Biotically sourced carboxylic acids are the decomposition products of lipids, phospholipids, and triglycerides, which are critical components of biological cells (Kates, 1964). Biologically controlled processes select for carboxylic acids of longer chain lengths as they have stronger intermolecular attractions and thus form stronger structures (Rawicz et al., 2000). In terrestrial biosignatures, carboxylic acids with a distribution around C16–C17 are indicative of a microbial source, C18–C22 of algae and >C23 of higher plants (Cranwell, 1974; Tissot and Welte, 1978; Perry et al., 1979; Bianchi, 1995). Biotic carboxylic acids also show a strong even-over-odd preference in their carbon number due to their extension through the addition of two methylene (C2H4 or CH2–CH2) units (Hartgers et al., 2000; Summons et al., 2008; Georgiou and Deamer, 2014).
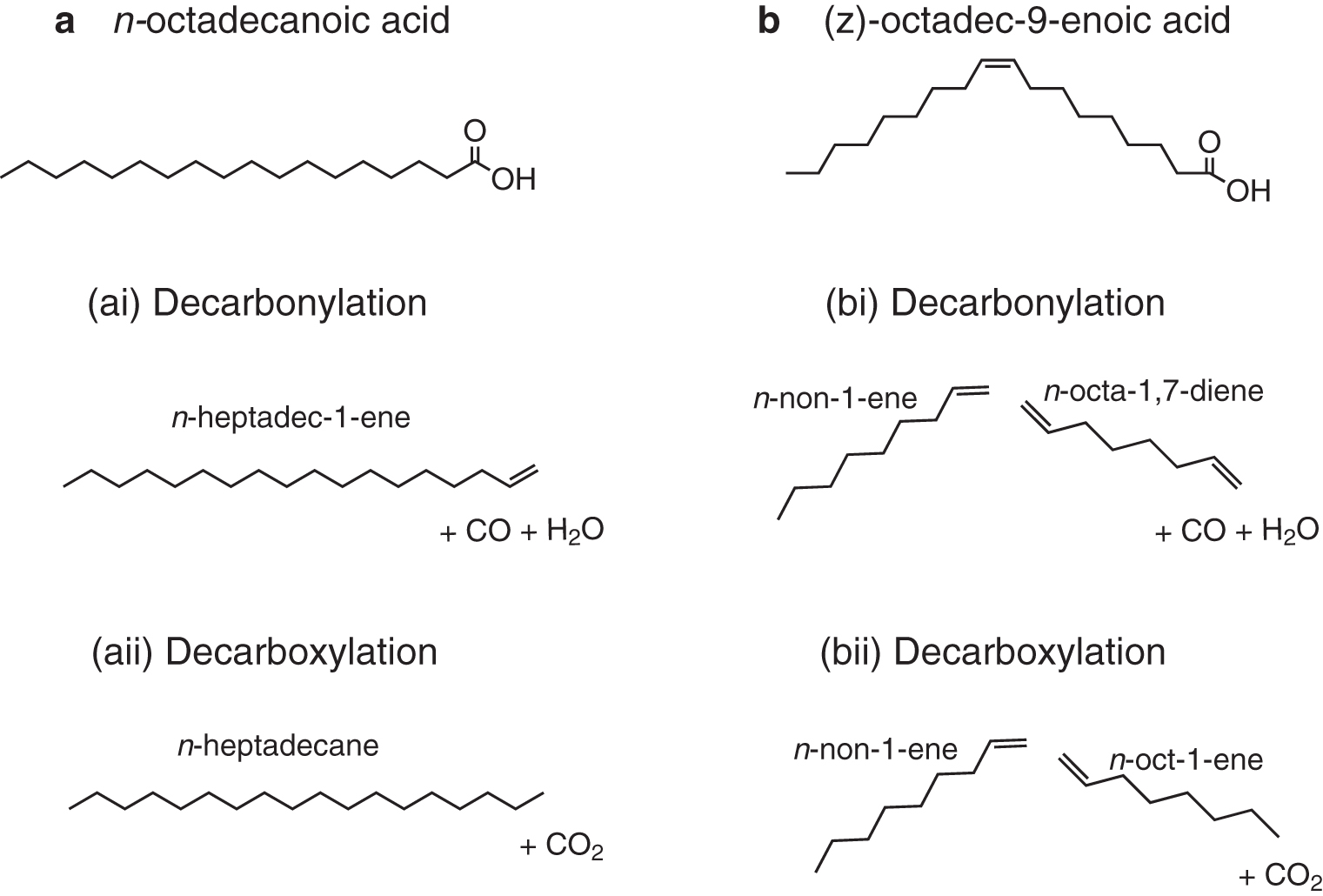
Unsaturated long-chain carboxylic acid may indicate the presence of recent (extant) life (O'Leary, 1962; Volkman et al., 1989; Alfaro et al., 2006), but they are highly labile, undergoing reactions at the sites of their double carbon bonds (through microbial degradation, autoxidation with the atmosphere, or other diagenetic processes) to quickly hydrogenate to saturated long-chain carboxylic acid, which are more stable over geological timescales (Parker and Leo, 1965; Matsuda and Koyama, 1977; Van Vleet and Quinn, 1979; Haddad et al., 1992). Chain length distribution patterns, however, are more resistant to alteration (Duda et al., 2018). Figure 1 shows saturated and unsaturated forms of C18 long-chain carboxylic acid.
Saturated
Further to these distribution patterns, abundances of specific carboxylic acids may be indicative of the metabolites of certain taxa (O'Reilly et al., 2017). For example, C18 polyunsaturated fatty acids are diagnostic of cyanobacteria (Allen et al., 2010; Willers et al., 2015; Sánchez-García et al., 2020), while methyl-branched unsaturated carboxylic acids may be indicative of sulfate reducers (Londry et al., 2004).
1.3. Pyrolysis of carboxylic acids
Under normal pyrolysis conditions, the breakdown of saturated carboxylic acids proceeds through a sequence of reactions starting with decarbonylation (Fig. 1a[i]) at low temperatures (the loss of a carbon and oxygen) or decarboxylation (Fig. 1b[ii]) at higher temperatures (the loss of the carboxyl group to form CO2 and the addition of a proton to the remaining alkene/alkane), and proceeding with cleavage of hydrocarbon bonds by β-scission (Hartgers et al., 1995). With unsaturated carboxylic acids, deoxygenation (via either decarbonylation or decarboxylation depending on temperature) occurs simultaneously with β-scission, which takes place preferentially at the sites of double carbon bonds (Fig. 1b[i], [ii]). The process results in a series of n-alk-1-enes and n-alkanes and carboxylic acids, with the carboxylic acids and any surviving internal alkenes becoming progressively depleted due to further breakdown as temperatures and reaction times increase (Asomaning et al., 2014a, 2014b).
The presence and position of the double bond influence the pyrolysis behavior of unsaturated carboxylic acids as the lower bond dissociation energy of the C = C bond (compared with C–H and C–O) means that initiation of the pyrolysis reaction involves homolytic cleavage around the C = C bond. This means that, for example as shown in Fig. 1b(i) and (ii), (z)-octadec-9-enoic acid will preferentially crack to form a n-non-1-ene and non-8-enoic acid, and further break down to a n-non-1-ene and n-octa-1,7-diene depending on temperature. A saturated carboxylic acid would eventually form a series of alkanes and terminal dienes/alk-1-enes with no preferential chain length (Hartgers et al., 1995). This preferential breakdown means that, in theory, by examining the pyrolytic products and their distribution, it should be possible to determine the carbon chain length of the parent carboxylic acid, its saturation state, and the positions of the double carbon bonds.
1.4. Pyrolysis of carboxylic acids on iron oxides
Mineral substrates can affect both the distribution and yield of pyrolysis products (Davis and Stanley, 1982; Espitalié et al., 1984; Karabakan and Yürüm, 1998; Faure et al., 2006a, 2006b; Fréty et al., 2014; Zafar et al., 2017). Thus, when pyrolyzing carboxylic acids adsorbed onto iron oxides, deviation from the theoretical products may provide information on the extent to which the substrate is influencing the breakdown products. This information can then be used to determine what organic species detected on Mars could be evidence of these interactions taking place during thermal decomposition analyses.
