
Abstract
The genomic diversity of bacteria and archaea in brines (BC1, BC2, and BC3) from two adjacent and perennially frozen Antarctic lakes (L16 and L-2) in the Boulder Clay (BC) area was investigated together with the metabolically active fraction of both communities, by analyzing the bulk rRNA as a general marker of metabolic activity. Although similar bacterial and archaeal assemblages were observed at phylum level, differences were encountered when considering the distribution in species. Overall, the total bacterial communities were dominated by Bacteroidetes. A massive occurrence of flavobacterial sequences was observed within the metabolically active bacterial communities of the BC1 brine, whereas the active fractions in BC2 and BC3 strongly differed from the bulk communities being dominated by Betaproteobacteria (mainly Hydrogenophaga members). The BC lakes also hosted sequences of the most thermally tolerant archaea, also related to well-known hyperthermophiles. Interestingly, RNA sequences of the hyperthermophilic genus Ferroglobus were retrieved in all brine samples. Finally, a high abundance of the strictly anaerobic methanogens (such as Methanosarcina members) within the active community suggests that anoxic conditions might occur in the lake brines. Our findings indicate perennially ice-covered Antarctic lakes as plausible terrestrial candidates for the study of the potential for extant life on different bodies of our solar system.
1. Introduction
Continental Antarctica hosts a number of cryosystems (e.g., ice-covered and subglacial lakes, permafrost, and glaciers) that conceal liquid hypersaline brine lenses (Murray et al., 2012; Dickson et al., 2013; Toner and Sletten, 2013; Dugan et al., 2015; Mikucki et al., 2015; Forte et al., 2016; Badgeley et al., 2017). Their unfrozen conditions, even several degrees below 0°C, are maintained by their high salt content. The genesis and mobilization of brines within Antarctic cryosystems remain poorly understood (Lyons et al., 2019). However, geophysical observations have demonstrated that brines are dynamic systems, with subglacial streams that can connect lakes and ponds with their movements (Wright and Siegert, 2012; Mikucki et al., 2015; Siegert et al., 2016).
Besides their geological relevance, brines are particularly interesting for better delineating and elucidating the functioning of briny ecosystems by the description of inhabiting life-forms, mainly microscopic (Papale et al., 2019). The latter aspect is also particularly attractive from a planetary geobiological perspective and becomes fundamental in the assessment of the habitability of other worlds within our Solar system (Preston and Dartnell, 2014).
The analogies between terrestrial sites and their extraterrestrial targets can rely on mineralogical, geochemical, and geomorphological features, as well as on physicochemical environmental parameters and conditions. Among them, the principal habitability criteria defined by NASA are addressed to extended regions of liquid water, intrinsically supporting life as on Earth (Hendrix et al., 2019). In this regard, liquid brines have been recently discovered below the ice of the south polar layered deposits on Mars (Orosei et al., 2018), in the subsurface of Pluto (Nimmo et al., 2016), on Jupiter's moon Europa, and Saturn's moons Enceladus and Titan (Postberg et al., 2011; Mitri et al., 2014). In particular, Enceladus drew astrobiological interest with the sighting of jets of fine icy particles and water vapor, which were observed first emerging from its south polar terrain and then feeding in a large plume. The plume contains simple organic compounds and notable levels of volatile species, such as N2, CO2, and CH4 (Porco et al., 2006; Neveu et al., 2020). Interestingly, Enceladus's plume might originate from a subsurface liquid water aquifer composed of NaCl, NaHCO3, Na2CO3, and K+ (Postberg et al., 2009 and reference therein) and possibly support the origin and evolution of life (McKay et al., 2008). Similarly, the putative brine ocean detected beneath the ice of Europa probably consists of hydrated minerals (Pappalardo, 2010).
The exploration of terrestrial analogues represents a unique opportunity to gain a “critical ground-truthing for astrobiological studies” also in terms of the detectability of biosignatures and potential for the long-term preservation of signs of past life (Preston and Dartnell, 2014). Terrestrial brines are “second-order analogues” as their classification is based on indirect or highly suggestive evidence that has the potential to be proven false. In turn, extremophiles are “analogues of third order” as no direct or indirect evidence that life exists anywhere other than on Earth is available (Preston and Dartnell, 2014). As stated in the study of Lyons et al. (2019), “Whether these sub-cryospheric oceans can sustain life today is a major question in astrobiology and planetary exploration.”
As potential astrobiological targets, the exploration of brines and their psychrophilic microbial inhabitants (in terms of both diversity and activities) on Earth become incredibly important for our comprehension of the boundaries of life on Earth and the development of scenarios for planetary habitability (Mikucki and Priscu, 2007; Burr et al., 2009; Mikucki et al., 2009; Murray et al., 2012; Kuhn et al., 2014; Preston and Dartnell, 2014; Ojha et al., 2015; Tregoning et al., 2015; Papale et al., 2019).
Previous surveys of the Boulder Clay (BC) (Northern Victoria Land, Antarctica) area revealed the presence of perennially ice-covered lakes with icing blisters and frost mounds (Guglielmin et al., 1997; French and Guglielmin, 1999, 2000). Moreover, a possible hydrostatic origin of the frost mound that occurs in the perennial frozen Lake 16 was suggested (Guglielmin et al., 2009). Some authors have speculated on the possible existence of Northern Victoria Land environments with similar chemical and physical features on the martian surface (Burr et al., 2009; Porcino et al., 2020), whereas subsurface origin of brines resembles physical conditions on the icy moons Enceladus and Europa, both characterized by a high habitability potential (Garcia-Lopez and Cid, 2017). Thus, brines from the BC lakes could represent a second-order terrestrial analogue environment and afford the opportunity to investigate planetary habitability by the exploration of diversity and activity of extremophilic micro-organisms.
To date, no attempts have been made to characterize the extant prokaryotic communities associated with these lake systems. Environmental DNA, which is preserved far longer in comparison with RNA, may originate from extracellular DNA or inactive microbial cells. Its exclusive use for describing the community structure might misinterpret the real prokaryotic community structure. Based on these considerations, we report on the overall genomic diversity of prokaryotes (bacteria and archaea) in brines from two adjacent lakes in the BC area compared with the metabolically active fraction of these communities by analyzing the diversity of bulk rRNA as a general marker of metabolic activity.
