
Abstract
Hydrogen peroxide has been postulated to be present on the surface of Europa and Enceladus. While it could represent a potential source of energy for possible life-forms, H2O2 may also interfere with a number of current detection technologies, including biosensors. To take advantage of the selectivity and portability of these devices, simple and reliable routes to degrade the potential H2O2 present should be developed and implemented to prepare for this possibility. Unfortunately, most of the current approaches for removing H2O2 are slow, may affect the sample, or could interfere with the performance of biosensors. To address these limitations, catalase was immobilized onto silica particles and used as a means to selectively decompose H2O2 prior to the analysis of common biomarkers with a biosensor. For these experiments, glucose,
1. Introduction
Since the presence of a liquid environment is considered a requirement for the development of life (as we know it), there is a growing interest in the exploration of extraterrestrial oceans. Among these so-called Ocean Worlds, Europa, a moon of Jupiter, and Enceladus, a moon of Saturn, are of particular importance. Both of these moons are thought to harbor a subsurface ocean potentially capable of supporting some form of biological life associated with the use of H2O2 as an energy source (Carlson et al., 1999; Gomis et al., 2004). Following this hypothesis, a number of reports have proposed that, if H2O2 is present below the icy shell and can be circulated through the subsurface oceans, it could indicate the presence of a radiation-driven ecosystem (Chyba, 2000; Dartnell, 2011; Ball and Brindley, 2015, 2019) similar to processes that are posited to have existed on Earth prior to photosynthesis (Haqq-Misra et al., 2011). It is also important to mention that, while several papers have pointed to the potential presence of H2O2 in other extraterrestrial bodies such as Mars (Encrenaz et al., 2012), many other groups have also provided alternative explanations for those observations and considered that neither H2O2 nor superoxide is required to explain the results collected by Viking LR and GEx probes (Houtkooper and Schulze-Makuch, 2007; Quinn et al., 2013). Regardless of the concentration, the potential presence of H2O2 in these samples represents a particular challenge for the detection of biomarkers, including amino acids, organic acids, and carbohydrates (Georgiou et al., 2016).
Historically, the standard technique applied for the detection of biomarkers on extraterrestrial bodies has been gas chromatography coupled to mass spectrometry (Pietrogrande, 2014). However, the environment believed to exist on Europa and Enceladus may also hinder the implementation of this approach because the H2O2 could interfere with the breakdown of organic molecules during the pyrolysis step (Benner et al., 2000; Navarro-González et al., 2006). To address this limitation, a number of liquid-based analytical techniques have been proposed (Blanco et al., 2017; Ribette et al., 2019). Out of those, immunoassays, electrochemical sensors (Thomson et al., 2020), and microfluidic capillary electrophoresis technologies hold the most promise in terms of power requirement, weight, sensitivity, and ability to detect biomarkers in model systems (Derveni et al., 2012; Sims et al., 2012; Willis et al., 2012; Butterworth et al., 2015; Mathies et al., 2017, 2019; Moreno-Paz et al., 2018; García-Descalzo et al., 2019; Lezcano et al., 2019). Unfortunately, even low concentrations of H2O2 present in the sample could interfere with all of these platforms because it may induce structural changes in the antibodies (required for selectivity in immunoassays) or through oxidizing the dyes used in fluorescence-based systems (Stockton et al., 2013). Therefore, addressing the potential presence of H2O2 seems to be a requirement to implement current methods to detect biomarkers on Ocean Worlds. In this regard, some of the most common methods to degrade H2O2 in biological samples are based on the use of metal catalysts, such as the Fenton reaction (Goldstein et al., 1993) or UV radiation (Gardner and Shama, 1998). While these methods are able to decompose H2O2, the kinetics and efficiency of these reactions can be significantly affected by the sample composition and/or may lead to significant temperature changes (Wang, 2008). Furthermore, methods based on UV degradation could potentially affect the composition of the sample due to the poor selectivity of the hydroxyl radicals formed, leading to the degradation of the analytes (Gardner and Shama, 1998; Wang et al., 2006; Ferreira Santos et al., 2019).
