
Abstract
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous in astrochemical environments and are disbursed into planetary environments via meteorites and extraterrestrial infall where they may interact with mineral phases to produce quinones important for origins of life. In this study, we assessed the potential of the phyllosilicates montmorillonite (MONT) and kaolinite (KAO), and the enhanced Mojave Mars Simulant (MMS) to convert the PAH anthracene (ANTH) to the biologically important 9,10-anthraquinone (ANTHQ). All studied mineral substrates mediate conversion over the temperature range assessed (25–500°C). Apparent rate curves for conversion were sigmoidal for MONT and KAO, but quadratic for MMS. Conversion efficiency maxima for ANTHQ were 3.06% ± 0.42%, 1.15% ± 0.13%, and 0.56% ± 0.039% for MONT, KAO, and MMS, respectively. We hypothesized that differential substrate binding and compound loss account for the apparent conversion kinetics observed. Apparent loss rate curves for ANTH and ANTHQ were exponential for all substrates, suggesting a pathway for wide distribution of both compounds in warmer prebiotic environments. These findings improve upon our previously reported ANTHQ conversion efficiency on MONT and provide support for a plausible scenario in which PAH-mineral interactions could have produced prebiotically relevant quinones in early Earth environments.
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs)—organic molecules composed of multiple aromatic rings—are one of the largest classes of compounds that comprise the organic inventory of the solar system and the observable Universe (Bada, 2001; Ehrenfreund et al., 2006; Iglesias-Groth et al., 2010; Roser et al., 2014; Materese et al., 2015). PAHs are ubiquitous in extraterrestrial environments (Allamandola et al., 1989; Galliano et al., 2008; Materese et al., 2015) and are presumed to arise principally from polymerization reactions of smaller organic precursors (e.g., acetylene) in circumstellar environments (Cherchneff et al., 1992). PAHs are similarly ubiquitous on the modern Earth where they are produced from the incomplete combustion of carbonaceous materials (e.g., fossil and nonfossil fuels) and, because of their stability, lipophilicity, and carcinogenic potentials, are considered priority environmental toxins (Lawal, 2017).
PAHs were also likely abundant on the thermally energetic primordial Earth whereupon they could have functioned as molecular precursors for seeding origins-of-life chemistries (Ehrenfreund et al., 2006; Ehrenfreund and Cami, 2010; Groen et al., 2012).
In addition to their abundance and importance on the modern and primordial Earth, PAHs have been detected in a myriad of astrobiologically interesting environments (Ehrenfreund et al., 2006), including carbonaceous meteorites (Becker et al., 1997; Sephton, 2002, 2005; Alexander et al., 2017), comets (Chyba et al., 1990), interplanetary dust (Allamandola et al., 1987; Chyba and Sagan, 1992), planetary atmospheres (López-Puertas et al., 2013; Cruikshank et al., 2014), circumstellar envelopes (Frenklach and Feigelson, 1989; Allain et al., 1997), and the interstellar medium (Puget and Léger, 1989; Gençağa et al., 2008; Ehrenfreund and Cami, 2010; Iglesias-Groth et al., 2010; Kwok, 2016).
Shock impact studies have also shown that PAH inventories comparable with those detected in carbonaceous meteorites are generated from the single-ring aromatic benzene molecule under simulated meteorite impact conditions (Mimura, 1995). Martian meteorite evidence also suggests appreciable PAH accumulations in early Mars environments (McKay et al., 1996), although PAH detection in current martian environments has remained challenging (Campbell et al., 2018). Detection of PAHs in astrobiologically relevant environments spanning such impressive spatial scales suggests that they are produced and transported throughout the cosmos, which in turn suggests that substantial quantities of PAHs, as well as their transformation products (e.g., oxy-PAHs), could seed prebiotic planetary environments and accumulate to concentrations sufficient to influence abiogenesis.
Within these environments, PAHs can interact with mineral phases, including phyllosilicate clays and metal oxides, to produce oxy-PAHs such as redox-active quinones (e.g., the 9,10-anthraquinone [ANTHQ] studied here) (Watson and Sephton, 2015). Phyllosilicates are common in terrestrial environments and have also been detected on, for example, Mars and the dwarf planet Ceres (King et al., 1992; Rivkin et al., 2006; Clark et al., 2007; Mustard et al., 2008). Clay minerals are of particular interest for astrobiological studies due to their capacity to exchange reactive cations with molecules entrained within their interlayer regions over a range of physicochemical conditions (Watson and Sephton, 2015; Juntunen et al., 2018). The cation exchange capacity of clays controls the surface charge distribution, which imparts unique reactive and/or catalytic capabilities to these geomaterials (Ertem et al., 2010; Jia et al., 2014, 2018).
These properties, combined with high surface areas, continue to motivate experimental efforts to characterize the extent to which mineral phases might act as reactants and/or catalysts in prebiotic systems (Ferris, 2005; Ertem et al., 2010; Hazen and Sverjensky, 2010; Brack, 2013; Watson and Sephton, 2015; Juntunen et al., 2018).
Quinones are oxy-PAH derivatives in which one or more of the hydrogen atoms in the aromatic ring are replaced by oxygen to form redox-efficient carbonyl groups (see Fig. 1A for a structure of the quinone studied here); quinones (e.g., ubiquinone and plastoquinone) are central to extant biochemistry and function in various redox states in electron transport chains essential for life (Taran, 2017; Milshteyn et al., 2019). In an early Earth context, PAHs have been hypothesized as precursors for the prebiotic synthesis of quinones; quinones have been proposed as plausible stabilizing agents and photochemical pigments in primitive membrane systems (Deamer, 1992, 1997; Groen et al., 2012).

General reaction scheme for the catalytic conversion of ANTH to ANTHQ on the mineral substrates, which is the same as that described by Juntunen et al. (2018)
In support of this idea, aromatic alcohols and quinones have been prepared from small PAHs, including naphthalene and anthracene (ANTH) in simulated astrophysical environments (Allamandola et al., 1988; Bernstein et al., 1999; Ashbourn et al., 2007; Materese et al., 2015), and quinones in particular are efficient at facilitating electron transport between electrodes composed of iron minerals relevant to origins-of-life studies (Taran, 2017). Milshteyn et al. (2019) demonstrated that meteoritic quinones can facilitate chemiosmotic gradients across lipid membranes via coupled redox reactions.
