
Abstract
Amino acids are fundamental to life as we know them as the monomers of proteins and enzymes. They are also readily synthesized under a variety of plausible prebiotic conditions and are common in carbon-rich meteorites. Thus, they represent a reasonable class of organics to target in the search for prebiotic chemistry or chemical evidence of life on Mars. However, regardless of their origin, amino acids and other organic molecules present in near-surface regolith and rocks on Mars can be degraded by exposure to cosmic rays that can penetrate to a depth of a few meters. We exposed several pure amino acids in dry and hydrated silicate mixtures and in mixtures of silicates with perchlorate salts to gamma radiation at various temperatures and radiation doses representative of the martian near-subsurface. We found that irradiation of amino acids mixed with dry silica powder increased the rate of amino acid radiolysis, with the radiolysis constants of amino acids in silicate mixtures at least a factor of 10 larger compared with the radiolysis constants of amino acids alone. The addition of perchlorate salts to the silicate samples or hydration of silicate samples further accelerated the rate of amino acid destruction during irradiation and increased the radiolysis constants by a factor of ∼1.5. Our results suggest that even low-molecular-weight amino acids could degrade in just ∼20 million years in the top 10 cm of the martian surface regolith and rock, and even faster if the material contains elevated abundances of hydrated silicate minerals or perchlorates. We did not detect evidence of amino acid racemization after gamma radiation exposure of the samples, which indicates that the chirality of some surviving amino acids may still be preserved. Our experimental results suggest serious challenges for the search of ancient amino acids and other potential organic biosignatures in the top 2 m of the martian surface.
Introduction
The search for chemical evidence of extinct or extant life on Mars is one of the primary objectives of the past (Viking 1 and 2), current (MSL—Mars Science Laboratory, Mars 2020), and future (ExoMars, MSR—Mars Sample Return) missions. Life, as we know it, is based on specific small organic molecules including amino acids, carboxylic (fatty) acids, polyols, and nucleobases. Many of these organic compounds can be formed by nonbiological chemical reactions and have been identified in carbonaceous meteorites (Glavin et al., 2018 and references therein). Martian meteoritic dust flux models predict significant delivery (∼up to ∼3 × 106 kg/year) of carbon-rich interplanetary dust particles (Borin et al., 2017) and, assuming an average carbon content of 10 wt % (Flynn, 1996), imply an interplanetary dust particles-borne organic carbon flux of 0.3 × 106 kg/year.
Thus, even if life never arose on Mars, a significant amount of organic carbon, including amino acids and other organic compounds found in meteorites, could have accumulated in the martian regolith and surface rocks over billions of years via exogenous delivery by comets, asteroids, and their fragments. Amino acids continue to be an important target in the search for evidence of both prebiotic chemistry and biochemistry on Mars as their relative distributions and enantiomeric compositions can be used to determine their origins (Glavin et al., 2020).
The presence of a variety of organic molecules in ancient mudstones in Gale crater, Mars, with abundances that range from tens to hundreds of parts per billion (ppb) of chlorobenzenes, dichloroalkanes, thiophenes, and other volatile aliphatic and aromatic hydrocarbons of martian origin was measured by the Sample Analysis at Mars (SAM) gas chromatography mass spectrometry (GCMS) instrument after heating samples to elevated temperatures (Freissinet et al., 2015; Eigenbrode et al., 2018; Szopa et al., 2020). However, no amino acids or fatty acids have yet been detected above the SAM GCMS instrument background after chemical derivatization of scooped aeolian material and a drilled mudstone (Millan et al., 2020). It is possible that the concentrations of these organic compounds in these samples were significantly reduced over time due to radiolysis by ionizing radiation.
Abiotic chemistry is currently the most parsimonious explanation for the origin of the organics that have been identified so far by SAM on Mars since none of the organic molecules detected and distributions observed is unique to terrestrial biology. It is therefore important to continue to search for organic molecules of potential biological relevance on Mars and conduct laboratory experiments to understand how ionizing radiation will change their original abundances, distributions, isotopic compositions, and chirality that will be critical for establishing their origin(s).
The cold and dry martian surface environment, weak hydrologic cycle, and lack of tectonic activity should have been ideal for the preservation of ancient organic molecules in the shallow subsurface rocks and regolith (Bada and McDonald, 1995). However, Mars’ thin atmosphere and lack of a global, strong magnetic field leave the entire martian surface and shallow subsurface poorly protected against ionizing radiation—solar and galactic cosmic rays (SCRs and GCRs) (Kminek and Bada 2006; Dartnell et al., 2007a; Pavlov et al., 2012).