Aiming to provide a fast, efficient, and selective method to degrade H2O2, this manuscript describes the possibility of integrating immobilized catalase as a sample pretreatment step, enabling the analysis of biological samples via the use of oxidase enzymes on microfluidic paper-based analytical devices (μPADs). This enzymatic approach was selected considering that catalase is not only selective but also features one of the highest turnover numbers in nature. Furthermore, dried catalase has been shown to maintain its enzymatic activity even in the presence of up to 3 Gy of γ radiation (Georgiou et al., 2016), which makes this enzyme well suited for extraterrestrial sample analysis. Similarly, aqueous samples maintain their catalytic activity when exposed to γ radiation; however, there is a more significant decrease in activity as the dose, dose rate, and total exposure time increase (Cuba et al., 2010). It is also important to note that the aqueous catalase samples exposed to γ radiation up to 50 Gy at a rate of 5.5 Gy h−1 were able to degrade 89% of the H2O2 in the sample at 27°C (Cuba et al., 2010). Given both the efficiency of catalase and its ability to maintain catalytic activity after exposure to γ radiation, this simple hydrogen peroxide removal method could aid in the selection of samples potentially containing the selected biomarkers that are to be confirmed by other techniques.
2. Methods
Catalase from bovine liver (240 kDa, isoelectric point = 5.4) was purchased in aqueous solution, with an activity greater than 40,000 units per mg of protein (Worthington Biochemical Corporation, Lakewood, NJ, USA). Hydrogen peroxide was purchased as 35% w/w (VWR International, Radnor, PA, USA) and diluted with deionized water (Milli-Q, Millipore Water Systems, Billerica, MA, USA). Fresh dilutions were prepared daily to ensure that the concentration of H2O2 did not fluctuate due to thermal or photolytic decomposition. Phosphate buffer solutions (PBS) were prepared by diluting sodium phosphate monobasic (Fisher Scientific, Pittsburgh, PA, USA) in deionized water to a final concentration of 100 mM. The pH of the solution was measured by using a glass electrode connected to a digital pH meter (Orion 420A+, Thermo, Waltham, MA, USA) and adjusted to 7.0 with 2 M NaOH (Sigma-Aldrich, Milwaukee, WI, USA). Lactic acid solution (≤85%), glucose, and
The μPADs used were fabricated by using Whatman 3MM chromatography paper (21427-395 VWR International, Radnor, PA, USA). To ensure uniform size, all μPADs were patterned with a 30 W CO2 laser engraver (Epilog Legend Mini Helix 24, Epilog Laser, Golden, CO, USA), set to 50% power and 50% speed (vector mode). The μPAD design was created with Adobe Illustrator 2019 (Adobe Inc., San Jose, CA, USA). The final design featured three detection zones, each with a diameter of 7.62 mm, and was labeled for identification of the respective oxidase enzyme deposited in the detection zones (G = glucose, L = lactate, and A = leucine) by using the raster mode (60% speed, 50% power) on the laser engraver. The detection zones were connected with small channels, width of 1.63 mm and length of 8.25 mm, in order to prevent the enzymes from mixing when the liquid sample was introduced. A channel (1.63 mm wide by 7.29 mm long) at the bottom of the design allowed the device to be used as a “dipstick” or as a more classical μPAD where sample solutions are pipetted directly onto the device.
Catalase was selected as a catalyst for the degradation of H2O2 in the original sample. Because even a small amount of the enzyme leaching to the detection zones would interfere with the analysis, the enzyme was immobilized onto silica particles, according to a previously reported method (Evans et al., 2014). Briefly, 0.1 g of silica beads was added to 10 mL of a solution of 5% v/v (3-aminopropyl) triethoxy silane (APTES) in ethanol (99.9% pure, LC grade) and thoroughly mixed for 3 h. The functionalized silica beads were then centrifuged at 9000 RPM for 4 min, had the supernatant removed, and were rinsed with 2 mL of fresh methanol in order to remove any remaining APTES in solution. Next, the silica beads were rinsed twice in 2 mL aliquots of 100 mM PBS with centrifugation steps between each rinse. The silica beads were then placed in 1.5 mL of fresh 100 mM PBS with 0.5 mL of a stock solution containing catalase. This solution was agitated to form a uniform suspension and incubated at 2°C for 30 min. The silica beads were then washed with fresh 100 mM PBS and finally stored in 2 mL of 100 mM PBS at pH 7.0 for a maximum of 24 h.