Importantly, among the quinones detected in their meteorite extracts was 9,10-anthraquinone (also known as 9,10-anthracenedione; ANTHQ; Fig. 1A), which is the focus of the present study. Discussion of the importance of ANTHQ for origins of life is perhaps a bit paradoxical as the compound exhibits appreciable toxicity at relatively low concentrations and, as noted, is thus considered a priority contaminant of the modern Earth. In this context, the compound has garnered considerable experimental attention primarily aimed at remediating its toxicity in environmental and biological compartments. However, ANTHQ also plays a prominent role in extant biology, being produced as a secondary metabolite in bacteria, fungi, lichens, and higher plant taxa; it is also known to function in key biochemical roles in, for example, present-day insects and higher plants (Singh and Chauhan, 2004).
Mars is a target of great astrobiological interest and current/future surface missions are interested in studying past and present habitability, as well as the possible presence of extant life (National Research Council, 2011; National Academies of Sciences Engineering Medicine, 2021). Phyllosilicate minerals are distributed over the martian surface (Banin and Rishpon, 1979; Mustard et al., 2008; Cuadros and Michalski, 2013; Bristow et al., 2018; Cuadros et al., 2019), but metal (particularly iron) oxide minerals predominate (Mitra et al., 2020) and have been proposed to be involved in facilitating abiotic reactions important for bioemergence (Fornaro et al., 2018).
With this in mind, we sought to advance our previous foundational study of montmorillonite (MONT)-catalyzed conversion of a PAH (ANTH
However, the recent report by Bizzarri et al. (2020) of quinone inventories attained from proton beam irradiation of hydroxynaphthalenes on meteorite substrates generally corroborates the thermal catalysis findings reported here.
To fill this knowledge gap, we assessed the potential of the clay minerals MONT (2 tetrahedral silica sheets:1 octahedral alumina sheet) and kaolinite (KAO; 1 tetrahedral silica sheet:1 octahedral alumina sheet), as well as the enhanced Mojave Mars Simulant (MMS), to facilitate the conversion of ANTH to ANTHQ (Fig. 1A). Upon establishing catalytic potential of the minerals, we quantified the kinetics of conversion to ANTHQ and loss of ANTH and ANTHQ over temperature regimes encompassing a wide range of environments relevant to astrobiological studies.
2. Methods
2.1. General
The methods described here are modifications of our previously reported methods developed to assess the kinetics of thermal conversion of ANTH to ANTHQ on MONT (Juntunen et al., 2018). In that study, larger volume 30 mL ceramic crucibles were used to incubate mixtures of ANTH and MONT. For the present study, much smaller volume 1 mL borosilicate glass reaction tubes were used to incubate mixtures of ANTH and the mineral substrates in pursuit of increased ANTHQ conversion efficiencies. Further methodological details are described below.
2.2. Reagents and substrate preparation
Reagent grade dichloromethane (DCM) and toluene were purchased from Sigma-Aldrich (Saint Louis, MO, USA) and used without further purification. Reagent grade ANTH and ANTHQ were purchased from Alfa Aesar (Tewksbury, MA, USA), prepared as 2 mg/mL stocks in toluene, maintained as 1% (m/m) powder mixtures for reactions, and stored in amber vials in a −20°C freezer throughout the study. All reagents and reactions were shielded from direct exposure to internal and external lighting. Samples and standards were handled under unlighted hoods, the sashes of which were lined with aluminum foil for additional shielding, and UV meters were used throughout the study to ensure that laboratory lighting (i.e., that outside the hoods) produced immeasurable UV flux under the hoods.
MONT (LOT. No. 10210586; Alfa Aesar), KAO (LOT. No. BCBV6863; Sigma-Aldrich), and MMS (The Martian Garden, Austin, TX, USA) mixtures were purchased and used as received. The mineral constituents of the MMS are as previously described (Beegle et al., 2007; Peters et al., 2008; Cannon et al., 2019). Using the EPA Method 9045D (EPA, 2004), we determined the pH of MONT, KAO, and MMS to be 3.65 ± 0.10 (n = 10), 5.62 ± 0.07 (n = 10), and 7.87 ± 0.03 (n = 10), respectively. Measurements of substrate pH were performed to provide basic sample characterization data and for comparison with forthcoming work. After an initial cleaning with Alconox detergent, followed by air drying, all glassware and reaction vessels were thoroughly solvent rinsed with DCM and toluene and then air-dried before use.
2.3. Preparation of substrates
Stock mixtures (∼3 g) of MONT, KAO, and MMS were mixed with 1% (m/m) ANTH and vortexed aggressively on a Vortex 2 Genie vortex mixer (Scientific Industries, Bohemia, NY, USA) at the maximum setting for 10 min. Twenty milligram aliquots of the mixtures were introduced into laboratory-fabricated reaction tubes (11.0 cm × 0.7 cm o.d.) prepared by cutting and then sealing one end of Fisherbrand™ borosilicate glass pipettes (Fisher Scientific, Waltham, MA, USA) with a butane torch (Supplementary Fig. S1).
To permit direct comparison with our previously reported ANTHQ conversion efficiency results obtained by using larger volume ceramic crucibles (Juntunen et al., 2018), reaction tubes were incubated in triplicate over the temperature range 25–250°C for the desired duration (0.25–11,520 min; Supplementary Table S1) in Fisherbrand™ Gravity Ovens (Fisher Scientific). All reactions were carried out at atmospheric pressure, which averages 0.993 atm due to the elevation of the laboratory. To quantify production and loss kinetics of ANTH and ANTHQ at temperatures beyond the limits of the Gravity Ovens, reaction tubes were incubated in triplicate over the temperature range 300–500°C for the desired duration (0.25–8 min; Supplementary Table S1) in a muffle furnace (Yamato Scientific, Tokyo, Japan).