In this study, the saturated n-octadecanoic acid (stearic acid) and the monounsaturated (z)-octadec-9-enoic acid (oleic acid) were examined as model carboxylic acids. Online flash pyrolysis-gas chromatography-mass spectrometry (GC-MS) was used to thermally decompose the carboxylic acids in the presence or absence of a variety of Mars-relevant iron oxides/(oxy)hydroxides. Comparing the effects of pyrolysis on both saturated and monounsaturated carboxylic acids of equal carbon chain lengths with a variety of Mars-relevant iron oxides/(oxy)hydroxides allowed the determination of the sources and mechanisms by which any pyrolysis products are formed. All carboxylic acids should behave in predictable ways when compared with these two model molecules, and so it should be possible to extrapolate the results to a wider range of carboxylic acid lengths, double bond positions, and ultimately their parent triglycerides. Analysis of the iron oxide/(oxy)hydroxide mineral substrates by XRD after pyrolysis will allow the reactions affecting the pyrolysis products to be determined. The results will provide guidance for the design of thermal extraction experiments on Mars and help interpretation of any data generated by life-detection missions to the Red Planet.
2. Methods
2.1. Sample preparation
Quartz sand (US Silica F-35) was used as a relatively inert substrate. Magnetite, hematite, and goethite were obtained from Sigma-Aldrich. Due to its instability, ferrihydrite dehydrates and recrystallizes rapidly at room temperature (Cudennec and Lecerf, 2006; Das et al., 2011); ferrihydrite had to be produced in the laboratory. Ferrihydrite was synthesized following the method detailed in Cornell and Schwertmann, 2003. XRD analysis (see below) confirmed the obtained ferrihydrite to be of the amorphous/least crystalline two-line ferrihydrite, as opposed to the more (but still poorly) crystalline six-line ferrihydrite, based on the number of broad peaks (two or six) exhibited in the XRD pattern (Cornell and Schwertmann, 2003; Kukkadapu et al., 2003). Eight grams of Fe(NO3) · 9H2O (Sigma-Aldrich, suitable for cell culture) was dissolved in 100 mL of deionized water and stirred with a magnetic stirrer (which had previously been cleaned by sonication in deionized water followed by 93:7 v/v dichloromethane [DCM]/MeOH solution). To this solution, 66 mL of 1 M potassium hydroxide (KOH) solution was continuously added to bring the pH of the resulting mixture to 7.5. The final volume of KOH solution was added dropwise, and the pH was continuously monitored (using a pH meter, which was rinsed with deionized water and DCM/MeOH between measurements). Dissolved salts were removed by adding deionized water, centrifuging and decanting the supernatant until the conductivity was <10 μS/cm as measured with a total dissolved solids probe (which was rinsed with deionized water and DCM/MeOH between measurements). The synthesized ferrihydrite was frozen, then placed in a Labcono freeze dryer at 0.133 mbar. Synthetic samples were ground gently in a solvent-cleaned agate pestle and mortar, wrapped in aluminum foil and stored at 4°C.
Quartz sand was cleaned by furnacing overnight at 450°C to remove any initial organic matter contamination. Due to their reactivity, the iron oxides magnetite, hematite, and goethite had to be cleaned through solvent extraction rather than heating; these minerals oxidize and/or dehydrate/dehydroxylate if furnaced in air at the temperatures required to combust organic contaminants (Ruan et al., 2002; Mazo-Zuluaga et al., 2003). Solvent extraction was carried out by using a standard liquid extraction protocol (e.g., Emmerton et al., 2012) as follows: 1–3 g of powdered sample was suspended in a 93:7 v/v DCM/MeOH solution and agitated in a sonic bath. The resulting suspension was then centrifuged, and the supernatant solution pipetted off. This process was repeated three times, and the materials were dried under a stream of nitrogen. As the ferrihydrite was produced in the laboratory by using clean materials and methods, and subsequently stored clean, it was deemed unnecessary to clean this highly reactive mineral further.
The cleaned substrates were spiked with solutions of either (z)-octadec-9-enoic acid or n-octadecanoic acid in pentane, which was subsequently evaporated off under a nitrogen stream, to produce quartz sand/iron oxide samples with known concentrations of carboxylic acid. To test detectability, spiked substrates at 50 ppm, 500 ppm, and 0.5% (z)-octadec-9-enoic acid by weight (wt %) were produced. n-Octadecanoic acid was only tested at 0.5 wt % and was not tested on the ferrihydrite substrate based on the unamiability of this substrate to detection of carboxylic acids as shown by the initial experiments with (z)-octadec-9-enoic acid. Unspiked quartz sand/iron oxide samples were also analyzed as control samples.
2.2. Pyrolysis GC-MS
Ten to twenty milligrams prepared samples were loaded into quartz pyrolysis tubes and held in place with quartz wool (both tubes and wool were cleaned by overnight furnacing at 450°C); (z)-octadec-9-enoic and n-octadecanoic acids were also pyrolyzed without a substrate with 0.4 (±0.1) mg of carboxylic acid supported by quartz wool.
Loaded samples were placed inside the platinum coil of a Chemical Data Systems 5200 Pyroprobe under helium and heated at a rate of 20°C m/s to the target temperature where it was held for 15 s. Each sample was run in replicate. Individual samples were run at 450°C or 650°C. These temperatures were selected because the usual range for flash pyrolysis of organic matter is 400–900°C, with most studies going no >700°C (Hartgers et al., 1994, 1995; Watson et al., 2012). Above this temperature range, fragmentation is too extensive and structural information is lost, except with highly refractory material (Matthewman et al., 2012; Sephton, 2012). Thus, 450°C and 650°C gave scope for comparing relatively “low” temperature pyrolysis where only “weak” (e.g., C–O–C) bonds should be broken and relatively “high” temperature pyrolysis where “strong” (e.g., C = C and C–C) bonds should break. The interface was run at 350°C but not turned on until after the probe had fired to avoid premature desorption of the spiked samples (Zafar and Watson, 2017; Zafar et al., 2017).
Separation and identification of products were achieved by using an Agilent Technologies 6890 gas chromatograph coupled to a 5973 mass spectrometer (GC-MS). The GC inlet was held at 270°C and operated in split mode (10:1) with a column helium flow rate of 1.1 mL/min. Separation was performed on an SGE BPX5 column (25 m × 220 μm × 0.25 μm). The GC oven was held for 2 min at 40°C and then ramped at a rate of 5°C/min to 310°C where it was held for 10 min. Mass spectra were acquired over a range of m/z 45–550. For gas phase analysis, to identify highly volatile low mass phases released, mass spectra were acquired over a range of m/z 10–150 (all other pyrolysis, interface, and GC settings were kept identical). Identification of peaks in the obtained chromatograms was carried out with the Agilent MSD Chemstation software package, and peak identification was achieved by comparison with the National Institute of Standards and Technology database, published literature and standards.
Data from the laboratory pyrolysis-GC-MS were assumed to be comparable with those from the Sample Analysis at Mars (SAM) and Mars Organic Molecule Analyser (MOMA) experimental setups. While differences in heating rate and environmental conditions between laboratory- and rover-based instruments (Table 1) could affect the distribution of pyrolysis products. Flash, as opposed to temperature ramped, pyrolysis can be advantageous in a laboratory analog experiment as it may provide a “worst-case scenario,” ensuring the coincidence of reactions of both the inorganic and organic phases.
GC-MS = gas chromatography-mass spectrometry; MOMA = Mars Organic Molecule Analyser; SAM = Sample Analysis at Mars.
2.3. XRD analysis
Representative XRD patterns of iron oxides were acquired before pyrolysis and after pyrolysis at 650°C with 0.5 wt % octadecenoic acid. A PANalytical X'Pert Pro MPD Alpha-1 X-Ray Diffractometer with an X'Celerator detector was used to analyze samples between 5 and 80° 2θ under copper radiation with a step size of 0.033° at a speed of 5°/min. Measurements were made at a current of 40 mA and a voltage of 40 kV. The diffraction patterns were analyzed using a X'Pert Highscore Plus program with references from the International Centre for Diffraction Data database.
3. Results
3.1. Pyrolysis-GC-MS
Control samples were analyzed by pyrolyzing quartz sand and iron oxide substrates postcleaning to determine the effectiveness of the techniques used (Supplementary Fig. S1). The only products detected were from column-bleed (siloxanes that were also detected in blank runs) and low molecular weight peaks from gas phases released during the thermal decomposition of the iron oxides. Product distribution and relative abundance were reproduceable between replicates. Release of adsorbed water and CO2 (the main low mass phases identified from all three iron oxides when the mass spectrometer was optimized for low mass compounds, data not shown) from ferrihydrite was orders of magnitude higher than the other iron oxides due to its adsorbent and hydrous nature.