2. Materials and Methods
2.1. Site description
The BC site (coordinates: 74°44′ S–164°01′ E) is located in the Northern Victoria Land, close to the Italian Antarctic Research Station “Mario Zucchelli” (∼6 km south from the station in a slope with southeastern exposure). It is a debris-covered glacier with several perennially ice-covered ponds characterized by the presence of icing blisters (Guglielmin et al., 1997, 2009; French and Guglielmin, 2000; Guglielmin and French, 2004). Surface features also include frost-fissure polygons and debris islands (French and Guglielmin, 1999). The site is exposed to katabatic winds from inland areas that produce numerous snow drifts. The soils here are mainly glacic haplorthels where no evidence of cryoturbation has been observed, and scattered mosses and epilithic lichens constitute the vegetation types (Cannone et al., 2008). The mean annual temperature is around −14.0°C. The mean annual ground temperature at both the surface and permafrost level is −16.5°C, whereas in the deepest monitor layer (3.6 m), the mean temperature is −17°C (Guglielmin and Cannone, 2012; Guglielmin et al., 2014). The site was selected for the construction of a semipermanent gravel runway for airfreight operations.
2.2. Sample collection
During a ground penetrating radar survey in November 2014, fluid brines were detected within two adjacent lakes in the BC area (Fig. 1). Brine samples (BC1, BC2, and BC3) were collected near the ice blisters of Lake 16 (L16; i.e., BC1 and BC2) and Lake L-2 (i.e., BC3) in November–December 2014. In particular, brines BC1 and BC2 were collected from two different sampling points and depths (2.5 and 0.9 m, respectively) of Lake L16, whereas brine BC3 was derived from 2.3 m depth of Lake L-2. All samples were collected in presterilized polycarbonate bottles by using sterilized peristaltic pump and tubing. BC1 was more saline (6333 ± 15 mg/L) than both BC2 and BC3 (37.6 ± 2.1 and 1583 ± 6.81 mg/L, respectively). pH values were 6.52, 7.85, and 7.02 for BC1, BC2, and BC3, respectively. At the depths of sampling, the surrounding ground temperatures ranged between −31.4°C and −0.9°C, with a mean value around −16.5°C (Azzaro et al., 2021).
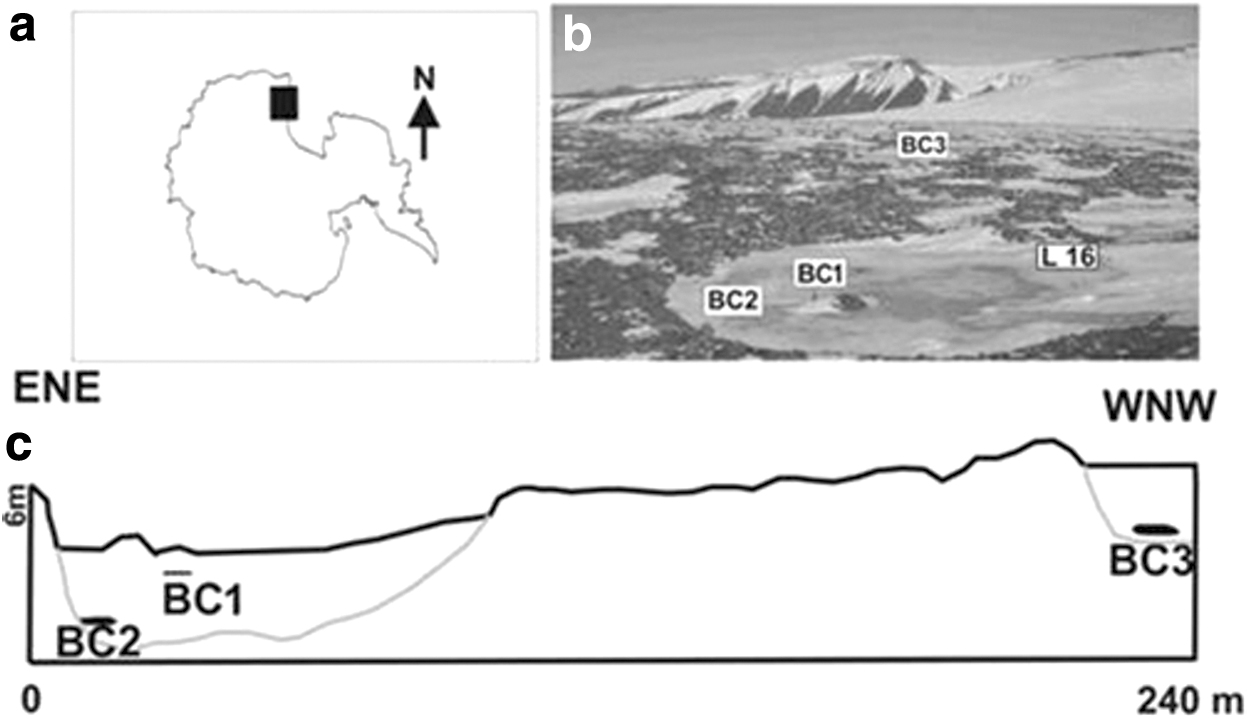
Study area.
2.3. Prokaryotic community structure and composition
2.3.1. DNA and RNA extraction and reverse transcription-polymerase chain reaction
Brine subsamples (i.e., 150, 450, and 600 mL for BC1, BC2, and BC3, respectively) were filtered onto 0.22 μm pore size polycarbonate filters (diameter 47 mm; Millipore). Membranes were directly frozen at −20°C until analyses. To avoid contamination, all instruments were sterilized by UVC radiation under laminar flow hood and washed with RNase away.
For the analysis of the total prokaryotic community, total DNA was extracted in duplicate from membranes by employing the PowerSoil DNA extraction kit (MoBio Laboratories, Carlsbad, CA, USA) according to the manufacturer's instructions. DNA concentrations and purity were quantified by using a NanoDrop ND-1000 UV-vis spectrophotometer (NanoDrop Technologies, USA).
For the analysis of the metabolically active prokaryotic community (MA-PC), total RNA was extracted in duplicate from membranes. A lysozyme solution (5 mg/mL) in TE buffer 1 × at pH 8 (10 mM Tris-HCl; 1 mM EDTA) was prepared and used for the release of RNA from inside the cells. The samples were incubated with this solution for 15 min at 37°C. Then, RNA was extracted using RNeasy Mini Kit (Qiagen) following manufacturer's procedure. The synthesis of cDNA was carried out from total RNA (4 μL) with a reverse transcriptase (RT)-polymerase chain reaction (PCR) using SuperScript III First-Stand Synthesis System (Invitrogen). Two reaction mixtures were prepared as follows: MIX 1 (RNA 4 μL; Random hexamers 50 ng/μL; dNTP mix 10 mM, RNasy free water until 10 μL) and MIX 2 (RT-buffer 10 × ; MgCl2 25 mM; DTT 0.1 M; RnaseOUT 40 U/μL; SuperScript III RT 200 U/μL). MIX 1 was subjected to the RT step, which involved incubation at 25°C for 10 min. MIX 2 and MIX 1 were then mixed (20 μL total volume), and incubation for 50 min at 50°C and 5 min at 85°C was performed. Until this step, the samples were incubated for a few seconds in ice, and 1 μL of RNase H was added, followed by 20 min at 37°C. After this, cDNA was isolated, and it was possible to pass to the PCR step.