Incubation temperatures were selected to encompass varied environments of astrobiological interest, including, but certainly not limited to, early Earth and Mars (0–85°C) (Kasting and Tazewell Howard, 2006; Ramirez et al., 2014), hydrothermal and subhydrothermal systems (25–500°C although at increased pressures) (Baross and Hoffman, 1985; Martin et al., 2008; Lee et al., 2009; Shibuya et al., 2010; Sojo et al., 2016; Barge and White, 2017; Deamer et al., 2019), subaerial hot springs (Kompanichenko et al., 2015; Milshteyn et al., 2018; Deamer et al., 2019; Des Marais and Walter, 2019; Kompanichenko, 2019; Damer and Deamer, 2020; Deamer, 2021), volcanic plumes (>100°C) (Delmelle et al., 2018), and meteorites (>25°C) (Sears, 1975; Hayatsu et al., 1980; Hinrichs and Lucey, 2002; Kimura et al., 2004).
Following incubation, reaction tubes were removed from the oven and allowed to cool to room temperature (∼10 min). The contents were transferred from the reaction tubes to 12 mL borosilicate glass culture tubes, 1.5 mL of 1:1 DCM:toluene (v/v) was added to extract the organics from the substrate, and the tubes were vortexed aggressively with a Multi Reax vortex mixer (Heidolph, Elk Grove Village, IL, USA) at the maximum setting of the instrument for 5 min. Extraction mixtures were centrifuged at 4000g in an AccuSpin 8C clinical centrifuge (Fisher Scientific) for 5 min to pellet the remaining mineral solids. The liquid extract was transferred to 2 mL screw top autosampler vials and adjusted to 1.5 mL final volume for direct gas chromatography-mass spectrometry (GC-MS) determination.
A general schematic of the sample preparation and analysis methods used in the study is shown in Supplementary Fig. S1. Extraction efficiency was assessed by mixing pure ANTH or ANTHQ with each substrate at 1% (m/m) followed by vortexing, solvent extraction, centrifugation, and direct GC-MS determination and quantitation as above. Extraction efficiency of ANTH was determined to be 97.3% ± 2.3% (n = 10), 93.7% ± 3.1% (n = 10), and 98.8% ± 1.4% (n = 10) for MONT, KAO, and MMS, respectively. Extraction efficiency of ANTHQ was determined to be 98.1% ± 4.9% (n = 10), 97.4% ± 4.2% (n = 10), and 101.5% ± 1.6% (n = 10) for MONT, KAO, and MMS, respectively.
2.4. Sample analysis
Standard curves of ANTH and ANTHQ were prepared via dilution from stock solutions in toluene such that a range of 0.1–750 and 0.1–100 ng of ANTH and ANTHQ, respectively, was injected onto the GC column in triplicate (Supplementary Fig. S2). All standards and sample extracts were analyzed with a QP2010 SE GC-MS (Shimadzu, Kyoto, Japan) equipped with an AOC-20s autosampler and Rxi-5ms capillary column (30 m × 0.25 mm with a 0.25 μm 5% diphenyl 95% dimethyl polysiloxane phase; Restek Corporation, Bellefonte, PA, USA).
One microliter of all standards and samples was injected onto the GC column. Helium was used as the GC carrier gas. The GC injector temperature was maintained at 250°C and operated in splitless mode with a helium flow rate of 1.15 mL/min. The initial GC column temperature was set to 110°C, held for 1 min, and then ramped to 320°C at 25°C/min with a 1-min hold at 320°C. The MS was operated in the electron impact ionization mode at 70 eV and 0.1 kV detector voltage. The ion source and MS interface temperatures were maintained at 260°C and 290°C, respectively. Mass spectra were obtained in full scan and selective ion modes, with the mass range 28–500 amu scanned at a rate of 3333 scans/s. Ions m/z = 178 and 208 amu were used for identification and quantification of ANTH and ANTHQ, respectively.
2.5. Data analysis
Analysis of total ion chromatograms (TICs) and mass spectra generated for all standards and samples was performed with GCMSsolution software (Version 4.11; Shimadzu, Kyoto, Japan). Compounds were first identified by comparison of their mass spectra with those of reference compounds in the NIST/EPA/NIH Mass Spectral Library (NIST 17, Gaithersburg, MD, USA), followed by analysis with authentic standards. Compound identities for ANTH and ANTHQ were confirmed by >95% similarity match to reference spectra followed by spiking pure compounds into unreacted and reacted samples as previously described (Juntunen et al., 2018). All statistical analysis and graphing of data were performed with Excel (Microsoft, Redmond, WA, USA) and Prism (GraphPad, San Diego, CA, USA) software.
3. Results
3.1. Development of improved methods for assessing ANTHQ conversion efficiency
In our previous study, the MONT-catalyzed thermal conversion of ANTH to ANTHQ was assessed by using larger volume ceramic crucibles, which yielded ANTHQ conversion efficiencies in the range of 0.027–0.066% over the temperature range assessed (25–250°C) (Juntunen et al., 2018). While this was a valuable proof-of-concept study, it became necessary to improve upon the relatively low conversion efficiencies of ANTHQ reported in this work.
To address this component of the method, we fabricated small-diameter (<1 cm) glass reaction tubes with internal volumes about 10% of those used previously. Use of the same preparation of ANTH with MONT as previously reported, but confined within a much smaller reactor geometry (and smaller sample volume), yielded ANTHQ production and conversion efficiencies as much as a hundred-fold higher than those attained with crucible incubation (Fig. 1B). This difference is presumably due to excess ANTH and ANTHQ vapor losses from the more open crucibles relative to the far more volumetrically confined reaction tubes. Because the procedure allowed for direct comparison among samples and between reaction vessels, the new reaction tubes represent an improvement to the method and were utilized for the remainder of the study.