H2O2 was quantified colorimetrically through the oxidation of either iodide (producing a red-brown color) or 3,3′,5,5′-tetramethylbenzidine (TMB, to generate a blue color). In both cases, images were taken with a CanoScan LiDE 7005 flatbed scanner (Canon Inc., Melville, NY, USA) after 15, 30, 45, and 60 min of exposure to the sample. Analysis of the color intensity and gradient was performed by Adobe Photoshop 2019 (Adobe Inc., San Jose, CA, USA) in a method similar to our previous work (McCann et al., 2017). In all cases, 5.0 μL of oxidase enzyme was pipetted onto the detection zone of the μPAD and allowed to dry for 15 min under ambient conditions. Afterward, 3.0 μL of 1% TMB in methanol was pipetted over the enzyme and allowed to dry for 15 min. An aliquot of 220.0 μL of sample was placed into a well on a 96-well plate. Samples were diluted with 30.0 μL of either fresh PBS (pH 7.0, control) or a suspension of catalase immobilized on silica beads (Cat-SiB). Once the μPAD dried, the injection region was dipped into the sample well until the sample wicked throughout the entire device (∼3 min); then the μPAD was placed in a Petri dish to dry under ambient conditions.
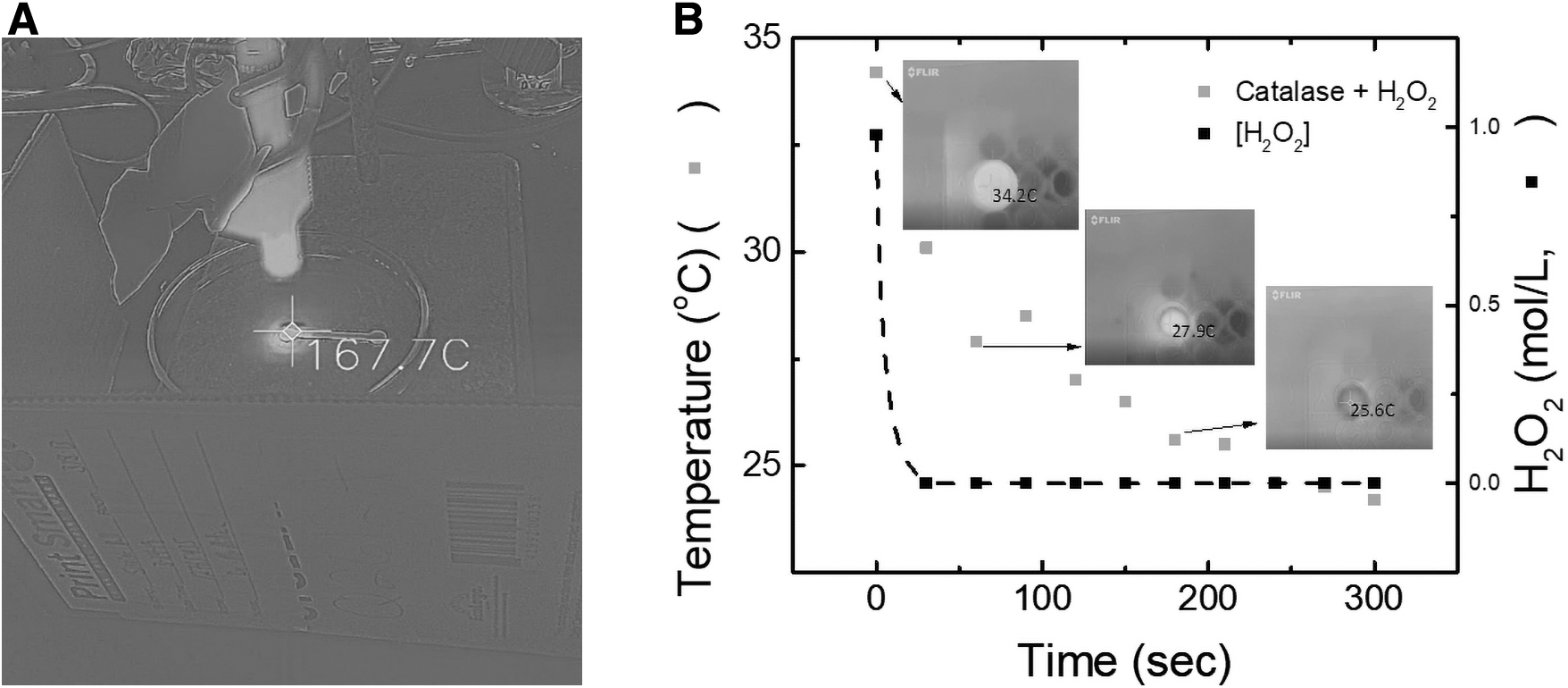
Enthalpimetry experiments were conducted by placing 100 μL of a 3.0% w/w H2O2 solution in one well of a 96-well microplate. An infrared camera (FLIRONE Pro iOS version, FLIR Systems Inc, Wilsonville, OR, USA) was attached to an iPhone X (Apple Tech Co., Cupertino, CA, USA) and positioned to observe the temperature change in the sample when catalase was introduced by focusing the camera in thermal MSX mode on the well with the peroxide sample (Lagasse et al., 2020). Once the recording of the sample was initiated, 100 μL of the catalase was injected into the sample well. The change in temperature was recorded by the IR camera, and 3 μL aliquots of the sample were removed every 15 s and spotted on 8.38 mm diameter circles of chromatography paper pretreated with a 2.0 M KI solution dried on the surface. These circles were dried under ambient conditions, scanned, and evaluated by the method previously described. The change in temperature was compared to the change in the intensity of the color produced from the oxidation of iodide to determine the relationship between temperature change and reaction progression.