3.1.1. (z)-Octadec-9-enoic acid
Figure 2 shows the total ion current chromatograms (TICs) produced by pyrolyzing (z)-octadec-9-enoic acid by itself and on the quartz sand, hematite, magnetite, goethite, and ferrihydrite substrates at 0.5 wt % at 450°C and 650°C.
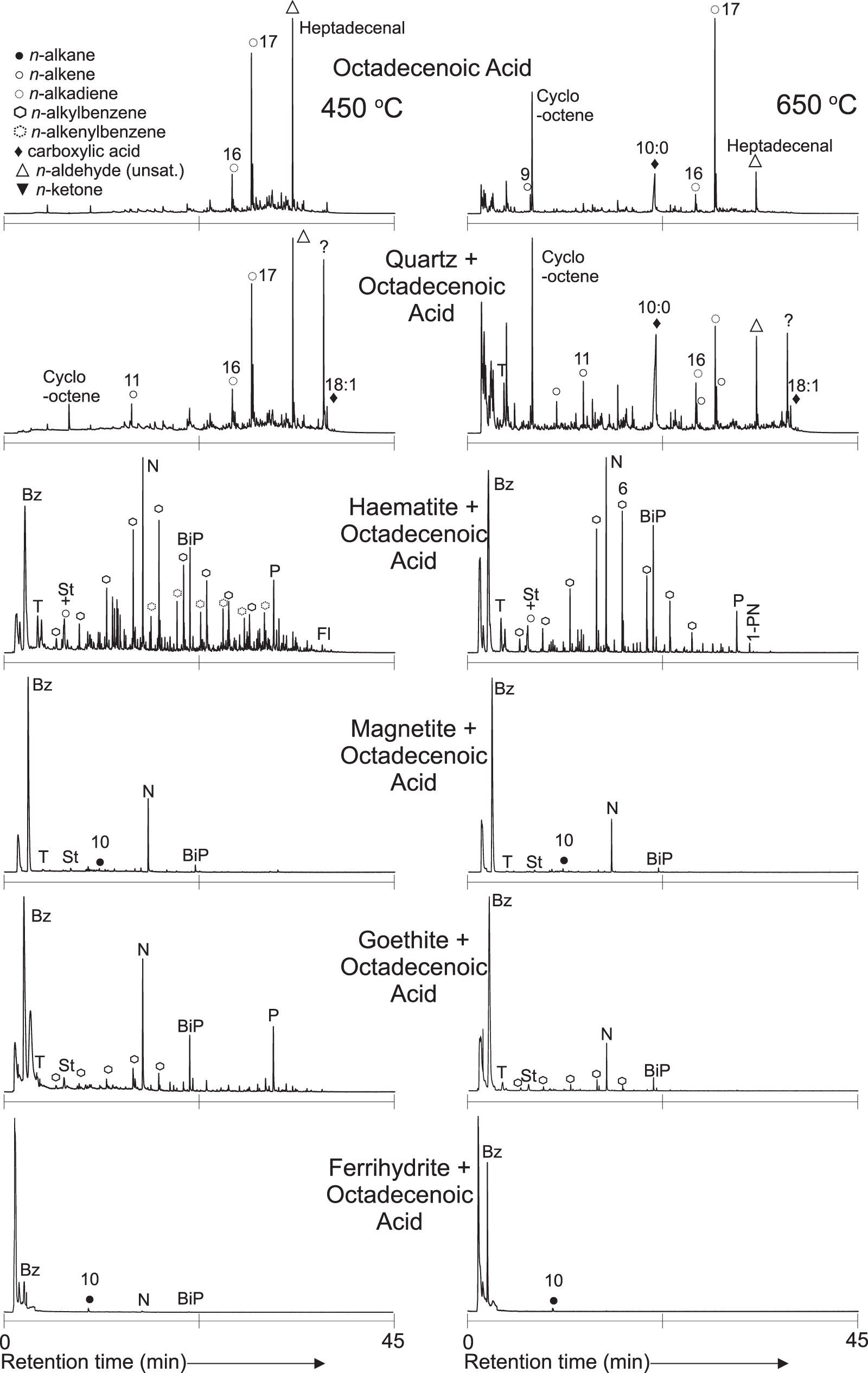
TICs of 0.5 wt % (z)-octadec-9-enoic acid pyrolyzed on various substrates. Numbers with symbols refer to the aliphatic carbon chain length of the molecule. Bz, benzene; T, toluene; St, styrene; N, naphthalene; P, phenanthrene; BiP, biphenyl; DBF, dibenzofuran; 9-F, fluorenone; PN, phenylnaphthalene; Fl, fluoranthene. TIC, total ion current chromatogram.
3.1.1.1. (z)-Octadec-9-enoic acid pyrolysis products
The main products of (z)-octadec-9-enoic acid when pyrolyzed, either by itself (Fig. 2, “octadecenoic acid”) or on a quartz sand substrate (Fig. 2, “quartz + octadecenoic acid”), were extremely similar, so they will be discussed together. The products were as expected based on previous studies of the pyrolysis of carboxylic acids (Zafar and Watson, 2017; Zafar et al., 2017; Santos et al., 2019). At both 450°C and 650°C, the signal was dominated by n-alkadienes, cyclic and n-alk-1-enes, and minor n-alkanoic acids and n-alkylbenzenes. A notable peak of the monounsaturated aldehyde heptadecanal was observed at both temperatures when (z)-octadec-9-enoic acid was pyrolyzed by itself.
At the lower (450°C) pyrolysis temperature, the products were dominated by products with 16–17 carbon atoms, particularly hexadeca-1,6-diene, heptadeca-1,7-diene, and heptadecanal (Fig. 2).
At the higher (650°C) temperature, the longer chain length fragments were less dominant. C8–C10 fragments, particularly cyclo-octene and n-decanoic acid, were greater in relative abundance at this higher pyrolysis temperature (Fig. 2).
Despite the similarities, differences were observed between (z)-octadec-9-enoic acid pyrolyzed on its own and on quartz in the relative abundances of products. With the addition of a quartz sand substrate there appeared to be both an increased survivability of the intact parent compound at both temperatures and a further increase in the relative abundance of lower molecular weight compounds at 650°C.
Pyrolyzing various concentrations of (z)-octadec-9-enoic acid on the quartz sand substrate (Supplementary Fig. S2) revealed nonlinear decreases in the response and diversity of products with decreasing concentrations. There was a decrease in the relative abundance of higher molecular weight products, especially heptadecanal and the parent (z)-octadec-9-enoic acid. When 50 ppm of (z)-octadec-9-enoic acid was pyrolyzed at 450°C, benzene was the only identifiable product.
3.1.1.2. (z)-Octadec-9-enoic acid on iron oxides
Figure 2 shows that when (z)-octadec-9-enoic acid was pyrolyzed on an iron oxide/(oxy)hydroxide substrate, the pyrolysis products were significantly different to those produced when the carboxylic acid was pyrolyzed by itself or on a quartz sand substrate. Aliphatic hydrocarbons and n-alkyl- and n-allylbenzenes (benzenes with n-alkene or n-alkane aliphatic side chains) were the major pyrolysis products observed when (z)-octadec-9-enoic acid was pyrolyzed in the presence of iron-bearing substrates. Supplementary Figures S7 and S8 show the distribution of aliphatic hydrocarbons and n-alkylbenzenes in the pyrolysis products of 0.5 wt % (z)-octadec-9-enoic acid spiked samples at 450°C and 650°C and reveal changes in chain length distribution and saturation state dominance between samples.
Figure 2 shows that of the iron-bearing substrates tested, pyrolysis of (z)-octadec-9-enoic acid on hematite exhibited the greatest diversity in pyrolysis products (Fig. 2 “hematite + octadecenoic acid”). When (z)-octadec-9-enoic acid was pyrolyzed on a hematite substrate, aromatic hydrocarbons and pairs of n-alkyl- and n-allylbenzenes dominated the pyrolysis products at both 450°C and 650°C. The aliphatic hydrocarbons were dominated by n-alkanes up to nonane (C9) but were dominated by n-alk-1-enes at higher carbon numbers (Supplementary Fig. S7). At 450°C, oxygenated species in the form of both aliphatic (particularly around C8–C11) and aromatic ketones were also present as minor products. When (z)-octadec-9-enoic acid was pyrolyzed on hematite, aromatization of products (in comparison with pyrolysis without substrate or on quartz sand) was increased at both 450°C and 650°C pyrolysis temperatures. A variety of aromatic moieties with PAHs up to four rings were detected.