3.2. Establishing conversion capacity of the substrates
In our earlier work (Juntunen et al., 2018), MONT was tested as the sole catalytic substrate due to its widespread use and availability. To increase the repertoire of substrates assessed when using the new sample reaction system, we explored the capacity of three mineral phases, relevant to both prebiotic chemistry in planetary environments and potentially habitable environments elsewhere (e.g., primordial Earth and Mars), to facilitate conversion of the representative PAH ANTH to the biologically important ANTHQ over simulated temperature regimes encompassing a wide range of environments of astrobiological interest. Substrates included two chemically distinct phyllosilicate clay minerals (the MONT and KAO) and a Mars surface soil analog (the MMS) (Fig. 1A and Supplementary Fig. S3).
Upon incubation, we observed distinct colorimetric changes in all three substrates over the temperature range indicative of reaction via electron transfer on and alteration of the substrates (Jia et al., 2014) (Supplementary Fig. S3). After incubation, extraction and GC-MS analysis of the substrates showed TICs and molecular ion (mass fragment m/z 208) peaks with an identical retention time as the authentic ANTHQ standard and displayed an identical mass fragmentation pattern (with mass fragments m/z = 208, 180, 152, 76 predominating spectra) (Fig. 2 and Supplementary Fig. S4–S7). Spiking of the same ANTHQ standard used in our previous study into these samples showed that the identified ANTHQ peak increased in area, no new peaks were observed in the GC domain, and the mass fragmentation pattern was unchanged, which further validated the identity of the product consistently observed here and in our previous work (Juntunen et al., 2018).
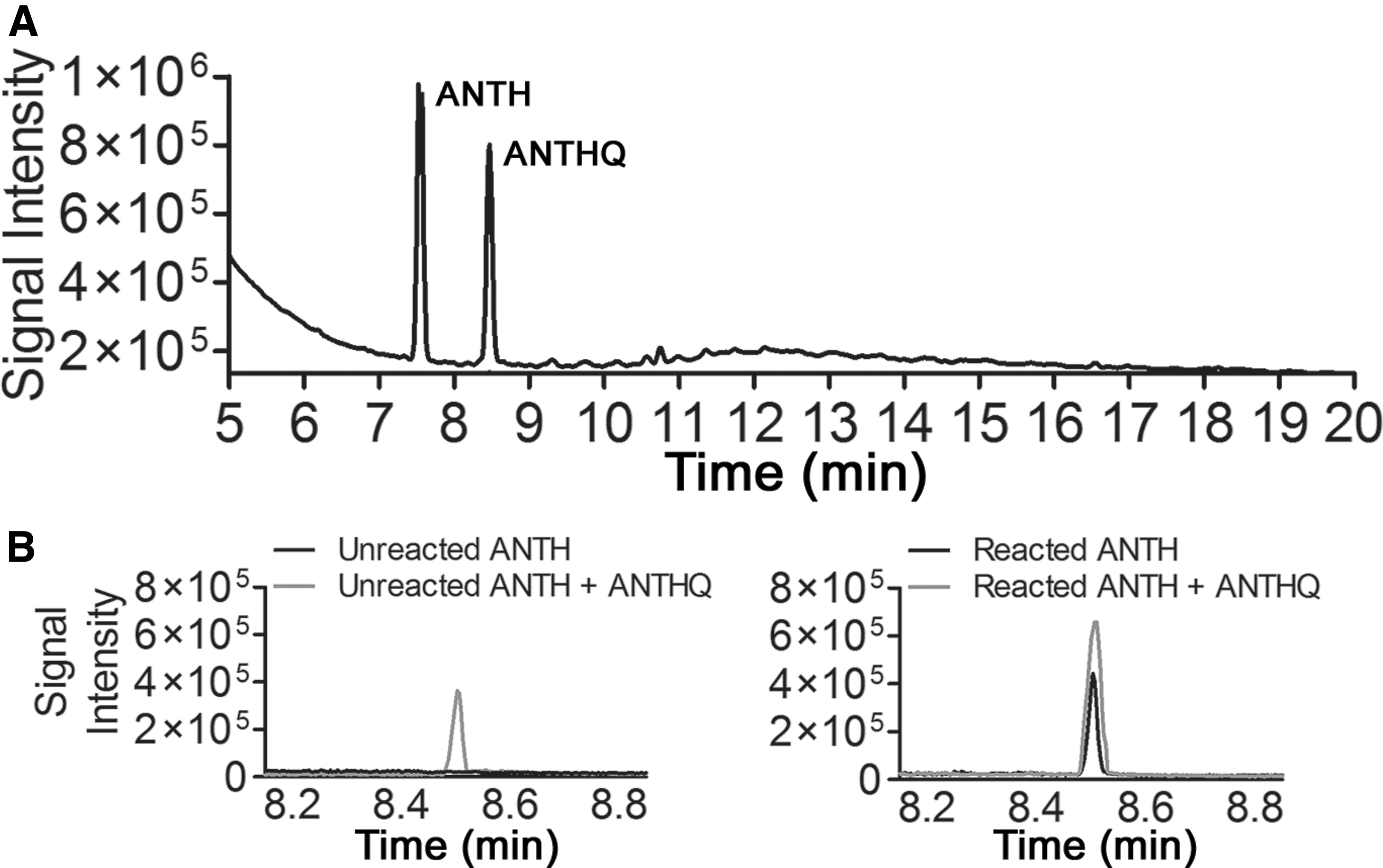
TIC of a representative sample extract showing the remaining doped ANTH (first peak at 7.5 min), and the ANTHQ product (second peak at 8.5 min) arising from catalytic conversion on a mineral substrate
Pure ANTH added to the reaction tubes and incubated over this range of temperatures in the absence of a catalytic substrate resulted in no appreciable formation of ANTHQ (i.e., below the established quantitation limit described in the Section 2 above), demonstrating the following: (1) that the reaction tubes themselves do not have catalytic capacity, (2) the mineral substrates are the catalysts, and (3) ANTH does not convert to ANTHQ in the absence of a catalytic substrate, even under the more extreme thermal conditions assessed. Taken together, these results indicate that the three substrates chosen for study catalyze the reaction of ANTH to ANTHQ seen previously in the improved reaction tubes.