The reaction mixtures were assembled at 0°C and contained 1–10 ng DNA, 5 × Phusion buffer, 150 ng of each forward and reverse primers (MWG, Germany), 0.1% of Bovine Serum Albumin, 10 mM of deoxyribonucleotide triphosphate (Polymed, Italy), 0.2 U of Taq polymerase (Phusion), and sterile distilled water to a final volume of 40 μL. Negative controls for cDNA extraction and PCR setup (reaction mixture without a cDNA template) were also used in every PCR run. cDNA concentrations and purity were confirmed with a NanoDrop ND-1000 UV-vis spectrophotometer (NanoDrop Technologies, USA).
2.3.2. 16S rRNA gene and cDNA amplification
The V1–V2 region of 16S rRNA gene of bacteria (primers 27f 5′-AGAGTTTGATCCTGGCTCAG-3′ and 338 5′-GCT GCC TCC CGT AGG AGT-3′) and the most conservative V3–V4 region of Archaea (primers ARC344f 5′-ACGGGGYGCAGCAGGCGCGA-3′ and 693r 5′-GGATTACARGATTTC-3′) were amplified. RNA and cDNA were pooled, and PCR amplification was carried out under the conditions described in the study of Papale et al. (2018). In brief, the PCR program was as follows: 95°C for 3 min, followed by 30 cycles of 94°C for 1 min, 50°C for 1 min, 72°C for 2 min, and a final extension step at 72°C for 10 min. The results of the amplification reactions were analyzed by agarose gel electrophoresis (1%, w/v) in TAE buffer (0.04 M tris-acetate, 0.02 M acetic acid, 0.001 M EDTA), containing 1 μg/mL of ethidium bromide. 16S rRNA gene and cDNA were quantified with a NanoDrop ND-1000 UV-vis spectrophotometer (NanoDrop Technologies, USA).
2.3.3. Tag pyrosequencing and postrun analysis
To reduce bias in massive sequencing, a two-step PCR protocol was applied: the first step consisted of a conventional PCR, and then amplicons were used as a template for the second PCR with barcoded primers for Ion Torrent sequencing. PCR products were purified by using the AgencourtAMPure XP (Beckman Coulter, Inc.) kit, according to the manufacturer's instructions, and then quantified using the Qubit dsDNA HS Assay Kit with Qubit Fluorometer 2.0 (Invitrogen; Thermo Fisher Scientific). Each purified product (20 ng) was pooled for emulsion PCR with Ion PGM Template OT2 400 Kit. Sequencing was performed on an Ion Torrent Personal Genome Machine™ (Life Technologies, USA) with Hi-Q sequencing chemistry used to reduce the problem of poor homopolymer sequencing (Churchill et al., 2016; Pereira et al., 2016) and Ion 314™ chip (all Ion Torrent reagents by Thermo Fischer Scientific) following manufacturer's protocols.
To assess the sequencing error type and rate in concert with the read quality score, the FastQC tool was adopted to analyze the reads. Subsequent to the quality control, raw data were analyzed using the bioinformatics analysis software MOTHUR (version 1.39.5). Barcodes and primers were identified with maximum one base error and trimmed off. Reads were cleaned by length (reads shorter than 200 bp were discarded) and by quality score using score quality windows (i.e., average 20 and size 10). Remaining sequences were aligned with the Silva reference files (release 128 full-length sequences and taxonomy references). Reads were denoized by using the
2.4. Statistical analyses
Significant differences in chemical parameters and taxonomical abundances between brine samples were analyzed by one-way analysis of variance (ANOVA) test and posthoc analysis (Tukey test) and carried out considering significant p < 0.05. Pearson's correlation analysis was also performed (MiniTab software, version 16.0). Principal component analysis (PCA) was computed on normalized data after clustering and Euclidean distance ordination (Primer-6; Plymouth Marine Laboratory, Roborough, United Kingdom).
Obtained OTUs for bacteria and archaea by 16S rRNA gene and cDNA amplicon sequencing were used to generate Venn diagrams with the R software version 3.0.1, VennDiagram package (Chen and Boutros, 2011). Heatmaps were constructed to display the presence and abundance of bacterial and archaeal genera in the brine samples, based on Bray–Curtis dissimilarities. The analysis was performed by using Heatplus and Gplots packages in R environment (R 3.4.4). The genera whose relative abundance was <1% were eliminated.
Diversity index calculation was performed after subsampling based on the lowest number of reads. A number of Alpha diversity measures were evaluated by Mothur software including the terminal richness estimation (Chao1). Mean values and differences between diversity indices were calculated and assessed for significance using one-way ANOVA (Excel software).
3. Results
3.1. Total prokaryotic community
Overall data from the 16S rRNA gene amplicon sequencing are reported in Table 1. After the trimming step, the number of high-quality bacterial and archaeal reads was in the ranges 11,396–17,780 and 10,018–14,161, and were distributed among 1057 and 11,217 OTUs, respectively. A number of bacterial (47) and archaeal (109) OTUs were shared among all brine samples (Fig. 2a, b).