To test the ability of transition metals in the decomposition of H2O2 via the Fenton reaction, solutions containing Fe(II), Fe(III), Co(II), and Cu(II) were prepared. Copper (II) chloride dihydrate, copper (II) acetate monohydrate, cobalt (II) acetate tetrahydrate, iron (III) oxide, and potassium hexacyanoferrate (III) were purchased from Sigma-Aldrich (Milwaukee, WI, USA). With the exception of iron (III) oxide, all other metal solutions were diluted to a concentration of 1.0 M in deionized water (Barnstead Nanopure Diamond, Thermo-Fisher Scientific, Waltham, MA, USA). As iron (III) oxide is insoluble in water, a suspension was created by placing 0.5222 g of the powdered iron (III) oxide (<5 μm particle size) in 1000 μL of ethanol. In all transition metal experiments, 10 μL of the solution was deposited on a central region of the μPAD and allowed to dry under ambient conditions. A 5.0 μL aliquot of 2.0 M KI was placed on the detection region of the μPAD to determine whether the H2O2 in the sample was decomposed, similar to the colorimetric method described above. Finally, a 10.0 μL sample containing the analyte and 3.0% w/w H2O2 was injected onto the μPAD, and the region of the μPAD with the transition metal was exposed to UV light at 365 or 405 nm by a Uvitron Skybeam LED Spot curing system (Uvitron International, West Springfield, MA, USA) with a 6 mm lens providing an intensity of 5980 mW/cm2.
To monitor the decomposition of H2O2, samples were analyzed by UV/vis spectrometry using a Genesys10 UV/vis spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) at 240 nm. The decrease in absorbance due to H2O2 decomposition was monitored as a function of time. The volume of the silica particle suspension was varied to determine the relationship between the volume of suspension and the initial rate of peroxide decomposition.