Pyrolyzing various concentrations of (z)-octadec-9-enoic acid on the hematite substrate (Supplementary Fig. S3) showed that as concentration was decreased the relative abundance of the minor aliphatic hydrocarbons, particularly unsaturated species, decreased relative to aromatic moieties. This effect was more pronounced at 650°C than at 450°C.
The products of (z)-octadec-9-enoic acid pyrolyzed on both magnetite (Fig. 2, “magnetite + octadecenoic acid”) and goethite (Fig. 2, “goethite + octadecenoic acid”) were similar. Compared with either quartz or hematite, these were defined by an overall lower variety and abundance of molecular species. Aromatic moieties, with n-alkylbenzenes and PAHs (moieties up to four benzene rings were tentatively detected), were increased relative to nonaromatic products. Hydrocarbon chain lengths of the aliphatic hydrocarbons and n-alkylbenzenes were further decreased (compared with quartz or hematite substrates). Unsaturated aliphatic compounds were rare, with alkanes dominating the aliphatic profiles at both temperatures (Supplementary Fig. S7). Aromatic ketones, including the symmetrical ketone fluoren-9-one, and benzaldehyde were minor products from both goethite and magnetite substrates. The observed transformative effects on the pyrolysis products of (z)-octadec-9-enoic acid were greater in the oxyhydroxide goethite compared with the oxide magnetite, with fewer aliphatic moieties detected from goethite.
Pyrolyzing various concentrations of (z)-octadec-9-enoic acid on the magnetite substrate (Supplementary Fig. S4) showed that as concentration was decreased alkanes became the dominant pyrolysis products relative to aromatic species.
The (z)-octadec-9-enoic acid pyrolyzed on ferrihydrite showed few identifiable peaks above detection limits in the TIC (Fig. 2, “ferrihydrite + octadecenoic acid”) and extracted ion current chromatograms (Supplementary Figs. S7 and S8 “ferrihydrite”). Small peaks around detection limits corresponding to benzene, decane, naphthalene, and biphenyl were the only pyrolysis products observed in the 0.5 wt % (z)-octadec-9-enoic acid on ferrihydrite data.
Detectable products from both the goethite and ferrihydrite substrates were significantly reduced at lower concentrations of (z)-octadec-9-enoic acid (Supplementary Figs. S5 and S6). At both 500 ppm and 50 ppm wt % (z)-octadec-9-enoic acid on the goethite substrate, only benzene was detectable, and (at the same concentrations) on the ferrihydrite substrate no products were identifiable.
3.1.2. n-Octadecanoic acid
Figure 3 shows the TICs produced by pyrolyzing n-octadecanoic acid by itself and on the quartz sand, hematite, magnetite, and goethite substrates at 0.5 wt % at 450°C and 650°C.

TICs of 0.5 wt % n-octadecanoic acid pyrolyzed on various substrates. Numbers with symbols refer to the aliphatic carbon chain length of the molecule. Bz, benzene; T, toluene; St, styrene; N, naphthalene; P, phenanthrene; BiP, biphenyl; DBF, dibenzofuran; 9-F, fluorenone; PN, phenylnaphthalene; Fl, fluoranthene.
3.1.2.1. n-Octadecanoic acid pyrolysis products
The main products of n-octadecanoic acid when pyrolyzed either by itself (Fig. 3, “octadecanoic acid”) or on a quartz sand substrate (Fig. 3, “quartz + octadecanoic acid”) were similar, so they will be discussed together. The products were as expected based on previous studies of the pyrolysis of carboxylic acids (Zafar and Watson, 2017; Zafar et al., 2017; Santos et al., 2019).
When n-octadecanoic acid was pyrolyzed at 450°C, the products were dominated by hydrocarbon species with carbon numbers of 16 or 17 (particularly hexadec-1-ene and heptadec-1-ene) and exhibited lower responses with decreasing chain length. Alkene/alkane pairs of aliphatic hydrocarbons were strongly dominated by the n-alk-1-ene (Fig. 3 and Supplementary Fig. S9) and peaked at C16–C17.
At the higher 650°C pyrolysis temperature, fragmentation of the aliphatic chain was more complete with shorter chain length n-alk-1-enes down to hex-1-ene (the lowest molecular weight detectable with the applied method) detected.
3.1.2.2. n-Octadecanoic acid on iron oxides
Figure 3 shows that pyrolysis of n-octadecanoic acid on the iron oxides/oxyhydroxides hematite, magnetite, and goethite produced a greater abundance of aromatic moieties (compared with no substrate or quartz). The dominance of longer chain lengths was also far less prevalent. With the addition of iron oxides, a wider variety of products were produced from the carboxylic acid. Of the nonaromatic products, alkanes were more dominant (when compared with the products of n-octadecanoic acid pyrolyzed by itself/on quartz), especially at the higher 650°C pyrolysis temperature. The apparent randomization of carbon chain lengths of the alkene/alkane pairs led to the loss of any clear distribution pattern in the products (Supplementary Fig. S9). A positively skewed distribution peaking at undecylbenzene (C17H28) is, however, preserved in the distribution of the n-alkylbenzene products (Supplementary Fig. S9) of both hematite and magnetite substrates. Ketones and aldehydes were produced as minor products from n-octadecanoic acid pyrolysis on both hematite and magnetite with C13–C19 aliphatic ketones (with tentative acetophenone) and C16–C17 aliphatic aldehydes on hematite and aromatic ketones fluoren-9-one and tentative identifications of benzophenone, acetophenone, and benzaldehyde on magnetite.
Goethite and magnetite appeared to promote the aromatization of n-octadecanoic acid relative to hematite, with a greater variety of aromatic molecules produced. A greater abundance of higher molecular weight aromatic species was also observed when n-octadecanoic acid was pyrolyzed on goethite as compared with magnetite, with PAHs up to four benzene rings tentatively detected.
3.2. XRD data
XRD data (Fig. 4) showed that phase transformations were occurring in the iron oxides during pyrolysis in the presence of 0.5 wt % octadecenoic acid. Hematite was partially reduced to magnetite [from iron (III) oxide to iron (II, III) oxide], although the transformation was incomplete—presumably due to the short (15 s) period of heating. Goethite was dehydrated to hematite and partially reduced further to magnetite, again, this transformation was incomplete. The goethite was seen to be an impure sample with quartz and possible clays present. Ferrihydrite is nanophase/amorphous prepyrolysis, which led to a lack of peaks in the XRD profile and shows the twin humps typical of two-line ferrihydrite (Janney et al., 2000; Kukkadapu et al., 2003) and on pyrolysis, in the presence of organic matter, this mineral transformed to more crystalline hematite with minor reduction to magnetite. Converse to the other reduction reactions, magnetite when heated in the presence of (z)-octadec-9-enoic acid was partially oxidized to hematite.
