
Abstract
The roots of biological homochirality remain unknown despite decades of study. A commonly proposed path includes an initial small enantiomeric excess that was amplified over time, but a satisfactory source of the excess and a plausible amplification process have yet to be described. We propose here a route to oligonucleotide homochirality from unactivated racemic sources based upon the facts that duplex structures are inherently homochiral and their synthesis from strands of complementary string nucleotide subunits is both uncommonly rapid and exergonic. Simulations employing available kinetic and thermochemical data in an iterated sequence of three equilibria in dry/wet cycles running from unactivated and racemic RNA monomers through oligonucleotides to duplex structures have shown that the exceptional association rate distorts the otherwise simple equilibrium string and overcomes the severe kinetic and stoichiometric barriers to the pairing of the statistically scant homochiral fractions. The simulations reveal widespread deracemization and the full conversion of racemic monomers to populations of L- and D-duplexes in a succession of growth in which the initially formed duplexes are replaced over time with increasingly larger descendants. This claim is open to experimental testing.
1. Introduction
With a history extending back well over a century to Louis Pasteur (Flack, 2009), the origin of biological homochirality has been described as “one of the great unanswered questions in evolutionary science” (Hicks, 2002). Amplification of chirally enriched prebiotic sources of monomers is commonly proposed as a threshold condition (Soai et al., 2014), but neither a satisfactory enrichment source nor a path to amplification has been realized. Template-directed ligation and chiral inhibition (Joyce et al., 1984) should be pieces of the puzzle, and indeed more than two decades ago Orgel and coworkers posited a template-based selection of one optical isomer from a racemic mixture of nucleotides (Kozlov et al., 1999). The issue, however, persists. Biological homochirality stands pointedly as an indispensable anchor for the path to life's start, but as Pizzarello and Lahav (2010) emphasized in their introduction to a series on the topic, after decades of study its origins remain elusive.
Given this disconcerted state, we have sought here to develop a scheme to oligonucleotide homochirality applying an Occam's razor approach. We presume a starting stock consisting of the racemic forms of the four RNA monomers, and we moreover consider no more than the native monomers themselves and include no activation. We were encouraged by the recent account of Higgs and coworkers who presented a scheme leading to homochiral RNA oligomers from racemic monomers based upon the fact that only homochiral strings can engage in template-directed ligation (Tupper et al., 2017). Utilizing a range of assigned rate constants in kinetic and Monte Carlo simulations, they showed that very rapid but highly endergonic pairing and esterification steps destabilize the reaction system and ultimately yield to symmetry breaking and a favoring of one of the enantiomers. Brandenburg (2019) and Chen and Ma (2020) similarly applied assigned rate constants in machine simulations in examinations of deracemization in RNA growth employing rapidly chiral exchanging monomers.
Our approach here employs a racemic RNA monomer starting point but is directed along a different path, utilizing available rate constants and recognized thermochemical values applied to a successive series of simple assembly equilibria. In addition to skirting activation, we avoid the notions of symmetry breaking and chiral exchange and explore avenues to the simultaneous production of populations of dextro- and levorotatory duplexes. The working constituents in the scheme are the homochiral strings that will necessarily be present at equilibrium but at vanishingly small concentrations. Thus were the scheme directed by conventional second-order rate constants their assembly to duplexes would take place at small and impracticable rates. To overcome that limitation we assembled a sequence fashioned from three facts: spontaneous deracemization is a recognized phenomenon (Soai et al., 2014; Buhse et al., 2021) 1 ; intrinsically homochiral duplex structures are a prominent component of RNA and DNA architecture (Joyce et al., 1984); and most significantly, duplex formation from a nucleotide string and its complement is both uncommonly rapid with rate constants orders of magnitude greater than those for common bimolecular reactions (Eigen, 1971) and substantially exergonic (Rauzan et al., 2013).
2. Background
Key kinetic features of our proposition arise from the rigid topology of the phosphodiester yoke as the backbone of the duplex structure. It impedes transesterification by the 2′-OH and curbs hydrolysis of N-glycosidic bonds, thereby obstructing hydrolytic disassembly. The transesterification suppression can be substantial (Hüsken et al., 1996; Mikkola et al., 2001) and likely resembles that for the internucleotide ester link in DNA, which with no neighboring 2′-OH group undergoes hydrolysis more slowly than RNA by a factor of about 10 4 at ambient temperatures (Wolfenden and Snider, 2001).