3. Results
To provide a direct comparison with established methods, the degradation of H2O2 was initially investigated by using the Fenton reaction (Goldstein et al., 1993) and UV radiation (Gardner and Shama, 1998). These initial experiments were performed with a solution containing 3% w/w H2O2, which mimics the maximum possible concentration expected on Mars, according to spectroscopy data (Encrenaz et al., 2012). As can be observed in Fig. 1A, the addition of Fe2O3 and UV radiation to the sample containing H2O2 led to a significant increase in temperature, reaching an average of 114°C within 30 s (maximum temperature 167.7°C). Even under these conditions, residual amounts of peroxide were observed, evidencing the limitations of the approach. While a clear color change was observed in the detection region, which correlated with the oxidation of KI, it was not possible to quantify the amount of peroxide remaining in the sample due to the scorching of the μPAD substrate (high temperatures produced). This further supported the preliminary conclusion that the Fenton reaction method would not be feasible for a μPAD system. On the other hand, when catalase was added to the solution containing H2O2, a fast degradation of hydrogen peroxide was observed (2H2O2 → 2H2O + O2) along with only a mild increase in the temperature (+10°C, Fig. 1B). As can also be seen in Fig. 1B, only 30 s was required for catalase to decompose the initial H2O2, supporting the suitability of the proposed approach (signal corresponding to remaining H2O2 within noise values).
(
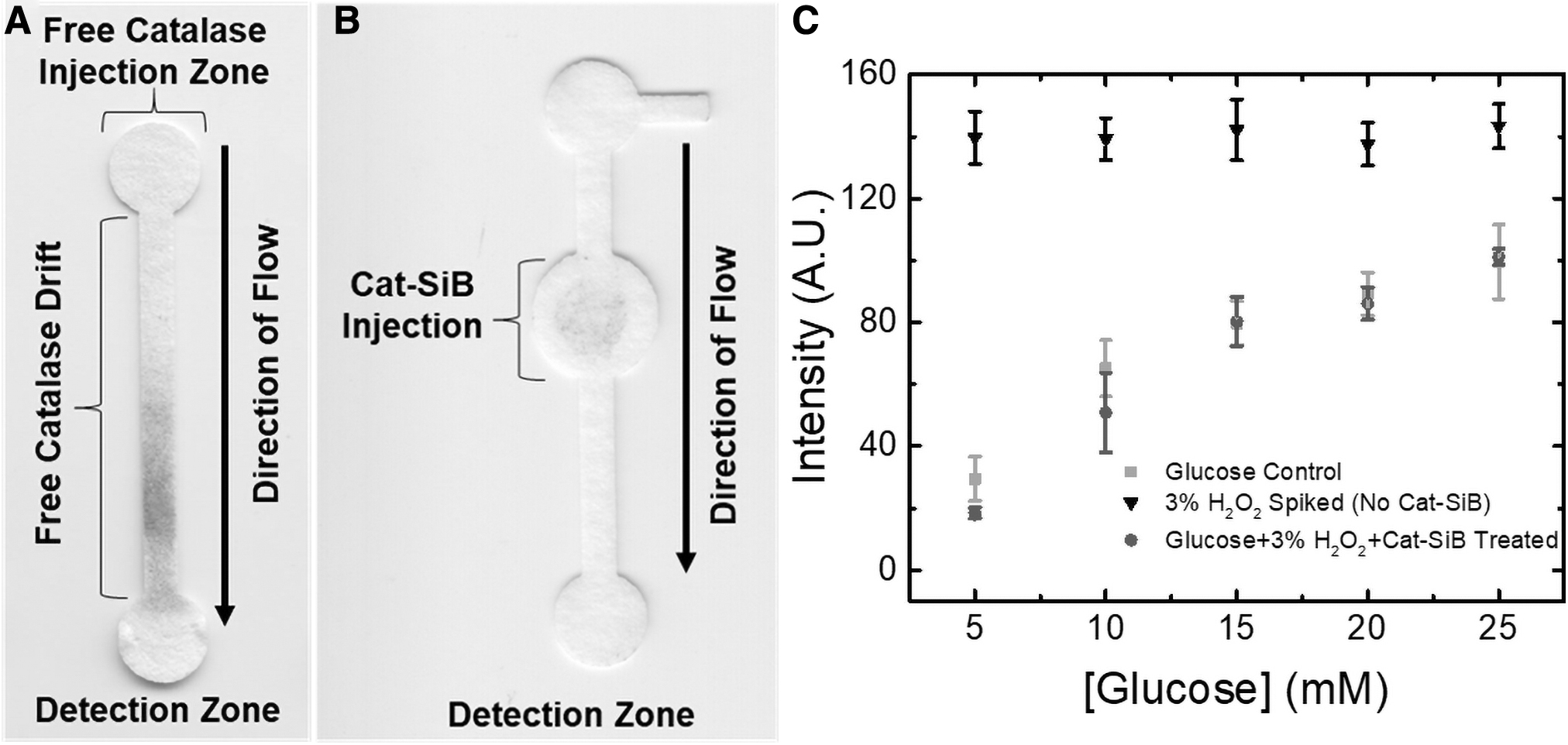
Next, the effect of the methodology selected to incorporate catalase into the μPADs was investigated. As the simplest approach, catalase was immobilized on the sample injection pad by simple adsorption. In this case, 15 μL of catalase was dispensed and allowed to dry for 15 min under ambient conditions. The efficiency of this approach was then investigated by dispensing 30 μL of a solution containing 3% w/w H2O2 and 10–100 mM glucose on the pad, allowing the sample to wick through the device, and finally quantifying the concentration of glucose in the detection spot of the device. According to these experiments (data not shown), it was observed that the enzyme was capable of rapidly decomposing H2O2. However, a significant decrease in the signal measured in the detection region was also observed. This issue was attributed to the migration of catalase along the channel and into the detection region. This behavior can be related to the high structural stability of the enzyme, which prevents