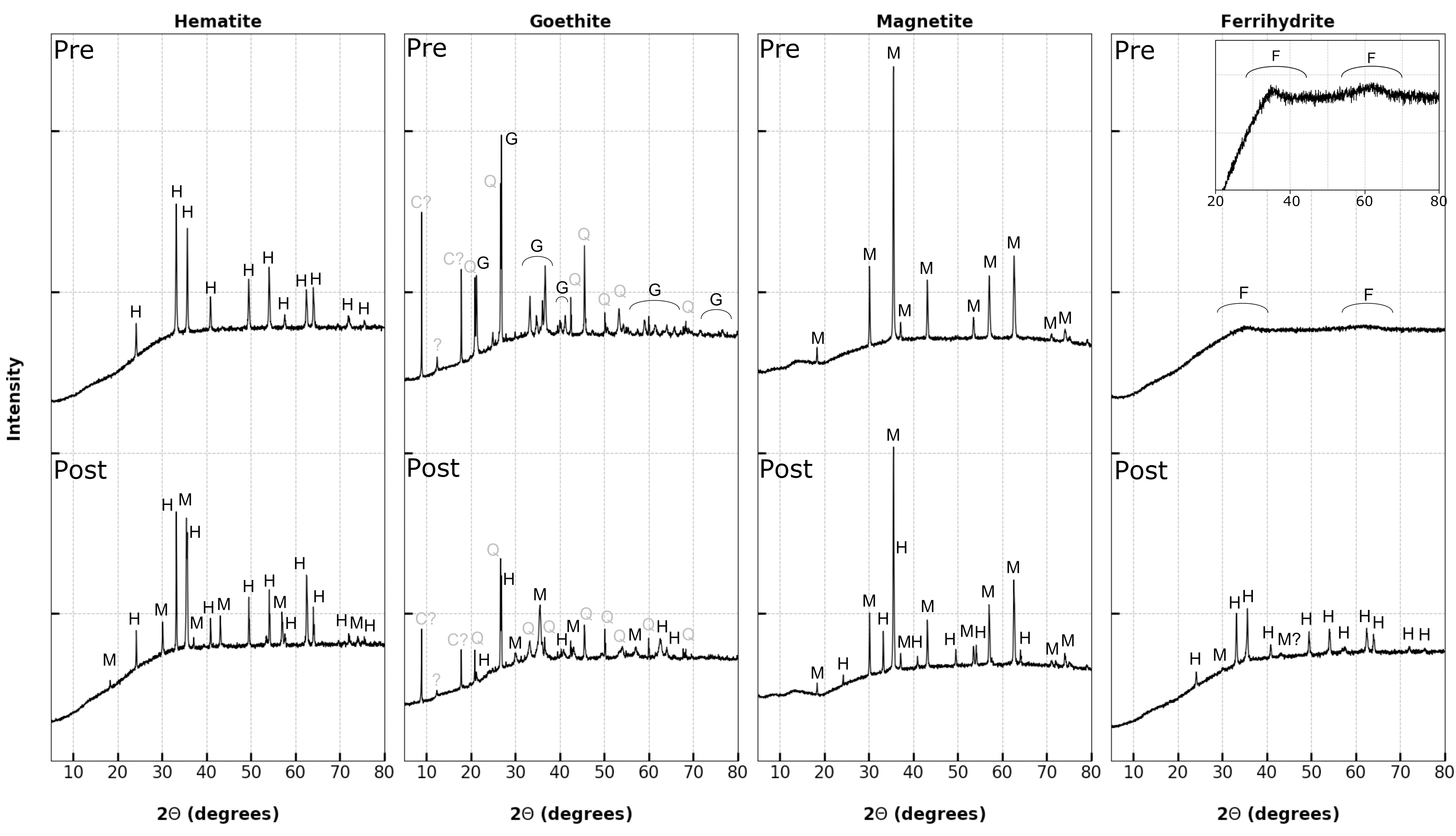
X-ray diffraction profiles of iron (oxy)hydroxides before pyrolysis (Pre) and after being pyrolyzed along with 0.5 wt % (z)-octadec-9-enoic acid (Post). Inset shows ferrihydrite with 3 × vertical axis exaggeration to emphasize twin humps typical of two-line ferrihydrite. Elevated background is due to Fe fluorescence under Cu X-ray radiation. C?, unidentified clay; F, ferrihydrite; G, goethite; H, hematite; M, magnetite; Q, quartz.
4. Discussion
4.1. Pyrolysis of carboxylic acids
When (z)-octadec-9-enoic acid or n-octadecanoic acid was (individually) pyrolyzed either without substrate or on quartz sand, the products were very similar, indicating that the quartz substrate had little effect on the pyrolysis products (Figs. 2 and 3). The pyrolysis products in all cases were as expected based on previous studies on the pyrolysis of carboxylic acids (Zafar and Watson, 2017; Zafar et al., 2017; Santos et al., 2019).
At the lower (450°C) pyrolysis temperature, the pyrolysis products of both carboxylic acids showed a dominance of moieties with 16–17 carbon atoms. This indicates that, at low temperatures, cleavage preferentially occurred through the loss of the carboxyl group. Decarbonylation was the dominant process of carboxyl group loss, as indicated by the dominance of the heptadeca-1,8-diene and hepta-1-decene peaks for (z)-octadec-9-enoic acid or n-octadecanoic acids, respectively. Chain scission was a secondary process at these lower temperatures for both the unsaturated and saturated carboxylic acids leading to shortened aliphatic chains.
At the higher (650°C) pyrolysis temperatures, for (z)-octadec-9-enoic acid pyrolyzed both without substrate and on quartz sand, the decreased abundance of longer chain length (C ≥ 16) fragments compared with shorter chain length (C ≤ 10) shows that this higher temperature enhanced homolytic C = C cleavage at the double bond or at the allylic positions (the sites adjacent to the unsaturated carbon atoms), due to the lower bond dissociation energies. The high abundances of cyclo-octene and n-decanoic acid produced at 650°C are the expected products of (z)-octadec-9-enoic acid undergoing cleavage at the allylic position. In contrast, n-octadecenoic acid, with no internal double bond to provide a weakness, produced a series of n-alk-1-enes and n-alkanes decreasing in abundance with decreasing carbon number from a peak at C17, indicating that chain scission primarily occurs at the end of this molecule. As these alkane–alkene pairs were strongly dominated by the n-alk-1-ene, it appears that decarbonylation was still the main process of carboxyl group loss, even at this higher temperature. Elevated peaks at hexadec-1-ene and tetradec-1-ene in the n-octadecanoic acid products may be due to secondary β-scission of the aliphatic chain on ionization within the mass spectrometer (Beyer and Walter, 1996). Aldehydes, such as heptadecenal, present in the products of both carboxylic acids, are intermediate products in the incomplete reduction of carboxylic acids (Fréty et al., 2014).
There is a tendency of the pyrolysis products of unsaturated carboxylic acids to cyclize, due to their doubly unsaturated carbon chains, after loss of the carbonyl group. This is evidenced by the abundance of cyclic and aromatic moieties in the products of (z)-octadec-9-enoic acid (Hartgers et al., 1995), particularly the obtained series of n-alkyl- and n-allylbenzenes (Fig. 2 and Supplementary Fig. S8). The n-alkylbenzene series observed in the pyrolysis products of n-octadecenoic acid (Fig. 3 and Supplementary Fig. S10) was less prominent than it was for (z)-octadec-9-enoic acid. This is presumably because the n-octadecanoic acid parent molecule lacked the internal double bond that aided aromatization in the case of the unsaturated acid (Hartgers et al., 1995).
The observed differences in relative abundances of products between (z)-octadec-9-enoic acid pyrolyzed by itself and on the quartz sand substrate are only minor but are of interest. There was an increased survival of intact (z)-octadec-9-enoic acid and increased relative abundance of higher molecular weight n-octadecanoic acid products on quartz. This may be due to the relatively poor thermal conductivity of the quartz shielding adsorbed carboxylic acids toward the center of the sample, so that they were subjected to lower temperatures and thermally desorbed intact.
Also of interest is the nonlinear decrease in detectable pyrolysis products as (z)-octadec-9-enoic acid concentration was reduced on the quartz substrate (Supplementary Fig. S2). Lower concentrations decreased the relative abundance of high molecular weight products, especially the heptadecanal and parent (z)-octadec-9-enoic acid, which were no longer detectable at the lower concentrations despite being among the most abundant species at 0.5 wt %. Our experiments demonstrated that even a nonreactive substrate will result in some surface effects; compounds that are released during heating can be actively reabsorbed onto the surface. This will preferentially affect the higher molecular weight compounds, especially those with functional groups that are readily attracted to surfaces like carboxylic acids. This effect on the distribution of products will be more pronounced at lower concentrations.
4.2. Pyrolysis of carboxylic acids in the presence of iron oxides
Figures 2 and 3 show that the presence of iron oxide/(oxy)hydroxide substrates had a clear effect on the pyrolysis of (z)-octadec-9-enoic acid.
Of the iron-bearing substrates tested, pyrolysis of carboxylic acids on hematite exhibited the greatest diversity in pyrolysis products. Pyrolysis of (z)-octadec-9-enoic acid at 450°C on hematite formed minor aliphatic hydrocarbon products comparable with those observed when pyrolyzing (z)-octadec-9-enoic acid on a quartz substrate at the higher temperature of 650°C, alongside many new alteration products (Fig. 2). In the aliphatic products (Supplementary Fig. S9, “hematite + octadecenoic acid”) at 450°C, there is still a dominance of dienes and alkenes over alkanes although there is a greater relative abundance of shorter chain products. This implies that decarbonylation and cleavage around the C = C double bond dominated pyrolytic breakdown from lower pyrolysis temperatures on a hematite substrate when compared with pyrolysis on a quartz substrate of the carboxylic acid by itself. At 650°C, the relative abundance of saturated verses unsaturated aliphatic hydrocarbons was increased. This implies that either decarboxylation took over as the dominant mechanism leading to the loss of the carboxyl group, or that secondary hydrogenation of the products occurred at this increased temperature. At 650°C, the abundance of aliphatics and n-alkylbenzenes over C12 was decreased. This reduction in carbon chain lengths indicates that bond cleavage occurred at the site of the unsaturated double bond more readily. This suggests that the presence of a hematite substrate has a catalytic effect, lowering the temperature necessary for cleavage of the double bond (compared with a quartz substrate or free carboxylic acid).