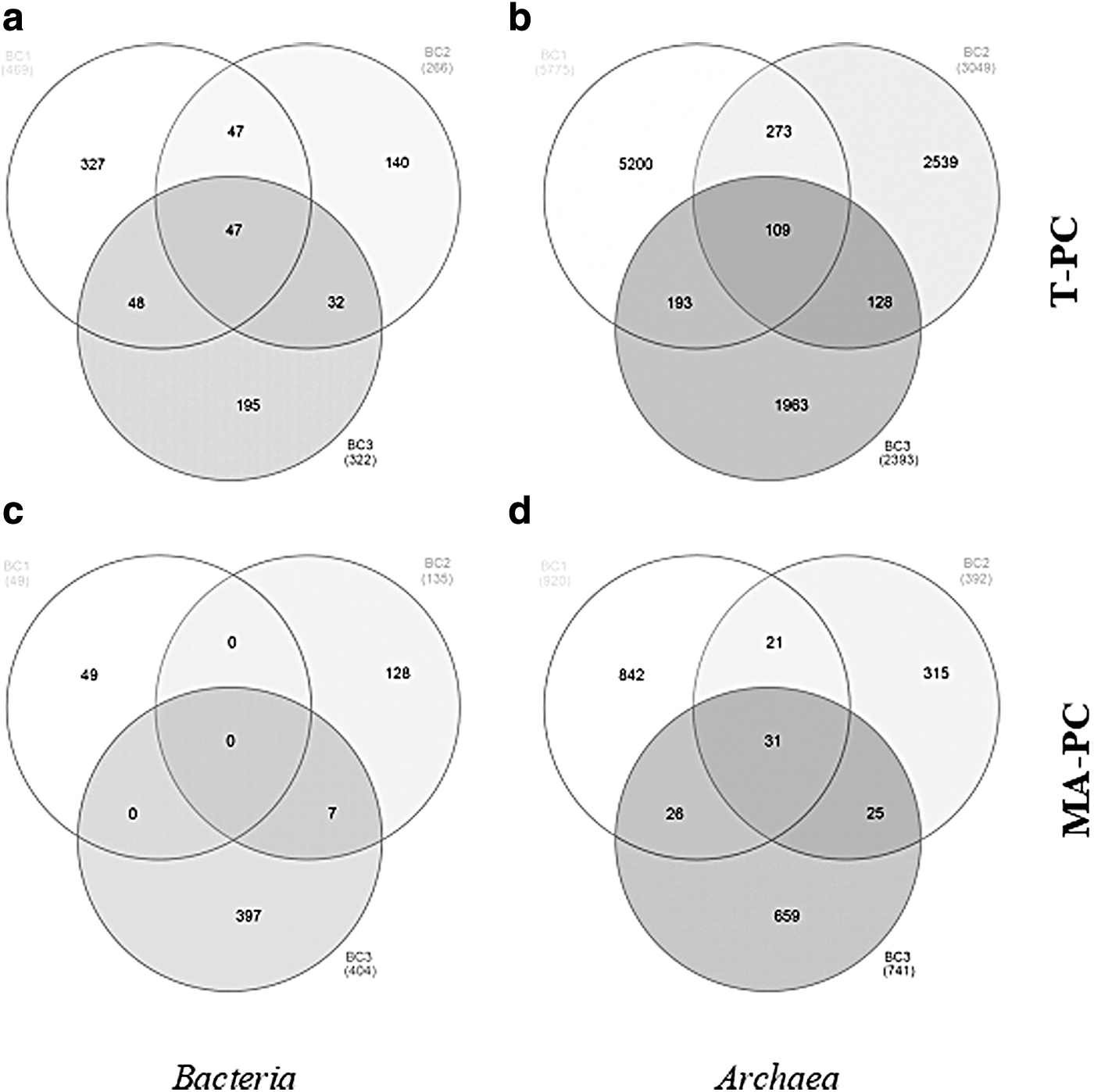
Venn diagram of OTUs retrieved in the total bacterial
Total and Metabolically Active Prokaryotic Community in Brines from Perennially Ice-Covered Antarctic Lakes
For the analysis of the T-PC, 16S rRNA gene sequences were generated from total DNA of bacteria and archaea. For the analysis of the MA-PC, cDNA sequences were generated from total RNA of bacteria and archaea.
MA-PC, metabolically active prokaryotic community; OTU, Operational Taxonomic Unit; T-PC, total prokaryotic community.
3.2. Total bacterial community
3.2.1. Lake 16 (brines BC1 and BC2)
Within the total bacterial community (T-B), Bacteroidetes predominated in both brines (79.7% and 52.7% of total sequences in BC2 and BC1, respectively) (Fig. 3a). In BC1, Bacteroidetes were followed in abundance by Proteobacteria (26.7%), Actinobacteria (2.7%), and Parcubacteria (1.6%). Proteobacteria (4.8%) and Actinobacteria (5.8%) were equally distributed in BC2, whereas Parcubacteria were not detected. Proteobacteria were mainly distributed among Alpha-, Beta-, and Gammaproteobacteria classes. Among them, Betaproteobacteria were particularly abundant in BC1 (14.3%).

Taxonomic phyla distribution observed in brine samples.
The relative abundances of other phylogenetic groups (i.e., Acidobacteria, Verrucomicrobia, Absconditabacteria, Gemmatimonadetes, Saccaribacteria, Epsilonproteobacteria, Planctomycetes, Peregrinibacteria, Microgenomates, Firmicutes, Cyanobacteria, and Chloroflexi; grouped in “Other groups” in Fig. 3a) were <1% each (ranging from 0.01% to 0.78%). Among them, the T-B in BC2 included only Gemmatimonadetes and Saccaribacteria. Conversely, Saccaribacteria sequences were the sole not detected in BC1. A number of sequences remained unclassified (14.7% and 9.6% in BC1 and BC2, respectively).
Between 7% and 17% of the total high-quality DNA bacterial sequences were classified at genus level (Table 1). Sequences were resolved in 32 (range 0.01–7.6%) and 6 genera (range 0.01–6.9%) for BC1 and BC2, respectively. Only genera occurring at ≥0.1% of the total bacterial sequences are reported in Fig. 4a and b and Supplementary Table S1. In line with the marked dominance of Bacteroidetes, most of the classified sequences at genus level were affiliated with this subphylum. In both brines, the dominant Bacteroidetes genus was Flavobacterium (∼7% of total sequences). In BC1, such genus was followed in abundance by Psychroserpens. Among Gammaproteobacteria, Salmonella accounted for 2.7% in BC1 and 0.2% in BC2, whereas Arcobacter (0.7%) was retrieved only in BC1. The genera Illumatobacter (among Actinobacteria; 0.1%) and Chloroploca (among Chloroflexi; 0.4%) also occurred in BC1.

Taxonomic genera distribution of the total and metabolically active bacterial sequences from
3.2.2. Lake L-2 (brine BC3)
As it was observed for the brines from Lake L16, Bacteroidetes predominated in the T-B of the BC3 brine (69.0% of total sequences), followed by Proteobacteria (18.4%) and Actinobacteria (4.0%). At subphylum level, Proteobacteria were mainly represented by Betaproteobacteria (17.7%) (Fig. 3a). The relative abundances of other phylogenetic groups (i.e., Gemmatimonadetes and Saccaribacteria; grouped in “Other groups” in Fig. 3a) were ≤1% each. The 8.2% of sequences remained unclassified.