A second element of our proposal is a setting employing dry/wet cycling in prebiotic hydrothermal ponds. Solute concentration and an increasingly prominent air-water interface during evaporation induce sizable changes in the properties of the aqueous medium and elicit a substantial unfavorable-to-favorable shift in the thermochemistry of phosphate/ribose esterification (Ross and Deamer, 2016, 2019; Nam et al., 2017; Ross 2018; Morasch et al., 2019; Damer and Deamer, 2020). Cyclic evaporating and filling can be viewed as two distinct reaction venues that respectively promote and reverse condensation chemistry, and at first glance it might appear that there should be no net effect of the cycling since products formed in the drying state should then simply revert to their original monomers upon dilution. As has been noted, however, the shift in the qualities of the aqueous medium in the swing from state to state results in marked kinetic and thermochemical disparities that in the end yield an overall net enrichment of the more stable condensation products (Kilburn et al., 2010; Ross and Deamer, 2016), and we submit that such cycles necessarily play a particularly strong role in the evolution of homochirality.
As we observed above, a fundamental obstacle in the assembly of homochiral structures from racemic monomers is the severe statistical barrier to suitable reaction rates. The solution of a racemic nmers assembled in a flat distribution of all possible oligomers from equimolar quantities of the four RNA monomers will comprise 4 n different sequences, and the homochiral fractions for each sequence and its complement are 1/8 n . Thus for a 1 mM solution of all racemic RNA 10mers the concentrations of a homochiral string of a given sequence and its complement will fall in the picomolar range. It then follows that for duplex formation from all 4 n /2 of the homochiral sequence/complement pairs in a solution of nmers at a molar concentration of cn mer M

and it emerges that the characteristic reaction periods for a conventional “rapid” k assoc of ∼100 M−1 s−1 (Mason and McConnell, 1994) extend nonetheless to hundreds of years and well beyond the hydrolytic lives of the reactants. 2
Our resolution to this obstruction lies in the unusually large second-order rate constant for duplex formation from a string and its complement at ambient temperatures, k assoc = 106 to 107 M−1 s−1 (Craig et al., 1971; Eigen, 1971; Pörschke and Eigen, 1971; Rauzan et al., 2013; Gleitsman et al., 2017). The value is no more than 2–3 orders of magnitude below the rates of encounter in aqueous media, and on that basis the characteristic periods of duplex formation from complementary strings at picomolar-like concentrations fall to no more than tens to hundreds of hours. As we shall see, a reduction of that magnitude promotes the formation of duplex structures from racemic sources to realistic intervals and presents the prospect of an effective and functional process.
The chemical basis of the brisk kinetics remains an interesting question (Gleitsman et al., 2017), and a factor that would seem to resolve the issue is an unexpected (and candidly counterintuitive) entropy-favored parallel self-alignment of rods that develops in cases of dense packing (van Anders et al., 2014; Frenkel, 2015). Of particular appeal in a life's origins context is its development prominently in evaporating systems (Jung et al., 2020). Further support arises from the predictive nearest neighbor models for duplex formation developed over the past two decades (Xia et al., 1998; SantaLucia, 1998; Lu et al., 2006) and represented by the additivity formulation in Eq. 2
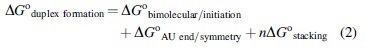
The first two terms encompass various thermochemical penalties of the process and sum to ∼5 kcal/mol, but the underlying feature of the formulation is the last term. It comprises a series of sequence-dependent stabilizing parameters for adjacent bases and their complements along a duplex string, with ΔG o stacking values falling in the range -0.9 to -3.5 kcal/mol and averaging -2.1 kcal/mol (Xia et al., 1998). 3 It thus emerges that successive WC bonding leading to duplex structures becomes exergonic for values of n beyond 3 or 4, a shift that explains the minimum nucleation length observed by Pörschke and Eigen (1971) and recently discussed by Gleitsman et al. (2017). Examples of an increasing proclivity with duplex length are reflected in the values in Table 1 taken from an extensive catalog provided by Xia et al. (1998).
Duplexes: Standard Gibbs Energies of Formation a
Taken from Xia et al. (1998).
3. Results and Analysis
3.1. A duplex formation model
It is thus evident that the combination of near-encounter rates and high exergonicity can promote the rapid assembly of unactivated monomers to thermochemically stable duplex structures. This factor, aligned with the observations by Higgs (1993) that tRNAs are thermodynamically favored over random oligomer sequences, is the principal component of the route to homochiral oligonucleotides from racemic RNA monomers offered here. The path is expressed in Eqs. 3–5, with Eq. 3 depicting overall oligomer assembly. Equation 4 is a subset of 3 involving solely homochiral oligomers denoted here by underscoring, and Eq. 5 portrays duplex assembly from those homochiral strings with the asterisk (*) identifying the corresponding oligomer complement.