Nevertheless, until recently, long-term exposure to cosmic radiation has not been recognized as a potentially major environmental factor for the preservation of organic compounds in martian surface rocks. Instead, recent and some future life search sampling strategies (MSL, Mars 2020) focus primarily on avoiding sampling martian surfaces that are exposed to ultraviolet (UV) and atmospheric oxidants, as both UV and atmospheric oxidants destroy organic molecules effectively (ten Kate et al., 2005). Although the total energy flux from UV is many orders of magnitude higher than the total energy of the cosmic rays at the martian surface, UV radiation penetration into the subsurface is blocked by the upper ∼1 mm of regolith (Ertem et al., 2017).
Oxidants and other reactive oxygen containing species that include peroxides, superoxide, and O2 − superoxide radical ion (Oyama et al., 1977; Yen et al., 2000; Encrenaz et al., 2004) have been detected or have been inferred to be present on Mars in both the atmosphere and regolith by previous missions. Atmospheric oxidants can penetrate the regolith through diffusion to depths of several centimeters and are estimated to be present at part-per-million levels in the near-surface regolith (Bullock et al., 1994; Zent and McKay, 1994; Yen et al., 2000; Lasne et al., 2016).
Another source of oxidants in the martian surface can come from perchlorate or chlorate salts. Perchlorate ions were directly detected on Mars by the Microscopy, Electrochemistry, and Conductivity Analyzer (MECA) instrument on Phoenix (Hecht et al., 2009), and evidence of the thermal decomposition of perchlorate salts was also found in samples analyzed by the SAM instrument on Curiosity in Gale crater (Glavin et al., 2013; Clark et al., 2021). Perchlorate ions (ClO4 −) by themselves do not react with organic molecules rapidly under Mars ambient temperature and pressure conditions (Urbansky, 1998). However, Quinn et al. (2013) found that, under gamma radiation, calcium perchlorate in a CO2 atmosphere decomposes and forms hypochlorite (ClO−), oxygen (O2), and chlorine dioxide (ClO2). In theory, those radicals could react with amino acids, but the rate of such reactions in the martian regolith and rocks has not been previously quantified.
The effect of UV and oxidants can be extended to tens and hundreds of meters of depths through mixing of the previously oxidized material during impact gardening of the martian surface. Oxidants can also be brought into the regolith to depths greater than several centimeters through mixing of wind-blown dust. However, on the ungardened rock surfaces (the sampling focus of both MSL, Mars 2020, and ExoMars), the effect of UV and oxidants will completely disappear within several cm from the surface.
To improve the chances of finding preserved organic matter that has been minimally exposed to UV radiation and oxidants, sampling of the ungardened ancient sedimentary rocks on Mars at more than a few centimeters of depth is thought to be required. Hence, both the MSL Curiosity and Mars 2020 Perseverance rover drills were designed to collect samples from a depth of ∼5–6 cm (Grotzinger et al., 2012; Farley et al., 2020). However, in contrast to UV radiation, primary cosmic ray particles (mostly protons and helium) will also produce cascades of secondary particles in the regolith (neutrons, gamma rays, energetic electrons, nuclear fragments, etc.), some of which in turn can penetrate effectively up to 2–3 m depth (depending on the rock density) (Dartnell et al., 2007a).
Both primary cosmic rays and secondary particles can alter organic material either directly or by producing oxidative radicals within the rock, which also can destroy or alter ancient organic molecules. Currently, only the ExoMars rover will have the capability to drill down to a 2 m depth for sampling, which may provide access to organic matter that has experienced less alteration by ionizing radiation.
Modeling studies by Pavlov et al. (2012) found that organic molecules with masses >300 Da would be degraded by cosmic rays in <300 million years in the top 5 cm of the martian surface, but smaller molecules with masses <100 Da might survive over 1 billion years. Based on the abundance of cosmogenic nuclides (3He, 21Ne, 36Ar), Farley et al. (2014) reported an average cosmic ray exposure age of the Cumberland mudstone sample in Gale crater of ∼80 million years. Similar noble gas measurements released from another mudstone in Gale crater called Mojave yielded a much longer cosmic ray exposure age estimate of ∼300 Myr to <1 Ga (Martin et al., 2017; Cohen et al., 2019).