Similar effects were observed in the pyrolysis of n-octadecanoic acid on hematite (Fig. 3). Compared with the products of n-octadecanoic acid pyrolyzed by itself or on a quartz substrate, the carbon numbers of aliphatic fragments are reduced, and there was a greater relative abundance of saturated products. Again, this shows that the presence of the hematite substrate had a transformative effect on the pyrolysis products, lowering the temperature necessary for increased levels of internal C–C bond cleavage and hydrogenation of products.
Aldehydes were produced during the pyrolysis of carboxylic acids both without substrate and on quartz through partial reduction of the carboxyl group (COOH) to a carbonyl group (C = O). With the addition of hematite, this reduction/hydrogenation was enhanced to produce a series of alcohols (hydroxyl functional group, –OH); this process is known to be catalyzed by metal oxide catalysts (Manyar et al., 2010). The observed ketones produced may be the result of the addition of methyl radicals to the carbonyl groups of aldehydes, or oxygen addition at the sites of terminal alk-1-enes produced after C–C cleavage has occurred.
The transformative effects of magnetite, goethite, and ferrihydrite were further pronounced than those of hematite, with the products of carboxylic acids pyrolyzed on these substrates bearing little resemblance to those of the carboxylic acids pyrolyzed either on quartz or by itself (Figs. 2 and 3). There was a clear increased severity of the transformative effects through hematite, magnetite, goethite, and ferrihydrite with the detectability, molecular diversity, and the average molecular weight of products decreased throughout this sequence.
In the products of both goethite and magnetite, the hydrocarbon chain length of both aliphatic hydrocarbons and in the aliphatic side chains of n-alkylbenzenes appear to have been less influenced by aliphatic bond position, suggesting hydrocarbon bond scission was more randomized.
The presence of aryl–aryl bonded moieties (e.g., diphenyl, diphenylnapthalene) in the pyrolysis products of both carboxylic acids on all iron oxide/(oxy)hydroxide substrates is significant. This suggests that the Fe3+ surface ions acted as electron acceptors, forming aryl free radicals, which could then react with other molecules to form dimers (e.g., biphenyl) and other, higher molecular weight polymers through oxidative coupling reactions (Grzybowski et al., 2013; Watson and Sephton, 2015).
The only detectable species in the TIC of (z)-octadec-9-enoic acid on ferrihydrite were benzene, naphthalene, biphenyl, and decane (Fig. 2). These were only observable in the 0.5 wt % concentration, which was the highest tested; lower concentrations showed no identifiable peaks (Supplementary Fig. S6). It may be that these simple aromatic species are the “end member” products of a near-complete transformation series where organic compounds are transformed into low molecular weight species such as CO and CO2. Gas phase data were collected, in separate experimental runs, however large releases of H2O from this particularly hydrous mineral phase swamped the signal (data not shown), so that these low mass phases were unidentifiable.
For the substrates to have had such a strong effect, the transformative reactions discussed must have occurred where the carboxylic acid and iron oxides were in contact. Thus, for efficient reactions to occur, the carboxylic acids may have adsorbed onto the iron oxide mineral surfaces. Fourier-transform infrared experiments to confirm adsorption were inconclusive even at high (5%) carboxylic acid concentrations due to the nature of the iron oxide substrates, although previous studies have shown that organic matter readily adsorbs to iron oxide surfaces through carboxyl groups (Gu et al., 1994). Carboxylic acids may act as negatively charged ions as the polar carboxyl group is attracted to the positive charge of the iron oxide (Wainipee et al., 2010; Chernyshova et al., 2011). The structure of ferrihydrite means that numerous free Fe atoms are situated on the crystal surface (Cornell and Schwertmann, 2003), and these are readily available to react with the OH group of the carboxylic acid, increasing the adsorption per unit area and thus increasing the rate of reactivity. Furthermore, the nanophase crystal size will add to the increased transformative effect of this substrate as there is a greater (by orders of magnitude) reactive surface area (Hiemstra, 2015) over which adsorption and therefore subsequent oxidative reactions may occur.
Unlike with the quartz substrate, as (z)-octadec-9-enoic acid concentration was decreased on the iron oxide/(oxy)hydroxide substrates, the abundance and diversity of pyrolysis products decreased relatively linearly. Hematite (Supplementary Fig. S3) and goethite (Supplementary Fig. S5) were the only iron-bearing substrates where any of the “normal” pyrolysis products of (z)-octadec-9-enoic acid, most notably unsaturated aliphatic hydrocarbons, were even detected as minor products at 0.5 wt %. The relative abundances of these unsaturated aliphatic hydrocarbons were greatly reduced, relative to aromatic moieties, as concentration was decreased. This shows that transformation of pyrolysis products was more complete at lower concentrations of carboxylic acid; at ≤500 ppm only benzene is detected from goethite. This is to be expected as at lower concentrations of carboxylic acid there will be a greater availability of reactive Fe sites and therefore increased potential for monolayer over multilayer adsorption of the carboxylic acid (and subsequently its pyrolysis products) on the reactive iron oxide/(oxy)hydroxide surface, leading to higher levels of mineral surface promoted transformation (Zafar and Watson, 2017). With decreasing (z)-octadec-9-enoic acid concentration on the magnetite (Supplementary Fig. S4) and ferrihydrite (Supplementary Fig. S6) substrates, abundances of the transformation products decreased until they passed below detection limits, suggesting that the same processes were occurring at all concentrations.
The increased transformative power of ferrihydrite means that the pyrolysis products of carboxylic acids (and other potential aliphatic biomarkers) will be more thoroughly altered in a substrate rich in this mineral. For a positive detection to be made in a sediment dominated by ferrihydrite, carboxylic acids must be present in much higher concentrations than in a similar sediment dominated by other iron oxide phases.
4.3. Transformation of iron oxides and mechanisms for the transformation of carboxylic acid pyrolysis products
The XRD data (Fig. 4) show that when heated in an inert atmosphere and in the presence of organic matter, hematite was reduced to magnetite; a simple redox reaction with the oxidation state of the iron changing from Fe3+ to Fe2+, Fe3+ [iron (III) to iron (II, III) oxide]. Minor surface water adsorbed onto the hematite during storage was released during heating (observed in low mass pyrolysis-GC-MS data, not shown), and this, alongside water gas shift reactions catalyzed by the iron oxide surface, would have produced free radical H• and OH• ions to promote bond cleavage in the hydrocarbon chains (Bukur et al., 2016). These radicals will have increased rates of bond scission and hydrogenation, leading to the observed decreased abundance of detectable molecules, increased saturation, and shorter aliphatic hydrocarbon chain lengths observed in the pyrolysis products of both carboxylic acids. Similar processes of free radical generation during the reduction of iron oxides/(oxy)hydroxides are well recognized as astrobiologically relevant methods of organic matter oxidation in Fenton-like reactions (e.g., Foustoukos and Stern, 2012; Shuai et al., 2019).
The presence of both Fe (II) and Fe (III) in the magnetite means that there are both oxidizing and reducing valence states available to react with the organic matter in different ways. While Fe (III) will scavenge H+ ions in the same way as hematite, promoting cyclization and aromatization, Fe (II) will react with the hydrogen poor aromatics produced during pyrolysis of the organic matter, oxidizing to Fe (III) and forming hematite (as shown by the XRD data in Fig. 4). This multistep redox process may explain the reduction in products between hematite and magnetite substrates as seen in Figs. 2 and 3.