surface-induced changes in the structure on the protein that would otherwise favor the adsorption of the enzyme onto the surface of the substrate. Fig. 2A shows a representative example of the displacement of catalase (stained with Coomassie Blue) in a simple μPAD, from the injection zone into the detection zone. As can be observed in Fig. 2A, the free catalase was transported from the injection region with the sample as it wicked across the device by capillary action. This caused a significant decrease in the resulting signal. It is posited that this decreased signal is a result of the catalase decomposing the H2O2 produced by the oxidase enzymes and a competitive inhibition of the oxidase enzymes.
(
To prevent this issue, catalase was immobilized on a solid substrate. In this case, silica microbeads were selected due to their uniform size (20 μm, which prevents their migration through the paper) and ability to be rapidly functionalized (Evans et al., 2014). As catalase does not readily adsorb to silica beads at neutral pH values, particles modified with APTES were used as the support (Cat-SiB). As can be observed in Fig. 2B, this strategy maintains the enzyme in the selected location and prevents its interference with the oxidases used to detect the biomarkers. Figure 2C shows the results corresponding to calibration curves obtained for glucose in the presence of catalase immobilized by different methodologies. It should be noted that, when catalase is not present, the initial H2O2 contained in the sample is able to reach the detection zone and react with the chromogenic agent, leading to a signal that is independent of the glucose concentration. On the contrary, when the sample containing glucose and H2O2 was treated with Cat-SiB, a trend matching that obtained with plain glucose was obtained. This indicates that catalase is not only active but also able to degrade the H2O2, avoiding the interference of both H2O2 (which would give a false positive) and catalase (which would give a false negative).
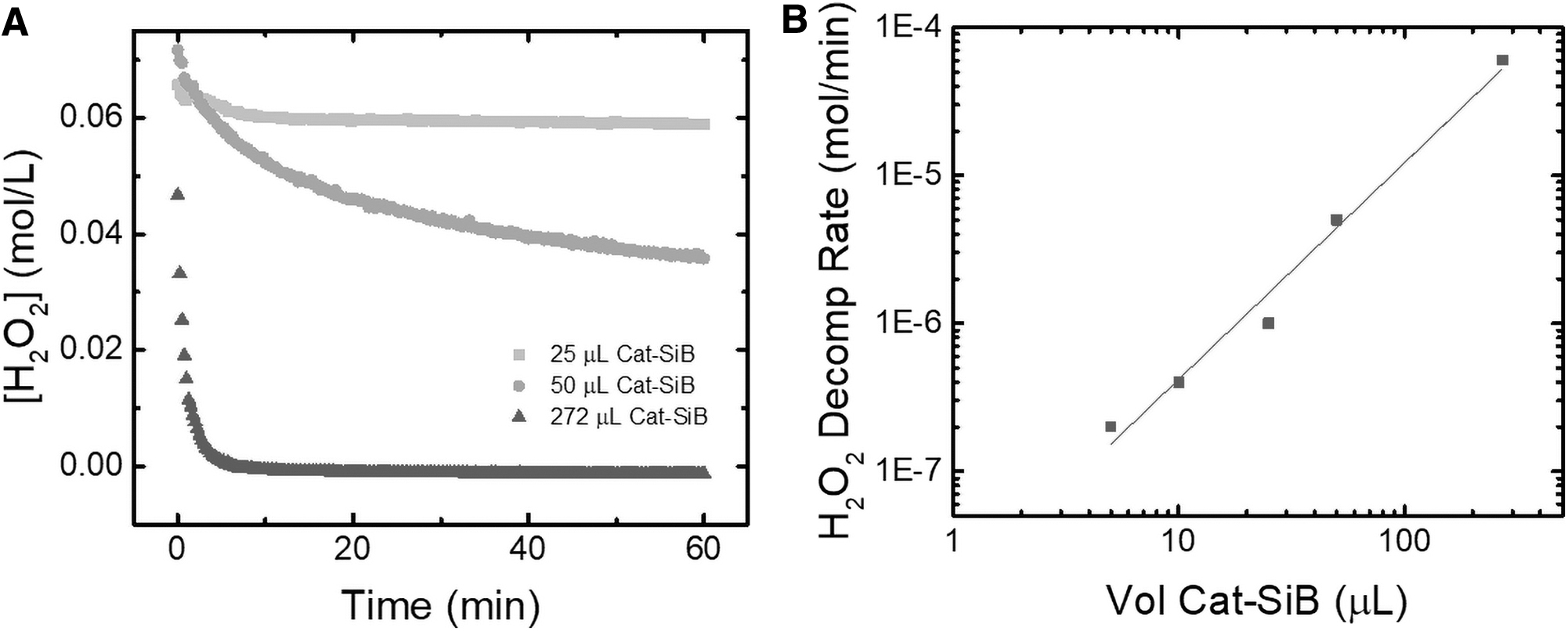
To study the kinetics of the Cat-SiB, the decomposition of peroxide was monitored at various volumes of the Cat-SiB suspension with UV spectroscopy. A comparison of the volume of Cat-SiB suspension to the total volume (v/v%) is shown in Fig. 3. It was found that a 7.5 v/v% of Cat-SiB to sample was adequate to fully decompose 65 mM H2O2 in less than 5 min. Additionally, due to the noncovalent bond between the APTES-coated silica beads and the catalase, it was found that an equilibrium process resulted in free catalase present in the buffer