3.3. Conversion kinetics of ANTHQ
After establishing that the desired reaction still occurred in the new system (and that it occurred more efficiently), it was necessary to determine the influence of temperature on ANTHQ conversion. In our previous work (Juntunen et al., 2018), the use of larger ceramic crucibles and a comparatively low-temperature incubator constrained our simulated temperature range to 250°C, despite the relevance of higher temperatures to the potentially important prebiotic environments described above.
In this work, all mineral substrates catalyzed ANTHQ conversion under temperature regimes encompassing a wide range of astrochemical environments (25–500°C), which doubled the thermal range assessed in our previous study of MONT catalysis (Fig. 3). Production rate curves were generated by assessing apparent rates of ANTHQ production in the linear range of conversion (0.5–11,520 min; Supplementary Table S1). Apparent rates of ANTHQ formation displayed sigmoidal relationships for MONT and KAO, while the MMS displayed a quadratic relationship (Fig. 3). Maximum apparent rates of ANTHQ formation were comparable on the MONT and MMS substrates, while those measured on the KAO substrate were approximately sixfold and eightfold lower than on the MONT and MMS, respectively.
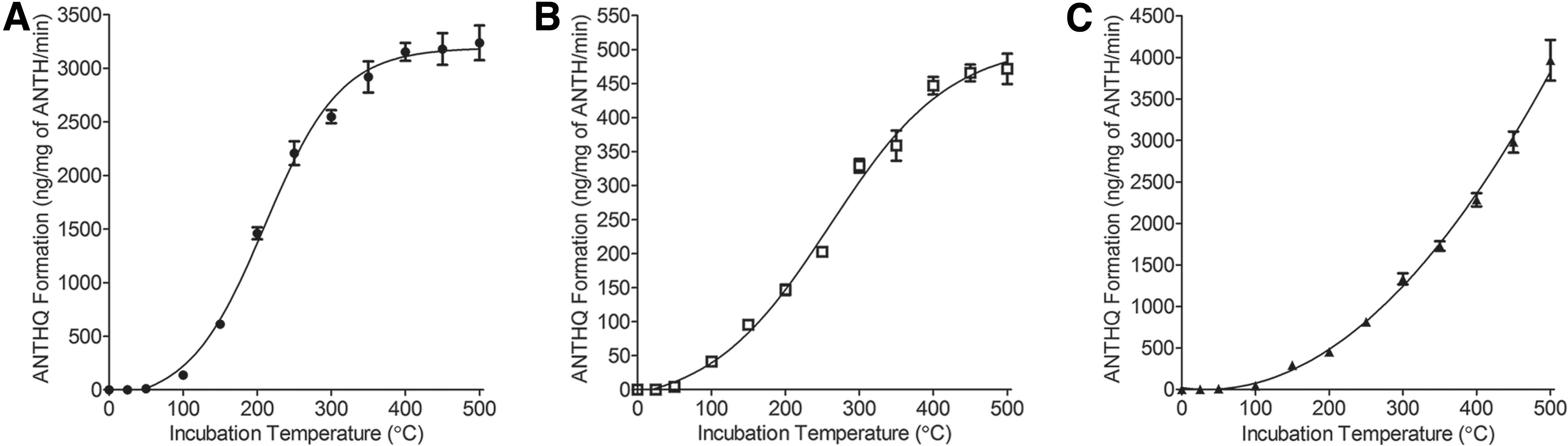
Apparent rates of ANTHQ formation on MONT (filled circles;
3.4. Conversion efficiency of ANTHQ
One of the drawbacks to the use of larger crucibles and lower temperatures in the previous study was the low ANTHQ conversion efficiency observed, which was maximal around 0.066% (Juntunen et al., 2018). Based on increased production rates in the improved reaction tubes and at higher temperatures, it was necessary to determine conversion efficiencies among the substrates. Mean ANTHQ conversion efficiencies attained under these new conditions were 1.90% ± 0.28%, 0.66% ± 0.094%, and 0.43% ± 0.052% for the MONT, KAO, and MMS substrates, respectively.
Mean conversion efficiencies were statistically different (p < 0.05) among the substrates as determined by a paired t-test (MONT:KAO, p = 0.000541; MONT:MMS, p = 0.000172; KAO: MMS, p = 0.0449). Maximum mean conversion efficiencies were observed at 350°C for each of the substrates (MONT: 3.06% ± 0.42%; KAO: 1.15% ± 0.13%, MMS: 0.56% ± 0.040%). The conversion efficiency values are much greater than those attained in the previous study and further validate use of the new reaction tube-based approach for this and future studies.
3.5. Loss kinetics of ANTH and ANTHQ
We hypothesized that losses of ANTH and ANTHQ, especially at the higher temperatures assessed, accounted for much, if not all, of the production rate curve trends observed. To test this, we assessed ANTHQ production and loss at the median temperature of 250°C over a longer incubation time of 60 min (Fig. 4A). This time was chosen to ensure that we captured and then far exceeded the linear range of production observed in previous incubations (i.e., 0.5–17 min; Supplementary Table S1) aimed at determining the rates of formation presented in Fig. 3.