In contrast to the simple redox reactions observed in hematite and magnetite, goethite and ferrihydrite undergo phase transformations involving exothermic reactions when subjected to these conditions (Cornell and Schwertmann, 2003). Goethite recrystallizes to hematite >250°C (Gualtieri and Venturelli, 1999), with the XRD data (Fig. 4) showing further reduction to magnetite. The metastable ferrihydrite readily dehydrates to either goethite or hematite, dependent on pH (Cudennec and Lecerf, 2006), and under the high temperature subjected to in these pyrolysis experiments dehydration to hematite [with minor further reduction to magnetite due to the presence of organic matter (Carlson and Schwertmann, 1981)] was observed.
Dehydration, dehydroxylation, and the recrystallization of these phases to hematite and magnetite, especially for the hydrous ferrihydrite, would have produced many free radical H• and OH• ions as water and hydroxyl ions react with free Fe atoms on the crystal surface and dissociate (Stanjek and Weidler, 1992; Cornell and Schwertmann, 2003). The two-way water gas shift reaction, also catalyzed by iron oxide surfaces, is a further source of H• radicals (Bukur et al., 2016). The increased temperature at the microscale, due to the exothermic energy release during dehydration and decarboxylation (Schwertmann and Fischer, 1973), may have also further increased the reaction rate of hydrocarbon bond thermal dissociation. It is possible that the formation of iron carboxylate salt complexes (Hartgers et al., 1995) and/or cross-linked carboxylic acid polymers (Fjällström et al., 2002) occurred on iron (oxy)hydroxides, which may have led to some of the randomization of product chain lengths observed; however, direct evidence of this process was overprinted by further degradation. The clay impurities in the goethite sample may also be affecting the pyrolysis products as clays have been shown to have catalytic effects on hydrocarbon breakdown (e.g., Davis and Stanley, 1982); however, as the clays are minor components of the sample, this should be secondary to the effect of the goethite.
4.4. Identifying the original carboxylic acid from its pyrolysis products
Based on the results presented here, it will be difficult to establish the provenance of the breakdown products of carboxylic acids pyrolyzed/thermally extracted in the presence of iron oxides. Saturated carboxylic acids appear to be more resistant to these transformative effects, due to not having a weak double C = C bond that is relatively susceptible to thermal breakdown and subsequent production of an extraterminal double bond, promoting further cyclization, aromatization, and hydrogenation. A saturated carboxylic acid is most likely to be identified by the presence of a cluster of breakdown products (predominantly n-alk-1-enes, benzenes with aliphatic side chains and potentially intermediate cycloalkanes) with C n−1 that of the original carboxylic acid. The products of unsaturated carboxylic acids will be much more ambiguous due to breakdown at the internal double bond and associated allylic positions, alongside chain scission from the end of the molecule, producing less of an obvious dominance of one carbon number in the pyrolysis products.
If the concentration of the carboxylic acid on the substrate is high enough, however, it may be possible to distinguish between a saturated and unsaturated carboxylic acid based on the distribution profile of the n-alkylbenzenes (Fig. 5). For saturated carboxylic acids, the total carbon number of the n-alkylbenzene molecules should increase in both value and abundance up to C n−1 of the original saturated carboxylic acid molecule. Molecules larger than this modal peak will be minor or absent owing to addition reactions occurring precyclization. For unsaturated carboxylic acids, the n-alkylbenzenes will be distributed about a modal total C n corresponding to the position of the double C = C bond ±1 (for breakdown at the allylic positions) and decrease relatively symmetrically from this point. However, as seen in the experimental data (Supplementary Fig. S8) there may be a shift to higher values, modal peak is ∼11–12 rather than 7–10, if breakdown at the double bond is incomplete with chain scission competitively occurring from the end of the molecules.

Cartoon showing distribution of n-alkylbenzenes in the breakdown products of unsaturated and saturated carboxylic acids. C n−1 corresponds to the total number of carbon atoms in the original carboxylic acid minus one (due to loss of carboxyl group); C n -(n ± 1) corresponds to the double bond position in the original unsaturated carboxylic acid molecule (±1 for breakdown at the allylic position).
4.5. Relevance to Mars
The minimal alteration of the pyrolysis products between the carboxylic acids pyrolyzed without a substrate and those pyrolyzed on quartz sand demonstrates that, in comparison with iron-oxide/(oxy)hydroxide-rich units, sediments rich in quartz will be less destructive to biosignature detectability when using classical pyrolysis techniques on Mars. This is in line with other work that has shown the amenability of silica-rich sediments to characterization of associated organic matter (Reinhardt et al., 2020). Hydrated silica (opal) has been detected at the ExoMars designated landing site of Oxia Planum (Carter et al., 2016), and there are reports of >3.5 Ga old organic molecules being preserved in terrestrial sedimentary hydrated silica (chert) deposits (Duda et al., 2018), making these deposits an ideal target.
However, with iron oxides/(oxy)hydroxides being ubiquitous across the martian surface, and their potentiality to be codeposited with (and aid preservation of) organic matter, it would be neither possible nor advisable to avoid them completely. Hematite, magnetite, and goethite have been detected (Evans and Adams, 1980; Bell et al., 1993; Klingelhöfer et al., 2004, 2005; Morris et al., 2006a, 2006b; Bish et al., 2013; Blake et al., 2013; Treiman et al., 2016) and, while not directly identified due to its poor crystallinity, nanophase ferrihydrite is believed to be a major component of the iron-rich amorphous soil component (Dehouck et al., 2017). Mössbauer data collected by the MER Spirit and Opportunity found evidence for poorly crystalline/nanophase iron oxide phases in samples from various localities at both Gusev crater and Meridiani Planum (Morris et al., 2006a, 2006b). All samples collected by Curiosity rover and analyzed by the Chemistry and Minerology (CheMin) XRD instrument have been shown to contain a significant iron-rich X-ray amorphous component (Blake et al., 2013; Vaniman et al., 2014; Treiman et al., 2016), which has been suggested to be dominated by a mixture of both ferrihydrite and amorphous silica (Rampe et al., 2016; Dehouck et al., 2017).
Oxidizing and dry conditions at the surface of Mars mean that in the uppermost regolith, hematite will be the most stable form of iron; however, exposed martian surface conditions are not amenable to habitability or the survivability of organic matter (e.g., Moores and Schuerger, 2012). On Earth, goethite is by far the most common iron oxide in soils and sediments due to its high thermodynamic stability (Schwertmann and Taylor, 1989). This is likely also the case in the martian regolith beneath the surface out of reach of oxidizing effects of ultraviolet radiation (Pollack et al., 1970), and ferrihydrite is likely to increase in concentration beneath the surface for the same reason. Based on the results of this study, this is likely to have a negative effect on organic matter detection and characterization efforts in drilled subsurface samples by the future ExoMars Rosalind Franklin and Mars2020 Perseverance rovers.
Ferrihydrite is metastable under oxic conditions and readily recrystallizes to either hematite or goethite depending on temperature and pH; at high temperatures, regardless of pH, hematite is formed, at low temperatures with high (>10) or low (<4) pH goethite will be formed, while hematite will form at neutral pH levels (Cudennec and Lecerf, 2006). However, if oxidizing conditions continue for an extended period hematite will end up as the dominant product regardless of temperature and pH (Das et al., 2011). Goethite dehydroxylates ∼200–300°C (Gualtieri and Venturelli, 1999; Gialanella et al., 2010); in a reducing environment with a high pH magnetite will be formed (Usman et al., 2013), in an oxidizing environment with neutral or acidic pH hematite will be formed (Walter et al., 2001). Magnetite will oxidize to hematite (Lagoeiro, 2004), while hematite may reduce to magnetite depending on the redox potential of fluids. Thus, hematite will be the most stable of the iron oxides in settings, which have been subjected to longer term, high temperature, oxidizing, and neutral/acidic fluid flow.
There is evidence of hydrothermal systems on Mars from in situ observations, martian meteorites, and remote sensing of mineral deposits. Brine water activities may have persisted all the way from Mars' early history to the present day (Chen et al., 2015), potentially providing a wide range of these environments to explore of varying age, in a suite of different host rocks representing a range of redox and pH conditions.