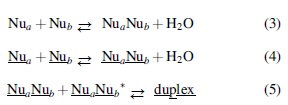
The productive functioning of the sequence develops from the opposing kinetic and thermodynamic interplay of Eqs. 1 and 2 wherein duplex stabilities grow with increasing n while their formation rates fall. The central feature and driver of the series is the extraordinary duplex formation rate in Eq. 5. Thus, throughout the process rate
5 for
>> rate
4 rev
, and as a result
3.2. Kinetic simulations
This prospect has been supported in simulations employing the Kintecus modeling package (Ianni, 2003), conducted for all values of a and b up to the 50mer at 25°C. 4 We employed starting solutions equimolar in unactivated racemic RNA monomers at overall concentrations of 2.3 × 10−2 M, the solubility limit of adenosine monophosphate at 25°C (Wang et al., 2010). The values for ΔG o 3 and ΔG o 4 were set to -5 kcal/mol in accord with the condensation-favoring evaporation portions of the wet-dry cycle, and from the data of Wolfenden and Snider (2001), k -3 = k -4 = 5.7 × 10−9 s−1. The parameters governing Eq. 5 were derived from Eqs. 1 and 2; the value of k 5 for emerges directly from Eq. 1 with k assoc = 107 M−1 s−1, and ΔG o 5 was developed from Eq. 2 with ΔG o stacking set at a conservative value of -1.5 kcal/mol.
The results displaying the effects of a ratcheted progression over a series of wet-dry cycles are presented in Fig. 1 and include profiles for the racemic oligomers up to the 14mer (in gray) and for representative homochiral duplex products from the 5-, 6-, and 8-10mers (in black). The figure shows from the earliest periods the rapid formation of duplexes to concentrations well beyond those of the insignificant starting levels of the homochiral components. Smaller duplexes emerge rapidly over periods of weeks, but they are short-lived and are successively replaced by increasingly larger and more stable duplexes at sizable concentrations, with the duplex products in the figure accounting for 24%, 49%, and 72% of the starting monomer feed after respectively 1, 10, and 100 years. It should be noted further that the kinetic parameters in these simulations apply to uncatalyzed chemistry; the reaction periods are therefore upper limit values, and the opportunity for catalysis is evident.
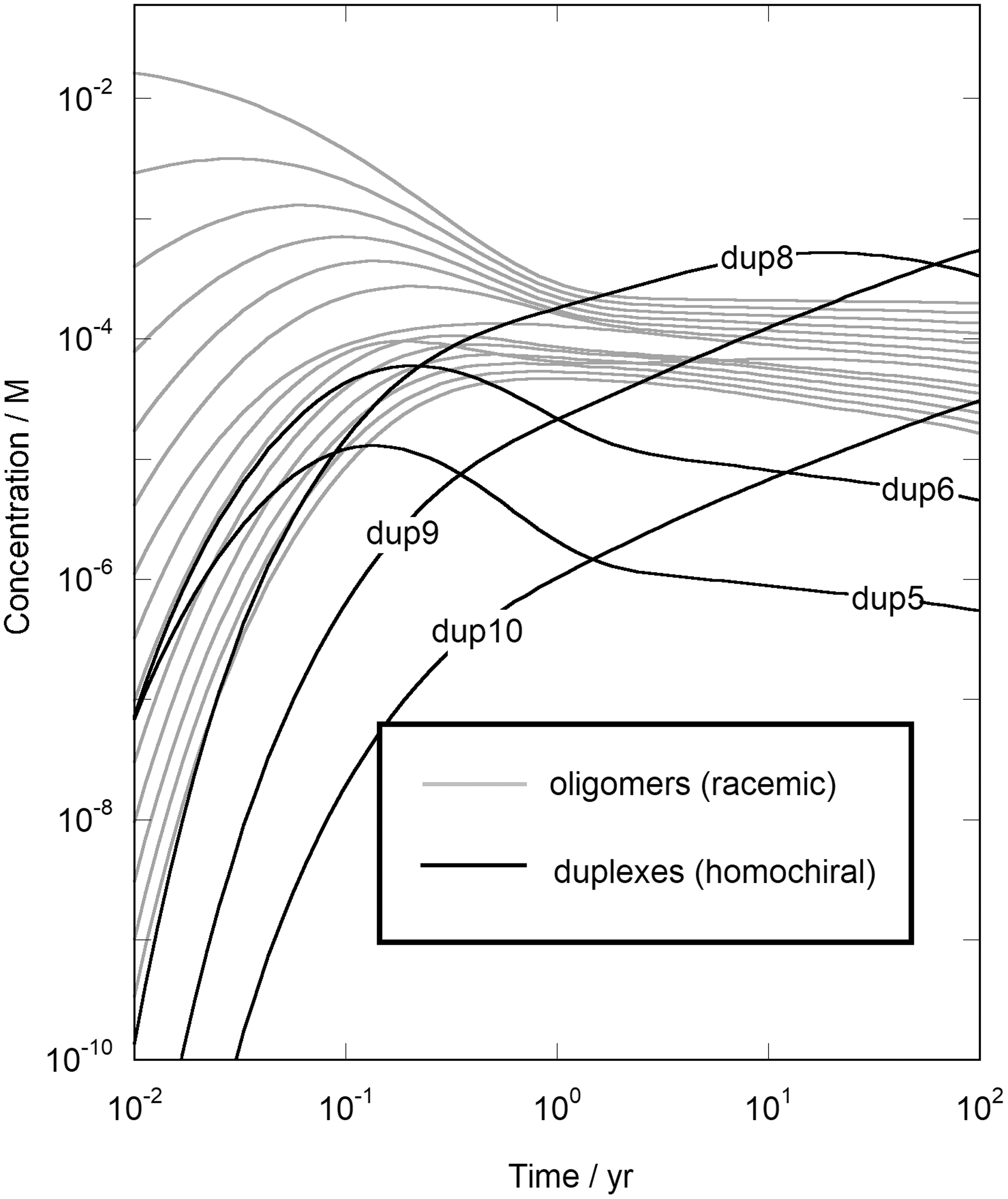
Duplex formation via Eqs. 3–5 from a starting solution of unactivated racemic RNA monomers at 25°C. The unpaired strands from the monomer up to the 14mer are shown in gray, and selected duplexes are presented in black.
It is apparent that the rate factors in Eq. 1 dominate initially and prompt the rapid production of the duplex products. The thermodynamic components in Eq. 2 are ultimately governing, however, and the early products are replaced over time by larger and increasingly more stable structures. The system continues to enrich itself through a sequence of wet-dry cycles; it remains racemic taken as a whole, but over the course of the process the components are partitioned on the basis of their handedness and in the end are fashioned into a pair of large, duplexed, and necessarily homochiral oligonucleotide populations.