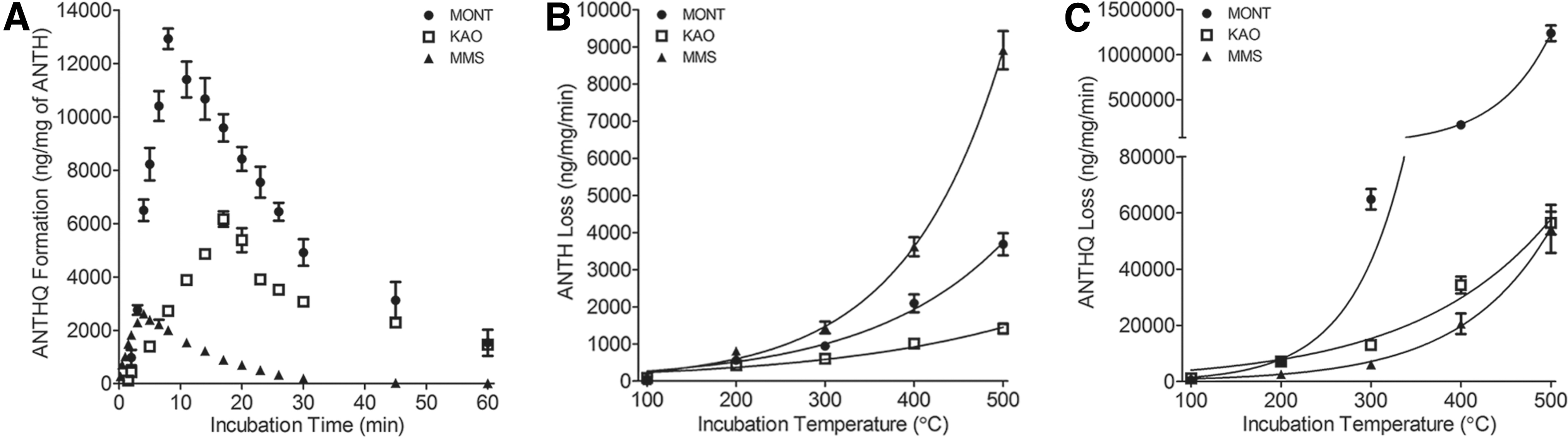
Apparent rates of ANTHQ formation and loss on the MONT (filled circles), KAO (open squares), and MMS (filled triangles) substrates at the median temperature of 250°C
As seen in those earlier incubations, apparent rates of ANTHQ formation on MONT far exceeded those observed on KAO and MMS at this median temperature up to the maximum linear production range of 17 min observed on the KAO. Under these conditions, maximum ANTHQ production rates were observed in the order MONT > KAO > MMS. Substrate pH occurred in the order MMS > KAO > MONT, suggesting a possible influential role thereof. As pH has been shown to strongly influence the catalytic capacity of minerals (especially clays; Joshi et al., 2009; Aldersley et al., 2011), more systematic assessments of pH effects on ANTHQ production on these substrates are underway in our laboratory.
On these same substrates, loss of ANTHQ was observed as early as 5 min on the MMS and as late as 17 min on the KAO (Fig. 4A). After 60 min of incubation, ANTHQ loss reached the limit of quantitation on the MMS, while the MONT and KAO retained ANTHQ well above that and to about the same extent. To determine whether diminishing ANTHQ formation was due to thermal loss or to the production of secondary reaction products arising from ANTHQ decomposition during incubation, we screened representative samples incubated over the temperature range using exhaustive GC-MS conditions (i.e., slow GC ramp rates [1°C/min] and long GC run times [120 min]).
We also extracted representative reacted substrates with 100% hexane (polarity index [PI] = 0.1), 100% DCM (PI = 3.1), and 100% methanol (PI = 5.1) and detected only a few putative reaction products, all of which were at or below our established signal-to-noise (S/N) >3 compound identification criteria used for all studies in our laboratory.
To gain insight into the ANTHQ formation trends presented in Fig. 3, and the process(es) by which loss may be occurring, we characterized the kinetics of ANTH and ANTHQ loss from substrates over the same temperature range used to assess apparent rates of ANTHQ production. Apparent rates of ANTH loss with temperature occurred in the order MMS > MONT > KAO, with loss rates becoming statistically different (p < 0.05) above 200°C (Fig. 4B).
In contrast, rates of ANTHQ loss were substantially higher with temperature than those of ANTH and occurred in the order MONT >> KAO > MMS (Fig. 4C), with loss rates converging to the same mean values (p > 0.05) for the KAO and MMS at 500°C. Loss rates of ANTHQ from the MONT were 25-fold higher at the highest temperature assessed. Collectively, these findings show that loss of ANTH occurs to a much lesser extent than loss of ANTHQ, and that maximum loss rates for both compounds differ with the nature of the substrate (i.e., maximum ANTH loss from MMS; maximum ANTHQ loss from MONT).
4. Discussion
As noted, the cosmic ubiquity and theoretically important role of PAHs for origins-of-life chemistries has been previously considered in some depth (Ehrenfreund et al., 2002, 2006; Ehrenfreund and Cami, 2010). Inspired by this problem and the dearth of studies thereon, to the best of our knowledge, we demonstrate here for the first time (at least in the case of KAO and MMS) a plausible pathway by which a biologically useful quinone can arise abiotically from PAH-mineral interactions relevant to prebiotic planetary environments of astrobiological interest (e.g., primordial Earth and Mars).
We further show (for all the substrates) the influence of temperature on the kinetic behavior of both production and subsequent loss thereon, as well as colorimetric changes indicative of altered redox states as the reactions proceed (Jia et al., 2014). The latter could be useful for remote sensing of PAH-mediated chemistries relevant to astrobiological missions to, for example, Mars. The results presented demonstrate that, while all of the substrates catalyze the conversion of ANTH to ANTHQ, they interact quite differently with each substrate as evidenced by the observed differences in production and loss. Such distinct behaviors are potentially attributable to, for example, differences in catalytic potential of the substrates, compound/substrate binding affinities, compound diffusivities, and physicochemical properties of the ANTH and ANTHQ over the energetic temperature regimes investigated here.
With regard to the latter, it is reasonable to presume that the kinetics of compound production/loss on the substrates is influenced by phase changes (ANTH MP/BP = 216/341°C; ANTHQ MP/BP = 285/377°C). However, several studies have reported on the oxidation behavior of ANTH over temperature ranges relevant to those studied here, but in the context of industrial syntheses (Subramanian and Murthy, 1972; Do et al., 1990; Bonfranceschi et al., 2002). As literature elucidating potential impacts of phase change on the kinetics of the mineral-catalyzed production/loss observed here is sparse, assessment of the influence of these parameters is an aim of our future studies.