While experimental work has shown that ferrihydrite can survive the short-term, low-temperature (<40°C), nonacidic aqueous alteration that is typical of the sediments of Aeolis Palus at Gale Crater, and ferrihydrite conversion to hematite may be retarded by the presence of amorphous silica even under high-temperature hydrothermal conditions (Dehouck et al., 2017). Even very brief, strongly acidic episodes of aqueous alteration will rapidly convert ferrihydrite into hematite or goethite; this is suggested to account for the lack of nanophase ferric oxides in the Burns formation at Meridiani Planum (Dehouck et al., 2017), where other phases such as jarosite are evidence for the presence of strongly acidic fluids (Klingelhöfer et al., 2004). As discussed above, the longer these conditions prevailed for, the more the intermediate goethite phase would have been replaced by hematite (Das et al., 2011). The presence of oxidizing and acidic brines is also evidenced from the mineral (and organic) assemblages in the Tissint meteorite (Chen et al., 2015; Steele et al., 2018; Jaramillo et al., 2019). These hydrothermal systems, coupled with the presence of reducing organic matter, may have provided a habitable environment with energy generation from brine–mineral interactions and the oxidation of iron (sulfate, chlorate, etc.) -bearing minerals (Fernández-Remolar et al., 2008; Steele et al., 2018). Concentrated regions of hematite-rich layers detected from orbit may be evidence of past or present large-scale active iron oxidation. On Earth, this is a process that is extensively mediated by chemolithotropic microorganisms (Weber et al., 2006; Fraeman et al., 2013).
While oxidizing environments are often associated with the poor preservation of organic matter (Sumner, 2004), this is not necessarily the case. Biomarker preservation in ferruginous sediments from recent and ancient deposits from acidic, oxidizing environments such as hot springs (Parenteau et al., 2014), acidic mine drainage (e.g., Rio Tinto) (Colín-García et al., 2011), and acidic streams (Tan et al., 2018) shows this not to be the case. Due to the preservation potential of iron oxides on organic carbon (Schelble et al., 2004; Oudemans et al., 2007; Lalonde et al., 2012), and the relative amenability of hematite (compared with goethite, magnetite, and ferrihydrite) to thermal decomposition analysis, these areas could be a prime target in the search for extinct martian life.
Thus, we recommend targeting areas where any initial ferrihydrite would have been converted to iron oxide phases that are more amenable to detection. Specifically, areas that show evidence of acidic hydrothermal fluid flow should be targeted. Localities with evidence of hydrothermal fluid flow should also have the added advantage of decreased concentrations of soluble perchlorates, further increasing the amenability of these sediments for thermal decomposition analysis of organic matter (Royle et al., 2018; Montgomery et al., 2019).
One concern with this strategy is that adsorbed organics on iron oxide surfaces may be significantly degraded by the recrystallization process. However, initial conclusions from experiments focusing on the degradation of soluble organic matter in ferrihydrite-rich environments suggest that ferrihydrite–magnetite recrystallization has little to no effect on the rate of degradation of organics (Tan, 2020). While these conclusions cannot simply be expanded to insoluble macromolecular organic or other iron mineral transformations, it does provide some insight on organic degradation during recrystallization processes. Nevertheless, the potential loss of ancient organic material due to the recrystallization of iron minerals must be discussed and considered before committing to a strategy that targets more stable iron minerals derived from ferrihydrite.
It must be considered that there are significant differences between the laboratory flash pyrolysis GC-MS setup used in this study and the SAM and MOMA flight modules. The much slower temperature ramps of the flown instruments, compared with the laboratory experiments described here (Table 1), will mean that any iron oxides/(oxy)hydroxides and putative organic matter in the analyzed sediments will be subjected to elevated temperatures for longer. Therefore, transformation reactions have more time to go to completion, raising the catalytic potential for processes associated with these reactions.
However, flash pyrolysis does represent a “worst case scenario,” forcing coincidence of iron oxide/(oxy)hydroxide transformation and pyrolytic breakdown of organic molecules, which will maximize the alteration/destruction of products (Royle et al., 2018). Under martian experimental conditions (ramped pyrolysis), thermal alteration of the iron oxide/(oxy)hydroxide phases may not coincide with the pyrolytic breakdown/thermal desorption of organic matter. The most labile/volatile phases are likely to be effected by surface effects and the release of water and free radicals from ferrihydrite [≥25°C (Cudennec and Lecerf, 2006; Das et al., 2011)] and goethite [≥200°C (Gualtieri and Venturelli, 1999; Gialanella et al., 2010)] dehydration and dehydroxylation, whereas macromolecular phases may be too refractory to thermally decompose until after these most reactive phases have recrystallized. Thus, the high, 500–820°C temperatures of release of organic molecules at Mojave and Confidence Hills (Eigenbrode et al., 2018) would mean that any goethite and ferrihydrite in these samples would have already dehydrated/dehydroxylated, and so only hematite or magnetite would still be available to catalyze reactions at these temperatures.
The catalytic potential of iron oxides/(oxy)hydroxides on the thermal decomposition of macromolecular or refractory organic matter, representative of meteoritic macromolecular matter, in contrast to the relatively labile monomers tested here, is therefore an interesting avenue to be explored by future studies.
Our data show that iron oxides promote the transformation of organic matter during pyrolysis. The transformative process can be so complete that biomarkers of extant life, such as medium- and long-chain carboxylic acids, may appear to have a nonbiological macromolecular source [e.g., meteoritic (Simmonds et al., 1969; Sephton et al., 2004)]. Detectability of any pyrolysis products of potential biomarkers in an iron-oxide-rich sediment will also be greatly reduced, increasing their concentration necessary for detection. This demonstrates that iron (oxy)hydroxide-rich areas present yet another complication for the search for potential biomarkers (and by extension life) on Mars. We have shown an example iron (oxy)hydroxide catalyzed breakdown and transformation of nonmacromolecular aliphatic molecules, during thermal decomposition experiments, could potentially be another way of forming pyrolysis products that appear to be from (abiotic) macromolecular material similar to those already detected on Mars (Eigenbrode et al., 2018). This means that in iron-rich sediments the structural information held in (potential biomarker) organic molecules will be lost due to the catalytic effects of iron (oxy)hydroxides.
The specific issues presented here are with classical pyrolysis-GC-MS as used by Viking Landers (Biemann et al., 1977), Phoenix's Thermal Evolved Gas Analyzer (Hoffman et al., 2008), and MSL-SAM (Mahaffy et al., 2012). However, both SAM and MOMA have wet chemistry capabilities with the ability to carry out derivatized pyrolysis-GC-MS. In these techniques, a derivatization agent is used to transform astrobiologically relevant organic molecules (including carboxylic acids) into species that are sufficiently volatile and amenable to GC column chromatography (Mahaffy et al., 2012). MOMA will have an advantage over SAM by having two derivatization techniques that occur at low temperatures N-methyl-N(tert-butyldimethylsilyl)trifluoroacetemide (MTBSTFA)/dimethylformamide (DMF) (≈250°C) and DMF-dimethylacetal (DMA) (≈140°C) (Goesmann et al., 2017). The iron oxide/oxy(hydroxide)-catalyzed reactions described here are likely to be slow at these low temperatures, especially with the DMF-DMA technique, which should be within the stability range of all but ferrihydrite dehydration.
MOMA will also have an advantage over previously employed thermal decomposition methods as its laser desorption–mass spectrometry capability will allow the targeting of heavier nonvolatile organic compounds (up to 1000 U), which may be present in the martian sediment. This technique is nondestructive, will preserve more structural information (Goesmann et al., 2017; Li et al., 2017), and is less likely to suffer from catalytic mineral surface reactions or the effects of oxidant species (e.g., perchlorates) as the nanosecond duration of the laser pulse mitigates heating of the substrate (Li et al., 2015; Goesmann et al., 2017).
5. Conclusions
Iron oxides act as both oxidative and reductive catalysts during the pyrolysis of carboxylic acids. Pyrolysis products are transformed, decreasing both the abundance and variety of molecular species detected. This effect must be taken into consideration when analyzing martian data in iron-rich sediments. Any carboxylic acids, or similar biomarker molecules, may be subjected to oxidative coupling and aromatization or hydrogenation and reduction as well as enhanced pyrolytic breakdown; this will be more pronounced with unsaturated than saturated carboxylic acids. The pyrolysis products of these carboxylic acids will appear very similar to mature hydrocarbons; thus, a recent biomarker molecule present at low concentrations could easily be mistaken for meteoritic macromolecular carbon if only classical pyrolysis GC-MS is used. The enhanced derivatization pyrolysis-GC-MS and laser desorption-MS capabilities of MOMA may provide a way of mitigating these effects. Therefore, iron oxides, along with the previously studied perchlorates and sulfates, are yet another complication for attempts to detect indigenous martian organic matter and the search for extraterrestrial life itself.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Science and Technology Funding Council/UK Space Agency grant ST/N000560/1.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.