Farley et al. (2014) also suggested that the Cumberland mudstone remained buried for billions of years and was exposed only “recently” by wind-driven scarp retreat in Yellowknife Bay. These cosmic ray exposure age results might seem promising for the prospects of detecting lower molecular weight (<300 Da) organic compounds in near-surface martian sediments. Pavlov et al. (2012) calculated the fraction of surviving organic molecules as a function of total ionizing radiation dose using radiolysis constants derived from gamma irradiation exposure of pure dry amino acid mixtures (Kminek and Bada, 2006). However, it is not clear whether the pure amino acid radiolysis constants derived from the Kminek and Bada (2006) experiments are an accurate representation of the actual degradation rates of amino acids in martian near-surface materials since ionizing radiation interactions with minerals will lead to the production of additional reactive O-containing species that could also degrade amino acids.
For example, Bonner et al. (1985) reported much higher gamma radiation degradation rates of solid L-leucine deposited on the clay minerals kaolin and bentonite and in aqueous solution compared with pure dry leucine. However, Bonner's work was never expanded to include other amino acids and was not conducted under martian-like conditions (e.g., cold temperatures and low atmospheric pressures).
In this study, we conducted several sets of irradiation experiments of a suite of amino acids and determined the effects of fused silica, water, perchlorate salts, and temperature on the individual amino acid degradation rates during gamma irradiation as an analog for cosmic ray exposure of amino acids on Mars.
Materials and Methods
Sample preparation procedure
Stock solutions of individual amino acids and mixtures (1 × 10−3 M) were prepared by dissolving solid amino acid standards (97–99% purity from Sigma-Aldrich) in Millipore Direct Integral 10 (18.2 MΩ, < 3 ppb total organic carbon) ultrapure water. All samples were prepared in 13-mm-diameter borosilicate glass test tubes. All glass tubes were wrapped in aluminum foil and then heated in a furnace at 500°C in air overnight before addition of samples. All samples were prepared in an ISO 5 High Efficiency Particulate Air laminar flow bench. Each set of experiments also had a procedural blank prepared in parallel.
Each glass tube was connected via a stainless steel 1/2 inch Swagelok Ultra-Torr fitting to a vacuum glass manifold with a liquid nitrogen trap and a turbo molecular drag pump. To reduce adsorbed water and oxygen in the sample tubes, each tube was evacuated until the pressure inside the tube reached ∼20 m Torr and then flame sealed with an oxy-propane torch. For the tubes that contained liquid water, the tubes were first frozen in liquid nitrogen before pumping and subjected to three freeze-pump-thaw cycles to remove dissolved air from the samples before flame sealing.
Irradiation
We used gamma irradiation as a proxy for cosmic ray exposure on the martian surface as has been done in previous studies (e.g., Bonner et al., 1985; Kminek and Bada, 2006; Quinn et al., 2013; Ertem et al., 2021).
The flame-sealed sample tubes were irradiated with gamma rays (∼1 MeV) from a 60Co source at a rate of 1.8 Gray (Gy)/s at the Radiation Science & Engineering Center (RSEC) facility at Pennsylvania State University and at the NASA Goddard Space Flight Center (GSFC) Radiation Effects Facility (REF). Two facilities were used in this study for scheduling reasons. During irradiation, the temperature of the samples was either at room temperature (approximately +20°C) or at “cold” temperatures (−50°C to −55°C), when samples were cooled by dry ice during irradiation. Note that the term “room” temperature means that the laboratory room's thermostat was set at +20°C, but no direct temperature measurements of the samples in the sealed tubes were conducted during irradiation. In the “cold” experiments where the samples were cooled with dry ice, the temperature of the irradiated sample tubes was recorded throughout the irradiation run.
Samples were exposed to total accumulated gamma doses of 0.5, 1.0, 2.0, and 4.0 MGy. The first three dosages were chosen for a direct comparison with the doses used in the prior study by Kminek and Bada (2006): 0.5, 1.0, and 2.0 MGy. A few sample sets were irradiated with a lower total dose of 0.2 MGy since amino acids in those samples degraded very rapidly, even by 0.5 MGy. Some samples were exposed to a higher gamma dose of 4.0 MGy based on modeling estimates of the expected dosages at the “freshly” exposed martian outcrop in Gale crater and the measured cosmic ray exposure age of the Cumberland mudstone.
A list of sample variables in this study is shown in Table 1.
Experimental Parameters and Variables Included in This Study.
Experimental Parameters and Variables Included in This Study.