About 45% of the total high-quality DNA bacterial sequences from BC3 were classified at genus level (Table 1) and were resolved in eight genera. Among them, only Flavobacterium occurred at ≥0.1% (44.7%) (Fig. 4c and Supplementary Table S1).
The PCA performed on the abundance of genera and chemical data (Azzaro et al., submitted) is shown in Fig. 5. Despite the fact that a significant statistical difference did not occur, as supported by the one-way ANOVA results, a clear clustering of BC1 brine is appreciable as shown in Fig. 5a. Regardless of the absence of significant differences, the SIMPER analysis was performed, highlighting a higher squared distance occurring between BC1 and BC2, mainly due to biological results, namely, Arcobacter and Salmonella abundances. As shown in Supplementary Fig. S1a, with regard to environmental parameters, BC1 and BC3 clustered together, whereas BC2 resulted as distantly ordered from them. Indeed, BC2 was characterized by a lower concentration of chemicals, NaCl, and a very low conductibility value. Among environmental variables, NaCl content and its potential influence on the prokaryotic communities in brines were deepened. Within the total bacterial communities, all taxonomic groups were positively correlated with salinity at genus level, with the only exception of Achromobacter members, which showed a slight negative correlation with NaCl concentration (Pearson coefficient r = −0.6) and were indeed present only in BC2 samples (even if at abundance <0.1%). Flavobacterium members were negatively correlated with salinity, even if not at a significant extent (Fig. S1b).

Principal Component Analysis (PCA) performed on abundance values and chemical parameters retrieved in the brine samples:
3.3. Total archaeal community
3.3.1. Lake 16 (brines BC1 and BC2)
As shown in Fig. 3b, the total archaeal community (T-A) within the BC1 and BC2 brines was dominated by Euryarchaeota, followed by Crenarchaeota (81.4% vs. 85.7% and 15.3% vs. 12.4%, respectively). A low percentage of reads were affiliated with Korarchaeota (0.3% vs. 0.8%) and the Ancient Archaeal Group (2.0% vs. 1.1%). The Marine_Hydrothermal_Vent_Group_1 occurred at 1.0% in BC1 only.
Among classified reads, ∼80% of sequences were assigned to an order, with Methanobacteriales, Methanopyrales, and Thermoplasmatales that were particularly abundant (≥10%) in both brines (Table 2). Furthermore, Methanomicrobiales (13.9% in BC1) and Methanosarcinales (12.5% in BC2) were also well represented.
Distribution of Archaeal Sequences in Known Orders
Higher percentages are in bold.
The number of sequences assigned to an order is in brackets. Percentages are referred to the number of good-quality sequences for each sample.
MA-A, metabolically active archaeal community; T-A, total archaeal community.
About 65% of the total high-quality DNA archaeal sequences were classified at genus level (Table 1). Sequences from BC1 and BC2 were resolved in 65 (range 0.01–20.4% of total sequences) and 54 (range 0.01–25.8%) genera, respectively. Only genera occurring at ≥0.1% of the total bacterial sequences are reported in Fig. 6a and b and Supplementary Table S1. In particular, among Euryarchaeota, the genera Methanopyrus and Methanothermus were particularly abundant in both brines BC1 and BC2 (between 13.1% and 25.8%). The genera Methanoplanus (among Euryarchaeota; 12.3%) and Sulfurisphaera (among Crenarchaeota; 4.9%) were also abundant in BC1, whereas Methanosalsum (among Euryarchaeota; 10.2%) and Aeropyrum (among Crenarchaeota; 4.3%) were well represented in BC2. Among less represented genera (range 1.0–4.0%), Aeropyrum and Ignicoccus (Crenarchaeota), Ferroglobus, Methanosphaera, and Methanosalsum (Euryarchaeota) occurred in BC1, whereas Desulfurococcus and Sulfolobus (Crenarchaeota), Ferroglobus, and Haloarcula (Euryarchaeota) were in BC2.

Taxonomic genera distribution of the total and metabolically active archaeal sequences from
3.3.2. Lake L-2 (brine BC3)
As shown in Fig. 3b, the T-A within the BC3 brine was mainly represented by Euryarchaeota (82.4% of total sequences), followed by Crenarchaeota (14.0%) and Ancient_Archael_Group (2.2%). A low percentage of reads were affiliated with Marine_Hydrothermal_Vent_Group_1 and Korarchaeota (0.3% and 1.1%, respectively).
Among classified reads, 61.6% of sequences were assigned to an order, but only Methanosarcinales occurred at ≥10% (Table 2).
About 51% of the total high-quality DNA archaeal sequences from BC3 were classified at genus level (Table 1) and were resolved in 61 genera, ranging from 0.01% to 19.5%. Only genera occurring at ≥0.1%, resulting in 26 genera, are reported in Fig. 6c and Supplementary Table S1. In particular, the genus Methanothermus among Euryarchaeota was the most abundant (19% of total sequences). The genera Methanosalsum and Methanopyrus were also well represented (7.5 and 4.2%, respectively). Aeropyrum, Sulfolobus, and Desulfurococcus (all Crenarchaeota), and Ferroglobus and Methanoplanus (both Euryarchaeota) occurred at a percentage ranging from 1.0% to 4.0%.
No significant differences were detected for the abundances of archaeal genera among samples. The PCA computed, considering the abundances of genera (occurring at ≥0.1% of the total sequences), confirmed this finding, with brines that clustered separately (Fig. 5c). Also, in this case, biological results had a greater weight, as was shown by the SIMPER analysis, according to which BC1 and BC2 samples showed the highest average squared distance, mainly due to Caldivirga, Methanocaldococcus, Methanospirillum, and Haloarcula abundances. The Pearson analysis applied to total archaea abundances and NaCl content values showed that there was a significant positive correlation for some taxonomic groups detected exclusively or at higher extent in BC1 (i.e., Caldivirga, Methanocaldococcus, Methanoplanus, and Methanospirillum; Pearson coefficient >0.9). Conversely, a strong inverse correlation was evidenced for taxonomic groups detected in all brines, with a highest extent in the BC2 (namely, Methanothermus, Methanosalsum, and Aeropyrum; Pearson coefficient >0.9).
3.4. Metabolically active prokaryotic community
Overall data from the cDNA amplicon sequencing are reported in Table 1. After the trimming step, the number of high-quality bacterial and archaeal reads was in the ranges 49–585 and 2531–2976, and were resolved in 588 and 2043 OTUs, respectively. A total of 31 archaeal OTUs were common to all samples, whereas any bacterial OTU was common to all samples (Fig. 2c, d).