The improved design of the reaction tubes over that used in our previous foundational assessment of the kinetics of conversion of ANTH to ANTHQ on MONT only allowed for higher rates of ANTHQ production and increased conversion efficiency (Juntunen et al., 2018). While both the crucibles used in our previous study and the tubes used here are open systems with ostensibly free exchange of gases, it is likely that the improved catalytic conversion attained by using the current tubes is due to the reduction in reaction vessel diameter and total volume of the reaction vessel and substrate.
In our previous work, attempts were made to quantify loss of ANTH and ANTHQ from MONT as a function of incubation temperature, but we were unable to obtain consistent measurements. We concluded that this was likely due to greater thermal losses (presumably via sublimation) permitted from the more voluminous and more open crucibles, which may have allowed compound escape in the gas phase at higher temperatures. The formation of ANTHQ and loss of both compounds increased as reaction temperatures rose; this trend changed at temperatures near and above their boiling points (341°C and 377°C for ANTH and ANTHQ, respectively). We note that the rates of loss at or above the boiling points are much higher than lower temperatures, while the rate of formation begins to level off in this range, except for ANTHQ formation on MMS.
It is possible that, because the structure of MMS is different than the MONT and KAO clays, some thermal protection may be conferred by the MMS, and the underlying physiochemical differences that modulate the kinetics of compound formation and loss are the subject of our ongoing investigations. It is also interesting to note that all three of the relevant substrates displayed catalytic efficacy, which may indicate that, in prebiotic environments in which these minerals (or their analogs) may co-occur, their interactions could result in combined effects on ANTHQ production efficiency therein.
In support of a potentially important role in seeding planetary bodies with biomolecular precursors, PAHs, along with their quinone derivatives, have been detected in a number of extraterrestrial chemical environments, including meteorites, comets, interplanetary dust particles, and the interstellar medium (Allamandola et al., 1987, 1988; Wing and Bada, 1991; Moreels et al., 1994; Becker et al., 1997; Botta and Bada, 2002; Krueger et al., 2004; Iglesias-Groth et al., 2010; Roser et al., 2014; Materese et al., 2015). Providing further support for this role, conversion of representative cosmic PAHs to quinones on interstellar ice analogs and on meteorite substrates under simulated astrophysical thermal and UV regimes has also been reported (Bernstein et al., 1999; Dworkin et al., 2002; Ashbourn et al., 2007; Allamandola, 2011; Bizzarri et al., 2020).
These latter studies are generally corroborative of our own findings and also suggest an important role for PAH-particle interactions in abiotic quinone synthesis. However, none assessed kinetics of quinone production, making direct comparison of study findings difficult. However, taken together, it is reasonable to conclude that the production of quinones, such as the ANTHQ examined in this study, via physicochemical processes acting on PAHs associated with catalytic particulate phases was likely an important step toward the emergence of quinone-based redox and chemiosmotic infrastructures essential for extant biochemistry. However, our understanding of their emergence and evolution, and especially their eventual uptake into emergent biotic systems, remains incomplete.
One plausible idea that motivates the current study is that redox-efficient quinones can arise in sufficient quantities to seed protometabolisms via PAH interactions with catalytic planetary minerals in the presence of labile oxygen reservoirs and thermal energy regimes.
Earth's atmosphere at the time of life's emergence, although devoid of significant O2, did contain other oxidant species, for example, CO2, H2O, and SOx, and NOx species (Ducluzeau et al., 2009; Trail et al., 2011; Wong et al., 2017), all of which might drive conversion of smaller PAHs to quinones under prebiotic conditions. The experiments presented here were conducted under ambient atmosphere for greater ease of laboratory experimentation and as proof-of-concept; however, we certainly acknowledge that these results are likely to differ in the presence of oxidant species other than O2.
Indeed, PAH oxidation to quinones could be driven by other geochemical oxidants (in gaseous, aqueous, and/or mineral phases) in addition to O2 (e.g., OH, NOx, Fenton and Fenton-like species, and silicates) (Vione et al., 2006; Usman et al., 2016; Maranzana et al., 2017; Potenti et al., 2018; Johansson et al., 2020). To further explore this possibility, studies of ANTH conversion to ANTHQ under atmospheric and mineralogical regimes relevant to high-priority astrobiological environments (e.g., Mars) are now underway in our laboratory.
Even so, there still could have existed geochemical microenvironments containing sufficient pools of O2 on the primordial Earth (perhaps as high as 5% derived from, e.g., UV photolysis of H2O, CO2, H2O2, and minerals), as well as on Mars (derived from, e.g., subsurface salt brines), to drive reactions analogous to those observed in this work (Borda et al., 2001; Selsis et al., 2002; Ślesak et al., 2012; Harman et al., 2015; Meadows, 2017; Petrescu, 2019).
The smectite MONT has been previously shown to have the capacity to catalyze astrobiologically important reactions (Ferris, 2005; Aldersley et al., 2011; Jheeta and Joshi, 2014; Joshi et al., 2015; Watson and Sephton, 2015; Juntunen et al., 2018; Zellner et al., 2020). However, apart from our previous study of MONT-catalyzed conversion of ANTH to ANTHQ (Juntunen et al., 2018), we are aware of only two reports of mineral-catalyzed conversion of PAHs to quinones in the context of origins of life (Watson and Sephton, 2015; Bizzarri et al., 2020). In the Watson and Sephton study, ANTHQ production from ANTH incubation on Fe-exchanged MONT was reported, but only anecdotally, while in the Bizzarri et al. study, hydroxynaphthalenes adsorbed to meteorite substrates were irradiated with high-energy proton beams to yield quinone inventories.
Although assessing ANTHQ production as such was not a primary study aim of either study, the findings of both are corroborative of those presented here.
While few studies of mineral conversion of PAHs to oxidized by-products appear in the astrobiology literature, the environmental toxicology and chemistry literature has previously recognized the catalytic potential of clay minerals to convert PAHs to more environmentally benign by-products (Jia et al., 2014, 2019; Li et al., 2015; Zhao et al., 2017).