Included L-serine, glycine, L-alanine, L-isovaline, α-aminoisobutyric acid, and 8-amino octanoic acid.
Samples were prepared at room temperature and then sealed under vacuum. Three sample sets were prepared as follows: • Amino acid mixture samples were prepared by adding 200 μL of the stock amino acid mixture solution (1 × 10−3 M each of L-serine, glycine, L-alanine, L-isovaline, α-aminoisobutyric acid, and 8-amino octanoic acid) to each test tube and drying under vacuum. • For fused silica material, we used fused silica FS-120 manufactured by H.P. Technical Ceramics (Sheffield, United Kingdom). Solid FS-120 was crushed with a porcelain mortar and pestle. Powdered FS-120 was passed through a 150 μm sieve and then baked at 500°C in air overnight to remove organic contaminants. • Amino acid mixture and fused silica samples were prepared by adding the stock amino acid solution (200 μL) to fused silica powder (∼1.2 g) and drying the mixture under vacuum. Amino acid mixture and fused silica plus water samples were prepared by adding the amino acid mixture (200 μL) to fused silica (∼1.2 g) and drying under vacuum, followed by the addition of 10 wt % water (∼0.1 mL of H2O). The water-containing samples were sealed, frozen, and stored under liquid nitrogen until they were placed in dry ice. Samples were irradiated at 0.2, 0.5, 1.0, and 2.0 MGy. Procedural controls for each sample set were prepared and analyzed in parallel and were not exposed to gamma radiation (0 MGy). Supplementary Figure S1 shows placement of the flame-sealed tubes with sample mixtures in a specially designed insulated cylindrical Dewar. The sealed sample tubes were irradiated in a vertical position in the Gamma Cell 220 60Co irradiator (RSEC). To maintain cold temperature during the irradiation, dry ice was replaced daily in the middle of the Dewar. The duration of the irradiation to reach the necessary dosages was adjusted to take into account partial shielding of samples by the Dewar itself and by dry ice (in the “cold” temperature runs). Two thermocouples were placed inside the Dewar at the top of the sample tubes and recorded temperatures of −50°C to −55°C during irradiation.
After the desired total dosage was achieved, samples were removed from the Dewar and placed in a −20°C freezer at RSEC. Once all samples were irradiated to the specified dosages, all samples were transported to NASA GSFC in a YETI haul hard cooler filled with dry CO2 ice. At GSFC, the samples were stored in a −80°C freezer until the tubes were opened for analysis. No attempt was made to analyze the volatile composition of the sample tube headspace after irradiation.
Irradiation (up to 2 MGy) performed at Penn State RSEC at room temperature
Four sample sets were prepared as follows:
• Individual glycine samples were prepared by adding 200 μL of a stock glycine solution (1 × 10−3 M) to each test tube and drying under vacuum.
• Glycine and fused silica samples were prepared by adding the stock amino acid solution (200 μL) to fused silica (∼1.2 g) and drying under vacuum.
• Glycine and fused silica samples were prepared by adding the stock glycine solution (200 μL) to fused silica (∼1.2 g) and drying under vacuum.
• Amino acid mixture and fused silica and perchlorate samples were prepared by vacuum drying solid magnesium perchlorate overnight to remove excess moisture before weighing, and then dissolving 0.1496 g of magnesium perchlorate in 6 mL of ultrapure water. A 1 mL aliquot of this solution was added to each tube along with 200 μL of the amino acid mixture and ∼1.2 g of fused silica.
The samples were dried and the powder vortexed. Samples were irradiated at 0.2, 0.5, 1.0, and 2.0 MGy. Procedural controls that were not exposed to gamma radiation (0 MGy) were processed in parallel with irradiated samples.
Irradiation (up to 4 MGy) at Penn State RSEC at room temperature
Room temperature irradiation experiments irradiated up to 4 MGy samples with solid individual amino acids were conducted to directly compare results with those of Kminek and Bada (2006). These sample sets were prepared at room temperature as follows:
• The individual glycine samples were prepared by placing ∼100 mg of solid glycine standard in each test tube.
• The individual L-alanine samples were prepared by placing ∼100 mg of solid L-alanine standard in each test tube.
• The individual L-isovaline samples were prepared by placing ∼10 mg of solid L-isovaline standard in each test tube.
The amino acids plus fused silica mixture samples were irradiated at 1, 2, 3, and 4 MGy. Procedural controls that were not exposed to gamma radiation (0 MGy) were processed in parallel.