The 0.4%, 0.9%, and 3.9% of the T-B resulted active in BC1, BC2, and BC3, respectively. The active archaeal community accounted for 21.0%, 19.2%, and 28.8% of the T-A, respectively.
3.5. Metabolically active bacterial community
3.5.1. Lake 16 (brines BC1 and BC2)
The metabolically active fraction of the bacterial community (MA-B) in brines from Lake 16 strongly differed as it was dominated by Bacteroidetes (73.5% of total sequences) in BC1 and Proteobacteria (83.8%; only represented by Betaproteobacteria) in BC2 (Fig. 3a). In BC1, active Proteobacteria accounted for 22.4% of total sequences and were represented by Alpha-, Beta-, and Gammaproteobacteria (16.3%, 4.1%, and 2.0%, respectively). No members of Actinobacteria and Parcubacteria (which were instead detected within the T-B) were within the MA-B of BC1. Conversely, in BC2 active Bacteroidetes and Actinobacteria accounted for 7.8% and 6.6% of total sequences, respectively. Between 1.8% and 4.1% of sequences remained unclassified. None of the minor groups detected among the T-B was represented within the MA-B.
A very low percentage of the total high-quality cDNA bacterial sequences was classified at genus level (∼7–8%) (Table 1), and it was resolved in few genera. Among them, only genera occurring at ≥0.1% of the total bacterial sequences are reported in Fig. 4a and b and Supplementary Table S1. Consistent with results obtained at phylum level, the only two genera retrieved in BC1, that is, Flavobacterium (among Bacteroidetes; 6.1%) and Pseudorhodobacter (among Alphaproteobacteria; 2.0%), were particularly abundant. Five genera were detected in BC2, with Hydrogenophaga (Betaproteobacteria) that accounted for the 3.6% total sequences, followed by Flavobacterium (Bacteroidetes; 1.8%), Polaromonas (Betaproteobacteria; 0.6%), and Chryseoglobus and Marisediminicola (Actinobacteria; 0.6% each).
3.5.2. Lake L-2 (brine BC3)
The MA-B in the BC3 brine was mainly constituted by Betaproteobacteria (72.5%) and Bacteroidetes (20.3%), followed by other Proteobacteria members (3.8%; including Alpha- and Gammaproteobacteria, and unclassified Proteobacteria) and Actinobacteria (2.2%). Only the 1.0% of total sequences remained unclassified (Fig. 3a).
About 5% of the total high-quality cDNA bacterial sequences from BC3 were classified at genus level (Table 1) and were resolved in five genera occurring at ≥0.1%. Consistent with results obtained at phylum level, Hydrogenophaga (Betaproteobacteria) was particularly abundant, accounting for 3.4%. The remaining four genera were Algoriphagus (1.0%), Hymenobacter (0.3%), and Lewinella (0.3%) among Bacteroidetes, and Polymorphobacter (0.2%) among Alphaproteobacteria (Fig. 4c and Supplementary Table S2).
The separation of BC1 (mainly due to the high abundance of Pseudorhodobacter members) was confirmed by the PCA computed on data of active genera, as shown in Fig. 5b. No significant differences occurred among brines in terms of metabolically active bacterial taxonomical groups. With regard to the metabolically active bacterial communities, an inverse correlation with salinity was obtained for Chryseoglobus, Polaromonas, and Marisediminicola members, which were detected only in BC2 (Pearson coefficient r = −0.6), and more strongly for Hydrogenophaga (Pearson coefficient r = −0.9) affiliates, which were not detected in BC1. Pseudorhodobacter abundance was strongly positively correlated with salinity (Pearson coefficient >0.9) (Supplementary Fig. S1c).
3.6. Metabolically active archaeal community
3.6.1. Lake 16 (brines BC1 and BC2)
Differently from the MA-B, the metabolically active fraction of the archaeal community (MA-A) was similar in BC1 and BC2, with its composition at phylum level that reflected T-A data. In fact, the MA-A within the BC1 and BC2 brines was mainly represented by Euryarchaeota, followed by Crenarchaeota and Marine_Hydrothermal_Vent_Group (70.9% vs. 73.6%, 24.4% vs. 21.2%, and 3.1% vs. 3.6%, of total sequences, respectively). A low percentage of reads were affiliated to the Ancient Archaeal Group and Korarchaeota (about one in both brines) (Fig. 3b).
Among classified reads, ∼70% sequences were assigned to an order, with Halobacteriales (12.4–15.7%) and Methanosarcinales (28.8–35.4%) that were better represented (≥10%) in both brines (Table 2). About 55% of the total high-quality cDNA archaeal sequences were classified at genus level (Table 1) and were resolved in 66 and 53 genera, ranging from 0.03% to 19.3% and from 0.03% to 27.1% of total sequences in BC1 and BC2, respectively. Almost all genera occurred at ≥0.1%, as shown in Fig. 6a and b and Supplementary Table S2. Among Euryarchaeota, Methanosarcina was particularly abundant in both brines, accounting for 19.7% and 27.1% of total sequences in BC1 and BC2, respectively. Additional few genera (all among Euryarchaeota) occurred at a relative percentage ≥1% (range 1–7%; i.e., Ferroglobus, Halobacterium, Halococcus, Halovivax, Methanosaeta, and Methanopyrus in both brines; Methanothermobacter and Pyrococcus in BC2 only).
3.6.2. Lake L-2 (brine BC3)
As was observed for BC1 and BC2, the MA-A in the BC3 brine reflected T-A data. The MA-A was mainly represented by Euryarchaeota (77.8% of total sequences), followed by Crenarchaeota (19.5%) and Marine_Hydrothermal_Vent_Group_(1.5%). Both the Ancient Archaeal Group and Korarchaeota occurred at a lower percentage (0.6% each) (Fig. 3b).
Among classified reads, ∼70 of sequences were assigned to an order, with Halobacteriales and Methanosarcinales that occurred at ≥20% (Table 2). About 54% of the total high-quality cDNA archaeal sequences from BC3 were classified at genus level (Table 1) and were resolved in 70 genera, ranging from 0.03% to 14.8%. Almost all genera occurred at ≥0.1% as it is reported in Fig. 6c and Supplementary Table S2. Methanosarcina and Methanosaeta (14.8% and 9.8%, respectively) among Euryarchaeota were the most abundant. Additional few genera (all among Euryarchaeota; exception was Caldivirga among Crenarchaeota) occurred at a relative percentage ≥1% (range 1–4%; i.e., Caldivirga, Ferroglobus, Halobacterium, Halococcus, Halorubrum, Halovivax, and Methanopyrus).