solution after approximately 24 h when the suspension was stored at 2°C. When the experiments were scaled down to the volumes for the microwell plates, it was found that the degradation of the 0.98 M H2O2 samples was completed within 5 min with no degradation of the analyte signal. Previous reports have defined one unit of catalase as the amount required to decompose 1 μmol of H2O2 per minute at pH 7.0 and 25°C. Based on the known volume of stock catalase used to prepare the Cat-SiB suspension and the rate of degradation, it was possible to determine the number of units of catalase required for this method (Felhofer et al., 2010). It was calculated that at a ratio of 7.5% v/v Cat-SiB a minimum of 60 units of catalase were present to achieve the necessary initial H2O2 degradation rate.
(
To show that the inclusion of immobilized catalase had no detrimental effects on the detection of the selected analytes, calibration curves of glucose,
4. Discussion
While the exact concentration of hydrogen peroxide on extraterrestrial bodies, such as Europa and Enceladus, remains a point of discussion and interpretation, some estimations have been given based on obtained spectroscopic information. For example, hydrogen peroxide was posited to be present in the leading atmosphere of Europa due to a 3.5 μm infrared adsorption band and inferred from ultraviolet reflectance spectra. Based off the Near-Infrared Mapping Spectrometer (NIMS) spectrum and comparison to laboratory samples of frozen hydrogen peroxide at different concentrations, Carlson et al. report a concentration of 0.13 ± 0.07% w/w (Carlson et al., 1999). While this interpretation is considered the most extensive, it is not indisputable quantitative evidence. Additionally, more recent investigations suggest lower concentrations of hydrogen peroxide (∼0.04% w/w) and argue that the original observations represent a local maximum (Hand and Brown, 2013). Similarly, a 3.5 μm IR band recorded on Enceladus by Cassini's Visual and Infrared Mapping Spectrometer (VIMS) has been contested as to whether or not it originated from hydrogen peroxide (Newman et al., 2007; Loeffler and Baragiola, 2009). Regardless of the interpretation, even minute concentrations of hydrogen peroxide could interfere with analysis of biomarkers. In our experiments, samples that contained 1% w/w (0.33 M) and were not pretreated with Cat-SiB still displayed a strong oxidation of the TMB and KI used as the colorimetric dye. This suggests that, if H2O2 is present on extraterrestrial bodies, some form of sample pretreatment would aid in the identification of biomarkers.