Irradiation (up to 2 MGy) at NASA's GSFC REF at room temperature
Samples were prepared at room temperature and sealed under vacuum. Amino acid mixture samples were prepared by adding 200 μL of the stock amino acid mixture solution (1 × 10−3 M each of L-serine, glycine, L-alanine, L-isovaline, α-aminoisobutyric acid, and 8-amino octanoic acid) to each test tube and drying under vacuum.
Sample extraction and analytical procedures
Sample extraction
After irradiation, each test tube was tested for leaks by placing the sealed samples in a 20 mm test tube that was filled with water and checked for air bubbles. Approximately 2% of the flame-sealed sample tubes cracked during irradiation/loading/transportation and were discarded before analysis. Intact tubes were then cracked open, and the samples were processed as described below.
The amino acid mixture samples were extracted by first reconstituting the sample in 200 μL of water. Then 100 μL of the solution was diluted with 900 μL of water and sealed and heated at 100°C for 24 h. The extract was then placed in a 10 mm test tube and dried under vacuum.
The solid individual amino acid samples were extracted by first dissolving the sample in 200 μL, and then 50 μL was diluted in 950 μL of water and heated at 100°C for 24 h. For the other individual amino acid samples, ∼10 mg out of each irradiated sample was extracted in 1 mL of water at 100°C for 24 h. After extraction, an aliquot was taken from each sample and placed in a 10 mm test tube and dried under vacuum.
The amino acid mixture and fused silica samples were extracted by first transferring the sample (∼0.6 g out of 1.2 g) to a vial with 1 mL of water and extracting at 100°C for 24 h. The samples were then centrifuged for 5 min at 3000 rpm. A 100 μL portion of the supernatant (100 μL) was transferred to a 10 mm test tube and dried under vacuum.
The amino acid mixture and fused silica and water samples were first dried under vacuum overnight. A portion (∼0.6 g out of 1.2 g) of the dried sample was then transferred to a vial with 1 mL of water and extracted at 100°C for 24 h. The samples were then centrifuged for 5 min at 3000 rpm. A 100 μL portion of the supernatant was transferred to a 10 mm test tube and dried under vacuum.
The amino acid mixture and fused silica and perchlorate samples (∼0.6 g out of 1.2 g) were transferred to an extraction vial with 1 mL of water and extracted at 100°C for 24 h. The samples were then centrifuged for 5 min at 3000 rpm. A 100 μL portion of the supernatant was transferred to a 10 mm test tube and dried under vacuum.
Hydrolysis and desalting
After extraction and dry-down, the samples were vapor hydrolyzed by the same procedure outlined in the work of Glavin et al. (2006) to allow analysis of the total hydrolyzed amino acid content. The samples were acid hydrolyzed under 6 M HCl vapor by heating at 150°C for 3 h. The acid-hydrolyzed, hot-water extracts were then desalted by using a cation-exchange resin (AG50W-X8, 100–200 mesh, hydrogen form; Bio-Rad), and the amino acids were recovered by elution with 2 M NH4OH [prepared from ultrapure water and NH3(g) (AirProducts) in vacuo].
Based on previously published results (Glavin et al., 2010), amino acid standards taken through the same hot water extraction, acid vapor hydrolysis, and desalting procedure showed no evidence of significant decomposition, racemization, thermal degradation, or carbon isotopic fractionation.
Derivatization and analysis
Two derivatization methods were used for analysis by liquid chromatography fluorescence detection time-of-flight mass spectrometry (LC-FD/ToF-MS). We used o-phthaldialdehyde/N-acetyl-L-cysteine (OPA/NAC) to separate amino acid enantiomers (e.g., Glavin et al., 2006), elucidate amino acid chirality, and investigate potential radioracemization during irradiation. OPA/NAC derivatization, however, is sensitive to the presence of salts, requires long (∼60 min) liquid chromatographic run times, and produces amino acid derivatives that rapidly degrade at room temperature. In contrast, derivatization with Waters AccQ•Tag (6-aminoquinolyl-N-hydroxysuccinimidyl carbamate) is much less sensitive to the presence of salts (Cohen and Michaud, 1993), allows for faster chromatographic runs of ∼10 min (Boogers et al., 2008 vs. Glavin et al., 2020, respectively), and has more stable derivatives. This allows for a sequence of many derivatized samples and calibration standards to be run automatically.