No statistical differences were evidenced between brines in terms of abundances of archaeal genera, and no separation occurred in the relative PCA (Fig. 5d). The spatial separation occurring between brine samples was mainly due to the abundance of a few taxonomical groups, namely, Haloferax, Pyrodictium, and Methanopyrus. Within the MAAs, Ignicoccus members were positively correlated with salinity (Pearson coefficient >0.9).
4. Discussion
The ongoing thought about whether life exists beyond Earth mainly derives from the exploration of life in terrestrial extreme environments. Organisms inhabiting them are mostly microorganisms as harsh conditions generally do not sustain other forms of life. In this regard, brine pockets can be considered as true microbial habitats where life is represented almost exclusively by microorganisms. Microbial diversity and activities play a significant role in the ecological functioning of these environments, supporting an environment that is more similar to the conditions found in icy worlds of our Solar system (Garcia-Lopez and Cid, 2017). In this study, the composition of the prokaryotic communities in brines of two Antarctic lakes was investigated with comparative analysis of the 16S rRNA genes and cDNA of 16S rRNA by pyrosequencing. The number of ribosomes, and the ribosomal RNA content, generally increases in actively growing cells, so that the ribosomal RNA can be used as an indicator of microbial activity in environmental communities (Moeseneder et al., 2005). However, also dormant cells can contain high number of ribosomes, the content of rRNA and growth rate are not directly correlated, and the rRNA concentration in active cells can differ significantly among taxa. These limitations [reviewed by Blazewicz et al. (2013)] may explain the discrepancies encountered when comparing our DNA- and RNA-based results. However, this approach furnished a more complete characterization of the prokaryotic communities inhabiting Antarctic brines within lakes in the BC area, allowing us to make both ecological and astrobiological considerations. Moreover, it is noteworthy that the sampling activities were carried out before the construction of the semipermanent gravel runway for airfreight operations at BC, next to the Italian Research Base “Mario Zucchelli,” so that our findings may constitute a baseline of a possible environmental monitoring activity aimed at evaluating, for preservation purposes, the potential shift in the prokaryotic community structure and composition in Antarctic lake brines after an anthropogenic impact.
As for Arctic cryopegs and brines from other Antarctic lakes, BC brines are characterized by permanently subzero temperatures, high salinity, and moderate isolation from the influence of external factors. Such mineral-enriched brines (Sannino et al., 2020) represent a model for indirectly investigating putative life-forms in free-water environments on extraterrestrial icy worlds. The potential analogue with other extraterrestrial environments could be related, at least in the case of BC1 brine, due to its chemical composition (Sannino et al., 2020). In fact, here chlorides are more abundant than sulfates, allowing a similarity with Enceladus (Postberg et al., 2011) and Mars brines (e.g., Knauth and Burt, 2002), even if the mechanism of deliquescence evocated for the latter cannot be considered here. Obviously, obtained data cannot be generalized and further insights are needed.
Analyzed brines shared bacterial members mostly affiliated with the same taxonomic groups, even if differences were encountered in their relative abundances within both the total and metabolically active bacterial communities. Overall, the total bacterial communities were dominated by Bacteroidetes. These chemo-organotrophic bacteria are involved in the detritus food chain and carbon cycling in aquatic ecosystems (Cottrell and Kirchman, 2000). The recovery of high number of flavobacterial sequences (mainly Flavobacterium), also within the active fraction, may indicate the presence of complex dissolved and particulate organic matter (including a number of biopolymers) in the analyzed brines, especially in BC1. In the latter brine, the genus Pseudorhodobacter belonging to Alphaproteobacteria was also well represented within the active bacterial community. Members of the family Rhodobacteraceae show a high phenotypic and ecological diversity (Pujalte et al., 2014). They are capable of utilizing several organic and inorganic compounds for their metabolism, and carry out sulfur and carbon monoxide oxidation, as well as aerobic anoxygenic photosynthesis. Most of the genera in the Rhodobacteraceae family have been poorly investigated. Recently, Pseudorhodobacter psychrotolerans sp. nov., isolated from Antarctic soil in King George Island (Lee et al., 2016), was reported as producers of secondary metabolites.
Surprisingly, in the brine BC1 samples, potentially pathogenic bacteria were identified within the T-B. The genera Salmonella (among Gammaproteobacteria) and Arcobacter (among Epsilonproteobacteria) include species that are considered emergent enteropathogens and potential zoonotic agents. The introduction of human-associated pathogens into Antarctic wildlife [e.g., Salmonella spp. have been isolated from Antarctic fauna, such as seals and seabirds; Cerdà-Cuéllar et al. (2019)] and waters could derive from the dramatic increase in human presence and activities (both research and tourism) in recent decades (Cunachi et al., 2016; Lo Giudice et al., 2019). However, members of both genera have also been reported to form symbiotic relationships with protists (Bleasdale et al., 2009; Jun et al., 2016), whose occurrence in the BC lakes has been observed (Diego Fontaneto, per. comm.).
Contrary to BC1, the MA-PCs in BC2 and BC3 strongly differed from the bulk communities being dominated by Betaproteobacteria, with a massive presence of members in the genus Hydrogenophaga (typically of freshwater origin). Their occurrence was reported in the ice column of an Antarctic coastal lake (Foreman et al., 2011). Our finding may suggest that brine pockets, independently if they were located next to the lake bottom or suspended, are fed by the drainage of melted ice. Hydrogenophaga species are facultative chemo-organotrophic or chemolithoautotrophic bacteria that can oxidize hydrogen, but not acetate, and are known to possess the ability to degrade a number of organic pollutants (i.e., methyl tert-butyl ether, benzene, and biphenyl) and cometabolize polychlorinated biphenyls.