Although some of these have reported on the kinetics of PAH conversion to various products on clay minerals, including the ANTH, MONT, and KAO reported on here (Li et al., 2015; Zhao et al., 2017), none has assessed the thermal kinetics of ANTHQ production from ANTH on these substrates, nor quantified ANTHQ formation or conversion efficiency. Rather, they assessed the kinetics of PAH/ANTH degradation within the context of environmental contamination.
The different foci of these studies thus render direct comparison of findings uninformative. Despite this problem, the ANTH loss kinetic trends observed on, for example, modified MONT and KAO by other workers are generally comparable with the concomitant ANTH loss kinetics observed in the current study (Jia et al., 2014; Li et al., 2015; Zhao et al., 2017). In this way, the results described here (and those reported in our previous study of ANTHQ conversion on MONT) (Juntunen et al., 2018), considered along with these environmental studies, support an important role for catalytic conversion of PAHs on mineral surfaces, regardless of the disciplinary context.
Informed by spectroscopic and other studies of ion-exchanged clays (Soma et al., 1985; Jia et al., 2014, 2018, 2019; Zhao et al., 2017; Liu et al., 2021), we have previously speculated about the underlying mechanism of ANTHQ production on MONT under simulated prebiotic thermal conditions (Juntunen et al., 2018). There, we posited that metal cations (predominantly Fe(III) and Al(III)) within the interlayer regions likely strip electrons from the least resonance stabilized central ring of ANTH to produce radical cations with strong affinity for O binding at the most reactive carbons (i.e., those at the 9 and 10 ring positions).
More generally, it is presumed that carbonyl formation would occur preferentially at the carbons of any PAH possessing a system of migrating pi sextets (Solà, 2013). Given their general structural similarities, we now hypothesize that a comparable oxidative mechanism likely operates within the interlayer regions of the KAO, but that electron abstraction is less efficient, presumably due to the absence of Fe(III) (or other efficient metal oxidizers) and the simpler 1:1 alumina octahedral:silica tetrahedral sheet configuration, which results in lower surface area, charge density, and cation exchange capacity relative to MONT (Jia et al., 2014).
More generally, the capacity of clays to facilely exchange cations is well known and many studies of their exchange within these uniquely reactive geomaterials have focused on Fe exchange due to its relatively high-standard redox potential (E o = 0.77 V). We have thus ourselves presumed that Fe(III) likely accounts for much of the conversion of ANTH to ANTHQ observed. Previous studies of ANTH conversion on Fe-modified MONT phases lend support to this idea (Jia et al., 2014; Watson and Sephton, 2015).
Based upon the results of our previous study of MONT-catalyzed ANTHQ production kinetics, we have also speculated that Al(III), despite its considerably lower standard redox potential E o = −1.66 V, and its presence at threefold higher concentrations than Fe(III), may drive more electron transfer, and thus greater ANTHQ conversion, than has been previously appreciated or reported. This may be due, in part, to a simple concentration effect, but other factors are likely also important. Support for a more prominent role for Al(III) in driving oxidation is provided by studies of organic compound binding to Al(III) ions located on the broken edges of the alumina sheets of clay minerals (Pedreira-Segade and Rogers, 2019).
While we remain uncertain of the underlying mechanism(s) governing ANTHQ production and the substrate constituents that ultimately influence catalysis, it is an area that our group is actively investigating and is the subject of forthcoming studies.
5. Conclusion
We have shown that three planetary relevant mineral substrates (MONT, KAO, and the MMS) catalyze the conversion of a PAH (ANTH
We attribute these gains to increased reactant contact and reduced loss of ANTH and ANTHQ during incubation, which suggests that similarly volume-constrained mineral microenvironments could produce larger pools of ANTHQ. Rate curves of apparent ANTHQ production were sigmoidal for MONT and KAO, but quadratic for MMS, indicative of differential catalysis on and losses of ANTH and ANTHQ from the substrates. Efficiency maxima for ANTHQ conversion (observed at 350°C) were 3.06% ± 0.42%, 1.15% ± 0.13%, and 0.56% ± 0.039% for MONT, KAO, and MMS, respectively.
Loss of ANTH and ANTHQ differed with substrate, but all loss rate curves were exponential, presumably due to sublimation. Loss trends suggest a viable mechanism for wider distribution of both compounds in prebiotic environments. Together, these findings support a plausible scenario in which PAH-mineral interactions produce quinone precursors that could drive bioemergence in prebiotic environments.
Footnotes
Authors’ Contributions
S.G.H., V.K., S.M.D., J.T.D., C.M.M., and C.M. conducted experiments, and assisted with data reduction/interpretation. J.M.W. and L.M.B. assisted with experimental design, data analysis/interpretation, and article preparation; P.V. and M.O.G. designed experiments, analyzed/interpreted data, and wrote the article.
Acknowledgments
We thank Dr. Dale Droge (Dakota State University [DSU]) for insightful conversations and financial assistance, and Nancy Presuhn and Pamela Lewis (DSU) for superlative administrative assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
M.O.G., S.M.D., L.R.D., C.M.M., and C.M. were supported by the National Science Foundation (NSF)/South Dakota EPSCoR RII Track-1: Building on the 2020 Vision: Expanding Research, Education and Innovation in South Dakota (Award OIA-1849206) and by the South Dakota Board of Regents. The work was further supported by Instituto de Astrobiología de Colombia undergraduate research awards to S.G.H. and V.K., and South Dakota Space Grant Consortium awards to L.R.D. and M.O.G. M.O.G. was further supported by DSU Faculty Research Initiative (FRI) and Supporting Talent for Research Trajectories (START) grant awards and by DSU's Office of the Provost.
Additional financial support was provided by the DSU College of Arts and Sciences as part of an undergraduate research-in-teaching initiative in M.O.G.'s Organic Chemistry I and II (CHEM 326 and CHEM 328) and Prebiotic Chemistry (CHEM 491) courses. P.V. was supported by faculty startup funds awarded by Southern Oregon University. J.M.W. and L.M.B.'s research was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration (NASA).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Abbreviations Used
Associate Editor: Nita Sahai
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.