4. Summary and Conclusions
We have described here a simple path to homochirality employing spontaneous deracemization within a small sequence of simple equilibria. The conversions require no activation and are based upon the authority that a single unusually rapid step can effect upon an otherwise straightforward linked equilibrium series. The magnitude of the kinetic action in that one step distorts the equilibrium tendencies; it directs the system through a sequence that in the end provides in the case here the spontaneous conversion of a mixture of unactivated racemic monomers to a bifurcated slate of increasingly larger homochiral products. The reaction course involves the direct intermediacy of duplex structures and requires no initial enantiomeric excess nor symmetry breaking. It is moreover exergonic and thus can yield readily to catalysis, and in contrast to activation-prompted oligomerizations that bear successively yield-declining oligomers, it provides size-ascending product sequences which are a required feature of a path to life (Ross and Deamer, 2019). As the solution is enriched with continued pond cyclings, the two populations should develop and advance independently, and we can expect that in time one of the two then emerges as the foundational chiral edifice of terrestrial life.
Finally, there is a straightforward way to test the proposal because it predicts that under certain conditions a cyclic condensation reaction will lead to spontaneous synthesis of homochiral oligomers from racemic mixtures of nucleotide monomers. Here we focused on wet-dry cycles that are plausible drivers of prebiotic polymer synthesis, but spontaneous polymerization also occurs in mixtures of chemically activated nucleotides such cyclic nucleotides (Morasch et al., 2014) and the imidazole esters used by Ferris et al. (1996). In previous work we have demonstrated that polymerization is enhanced in eutectic mixtures of activated nucleotides incubated at -18°C, with oligomers up to 17mers as products (Monnard et al., 2003). We predict that if such reactions were performed with racemic mixtures of activated nucleotides, the products will increasingly incorporate homochiral duplex structures. However, some version of cycling must be included so that the reaction does not simply run to equilibrium as the activated monomers are hydrolyzed. One possibility is to cycle between the low temperature where polymers are synthesized, then increase the temperature to the point where shorter duplex strands melt and hydrolyze while longer duplex strands do not. Fresh activated monomers are added at this point to replace those lost by hydrolysis, and the mixture then undergoes another freezing cycle. This process represents a molecular version of Darwinian selection that will drive the products toward ever-longer increasingly homochiral duplex molecules.
Footnotes
Associate Editor: Christopher McKay