Any cold environment on Earth is likely not a habitat suitable for thermophiles. However, even in Antarctica, different geothermal sites (e.g., thermal springs, fumaroles, hot soils, and hydrothermal vents) provide ideal environments for the growth of thermophilic and hyperthermophilic microorganisms. The most thermally tolerant archaea (e.g., Aeropyrum and Sulfurisphaera), also related to well-known hyperthermophiles (e.g., Methanopyrus, Ferroglobus, and Methanothermus), have been identified within the T-A and, even if at a lesser extent (1–7%), within the active one. Papale et al. (2019) supposed that thermophilic and hyperthermophilic species, or their DNA, in saline Antarctic brines in the Tarn Flat area (Northern Victoria Land), where no geothermal activity has been reported in the past, might derive from deep circulation of brines, based on the hypothesis of Forte et al. (2016). Regardless of their origin, the occurrence of thermophilic RNA sequences in the cold BC brines leads to some considerations. At first glance, this could suggest that thermophilic micro-organisms might not be transient contaminants but potentially active members of the briny community, which implies that the biogeographical distribution of active thermophiles might be wider than previously recognized. However, RNA sequences related to the hyperthermophilic genus Ferroglobus, to date consisting of the single species Ferroglobus placidus (Hafenbradl et al., 1996), were retrieved in all brine samples. Interestingly, Ferroglobus can grow only between 65°C and 95°C (with an optimal growth temperature of 85°C), and it is the first (and sole) hyperthermophile discovered to grow anaerobically by oxidizing aromatic compounds (e.g., benzoate, phenol, and benzaldehyde) to CO2 coupled to the reduction of ferric iron Fe(III) to ferrous iron Fe(II) in the dissimilatory iron reduction (mainly occurring in geothermally heated environments). According to Smith et al. (2015), it is probable that F. placidus-like organisms populated early Earth, when photochemical oxidation of Fe(II) continuously formed Fe(III) in archaean seas and hydrothermal vent fluids, and may be responsible for iron banded formations that are often visible in ancient rocks. The phylogenetic tree (which is shown in Supplementary Fig. S2) constructed using Ferroglobus sequences from this study and open-source sequences on GenBank confirmed that at least two OTUs (i.e., OTU559 and OTU786) were strongly related to F. placidus AEDII12DO (strain DSM10642) isolated from shallow hydrothermal vents at Vulcano Island (Aeolian Islands, Italy) by Hafenbradl et al. (1996). Conversely, two additional OTUs (i.e., OTU1193 and OTU1267) clustered separately (similarity <96%), suggesting that sequences may be related to new species. Now, the question is how such cold environment (temperature never raises over 0°C) into which these thermophilic organisms are delivered could sustain their growth. Cockell et al. (2015) suggested that, for an Icelandic rocky environment, temperature-induced niche differentiation may permit the presence of thermophilic organisms in a cold environment as well. In our case, these types of temperature differentiations are hardly expected, and no evidence of geothermal activity is known in the BC area. However, wind-transported ashes from relatively recent eruptions (past 35,000 years) of Mounts Melbourne and Erebus or fumarole activities can occur in the massive ice that is present below the bottom of the lakes and in the morainic sediments that surround the lakes (Broady et al., 1987; Nicolaus et al., 1996). Thus, it is more plausible to assume that the briny environment, probably providing a stable and nonreactive environment, has contributed to long-term preservation of both microorganisms and nucleic acids.
Beside ecological implications, the detection of sequences related to methanogens and haloarchaea, in addition to some physical and chemical features of brines, mimic both environmental conditions and microbes that could survive on extraterrestrial icy planets. The high abundance of the strictly anaerobic genus Methanosarcina within the active community suggests that anoxic conditions might occur in BC lake brines, or more likely, analyzed brines might be in contact with a deeper brine circulation system. This makes such sites attractive as analogues for Europa and Enceladus (Preston and Dartnell, 2014). Interestingly, such methane-generating archaea produce methane by using all three known metabolic pathways for methanogenesis, that is, metabolizing the methyl group of acetate (acetoclastic pathway), carbon dioxide, and hydrogen gases (hydrogenotrophic pathways) and methylated one-carbon compounds (e.g., methylamines, methanol, and methyl thiols; methylotrophic pathways). Conversely, other Methanosarcinales, such as Methanosaeta members (also well represented within the active archaeal community), or Methanopyrales are acetoclastic and hydrogenotrophic, respectively. The occurrence of active methanogens in Antarctic lake brines may indicate the adoption of peculiar adaptation strategies to surmount subzero temperatures and high salinity, making them plausible candidates for extraterrestrial life. Recently, Mickol et al. (2018) demonstrated that nonpsychrophilic methanogens (i.e., Methanobacterium formicicum and Methanothermobacter wolfeii) resumed their metabolism (including methane production) during a long-term exposure to freeze–thaw cycles (temperature changed between −80°C and 37/55°C) when warmer temperatures were achieved.
Methanogenesis under anaerobic conditions has been widely accepted as the major mechanism of production of atmospheric methane. However, bacteria could also produce methane as a byproduct in oxic conditions, utilizing certain methylated organic compounds. For example, Li et al. (2019) reported on different bacterial isolates (e.g., Algoriphagus members) from the Antarctic Lake Bonney that were capable of demethylating methylphosphonate through C-P lyase pathway, with the subsequent generation of methane.
Halophilic archaea, as well as methanogens, are unanimously chosen as model organisms in exobiological or astrobiological studies due to their potential adaptation to the extreme conditions that are prevalent in extraterrestrial bodies (Williams et al., 2017; Sundarasami et al., 2019). They are able, in fact, to cope with simultaneous environmental stresses, such as high salinity, but also desiccation, temperature, and pH variability, and UV radiations (Leuko et al., 2015). Haloarchaea in the order Halobacteriales were well represented among the active archaeal communities in the analyzed brines. Their occurrence was not surprising as they are extreme halophiles that require at least 1.5 M NaCl for growth and possess a salt-in strategy to front the osmotic challenges associated with life in hypersaline environments (Grant et al., 2001).
5. Conclusions
The genomic diversity of the total and metabolically active prokaryotic assemblages in brine pockets from two lakes lying in the BC area (Northern Victoria Land) was explored for the first time. Along with the predominance of bacterial phylogenetic groups (e.g., Bacteroidetes and Alphaproteobacteria) also within the active fraction that are typically found in freshwater systems or in environments rich in organic compounds, this study furnished a number of findings that merit further investigation, such as the occurrence of (hyper)thermophilic sequences. Finally, our results demonstrate that methanogenic archaea participating in three different methanogenic pathways are present in Antarctic lake brines and could be active at in situ conditions. This finding, together with the occurrence strictly of anaerobes and halophiles, also add interesting elements useful for the comprehension of potential extraterrestrial life under extreme multifaceted environmental constrains. More thorough sampling and analysis of different brine types are needed to reach a comprehensive understanding of the diversity and function of microbial communities that inhabit such peculiar extreme habitats.
Footnotes
Acknowledgments
The authors thank all the staff at “Mario Zucchelli” Station, Dalle Fratte M., and Forte E. for the logistic help and support, which made the expedition possible.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the National Antarctic Research Program (PNRA), Italian Ministry of Education and Research (Research Projects PNRA 2013/AZ1.05; PNRA 2016_00194-A1; and PNRA 2018_00186-E).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
Associate Editor: Radu Popa
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.