It is well known that transition metals and ultraviolet irradiation catalyze the decomposition of hydrogen peroxide to form hydroxyl radicals (Weiss, 1935). This process is well established in environmental fields for the treatment of wastewaters as the hydroxyl radicals rapidly oxidize the natural organic compounds found therein (Wang et al., 2006). While metal-catalyzed peroxide decomposition is effective, with rate constants ranging from 7.1 × 102 s−1 for platinum to 4.57 × 10−4 s−1 for iron III oxide (Yeremin, 1979; Lousada et al., 2013), they present several drawbacks for biological samples including the decomposition of organic molecules. While the adsorption of the peroxide onto a solid metal catalyst has been found to be a relatively fast process, the decomposition of peroxide is a relatively slow process that lasts for hours. This large reaction time can increase radical formation due to stabilization of the adsorbed species on the oxide surface. These usually short-lived species can then self-terminate to reform H2O2 (Mancuso et al., 1999; Lousada et al., 2013). Additionally, these Fenton-like processes are optimized for acidic media (Huang et al., 2001), which is in contrast to the neutral or alkaline conditions proposed on Mars, Europa, and Enceladus (Soffen, 1976; Kempe and Kazmierczak, 2002; Glein et al., 2015). Not only would lowering the pH of samples be labor intensive, but it could also potentially negatively affect any organic molecules in the sample. Additionally, it was found that iron III oxide immobilized on chromatography paper in combination with a 365 nm high-intensity light source significantly increased the temperature to a maximum of 167°C. This not only burned the μPAD substrate but also led to a potential loss of analyte by exothermic means. Thus, despite the widespread use of transition metal oxides and UV light for peroxide decomposition, Fenton-type reactions and UV decomposition methods were deemed incompatible for biosensors.
Alternatively, enthalpimetry experiments proved that free catalase could decompose 3% w/w H2O2 at a neutral pH, in under 30 s, with only a 10°C temperature increase. While small volumes (100 μL) of free catalase were capable of rapidly decomposing H2O2, a significant decrease in the signal in the detection region was observed when free catalase was applied to μPADs. It was determined that the catalase was transported via capillary action into the detection region, thereby decomposing the H2O2 produced by the analyte-enzyme reaction and weakening the resulting signal. By immobilizing the enzyme on silica microbeads, it is possible to eliminate this signal degradation. While the efficiency of the catalase immobilization process is relatively low (approximately 90% of units lost) due to the reversible nature of electrostatic interactions, the method was capable of decomposing 3% w/w H2O2 in under 5 min with no negative impact on the biomolecule analytes. Furthermore, the immobilization onto a solid substrate allows compatibility with common liquid sampling techniques and possible integration into current spaceflight technologies. A main advantage of the Cat-SiB method is that it converts the homogeneous catalyst of catalase solution into a heterogeneous catalyst, which can be removed from samples.
5. Conclusions
The proposed method of eliminating H2O2 that is present in a sample by utilizing silica particle immobilized catalase was shown to be effective. It is first necessary to immobilize the catalase on the surface of silica beads, which are then placed in a suspension and injected into the sample. The method degrades up to 3% w/w H2O2 in a sample with no adverse effects on the analytes. This concentration of H2O2 is more than double the potential concentration on extraterrestrial environments, which makes the enzymatic degradation of H2O2 by catalase a potential method for the identification of biologically relevant molecules.
Footnotes
Acknowledgments
The authors would like to acknowledge partial financial support for this project from Clemson University.
Abbreviations Used
Associate Editor: Christopher McKay