However, LC-FD/ToF-MS analyses of AccQ•Tag amino acid derivatives are less sensitive than OPA/NAC derivatives, and amino acid enantiomers cannot be separated after AccQ•Tag derivatization (Simkus et al., 2019). As a result, we typically use OPA/NAC derivatization for the analysis of samples for which high sensitivity and/or enantiomeric resolution are required. We typically use AccQ•Tag when chiral resolution is not required (Dworkin et al., 2018), when the samples contain large amounts of salts or other compounds, or when expediency is required.
OPA/NAC derivatization and analysis
Except as noted below, all reagents were purchased from Sigma-Aldrich. A stock amino acid standard solution (1 × 10−6 M) was prepared by mixing individual amino acid standards (97–99% purity) in Millipore Milli-Q Integral 10 (18.2 MΩ·cm, <3 ppb total organic carbon) ultrapure water (Figs. 1 and 2). The OPA/NAC reagent used for amino acid derivatization for the LC-FD/ToF-MS measurements was prepared by combining 300 μL of 0.1 M OPA (0.1 g OPA in 7.5 mL Optima Grade methanol), 670 μL of 0.1 M sodium borate buffer (pH 9), and 30 μL of 1 M NAC (0.408 g NAC in 5 mL of ultrapure water). A 0.1 M hydrazine (NH2NH2) solution was prepared by vacuum distillation of concentrated anhydrous hydrazine (98% purity) and subsequent dilution in ultrapure water. The HCl was quadruple-distilled, and the ammonium formate buffer used in the LC-FD/ToF-MS analyses was prepared by ammonia titration (95% purity) of a 50 mM formic acid solution to pH 8 and the addition of 64 μL of Optima Grade methanol (Glavin et al., 2010).
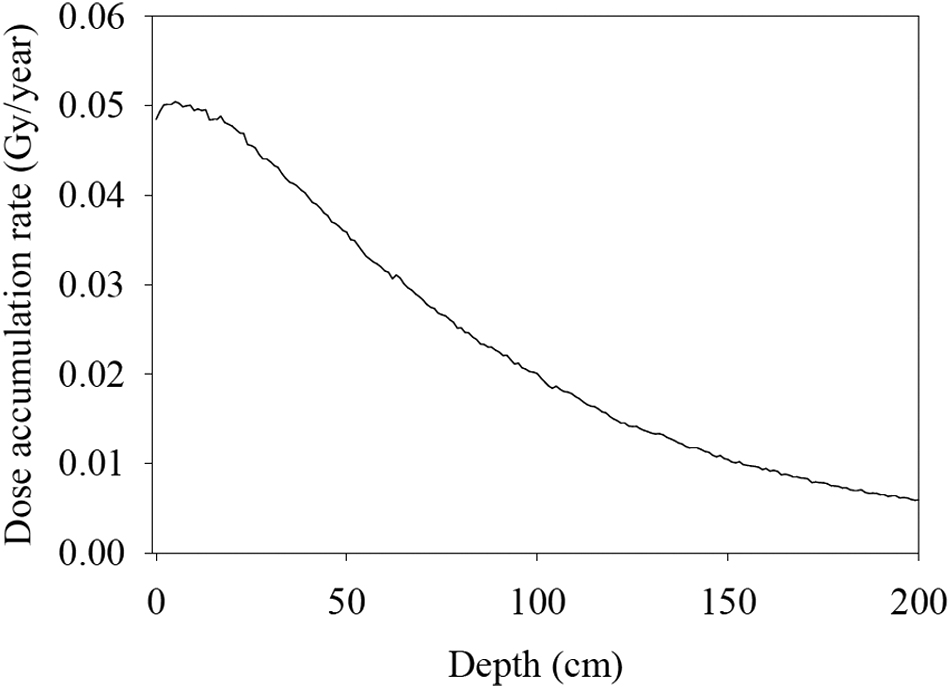
Radiation dose accumulation rate versus rock depth at the Cumberland mudstone drill location. In this plot, the atmosphere above the rock was assumed to be 7 mbar of CO2. Rock density was set at 2 g/cm3. Elemental rock composition was taken from Farley et al. (2014). This simulation assumed exposure of the martian surface rocks to GCRs only because our primary concern is a depth of 5–6 cm—MSL and Mars 2020 maximum sampling depths. However, as pointed out in Pavlov et al. (2012), the SCRs’ contribution into exposure is dominant in the top 2 cm. GCRs, galactic cosmic rays; MSL, Mars Science Laboratory; SCRs, solar cosmic rays.
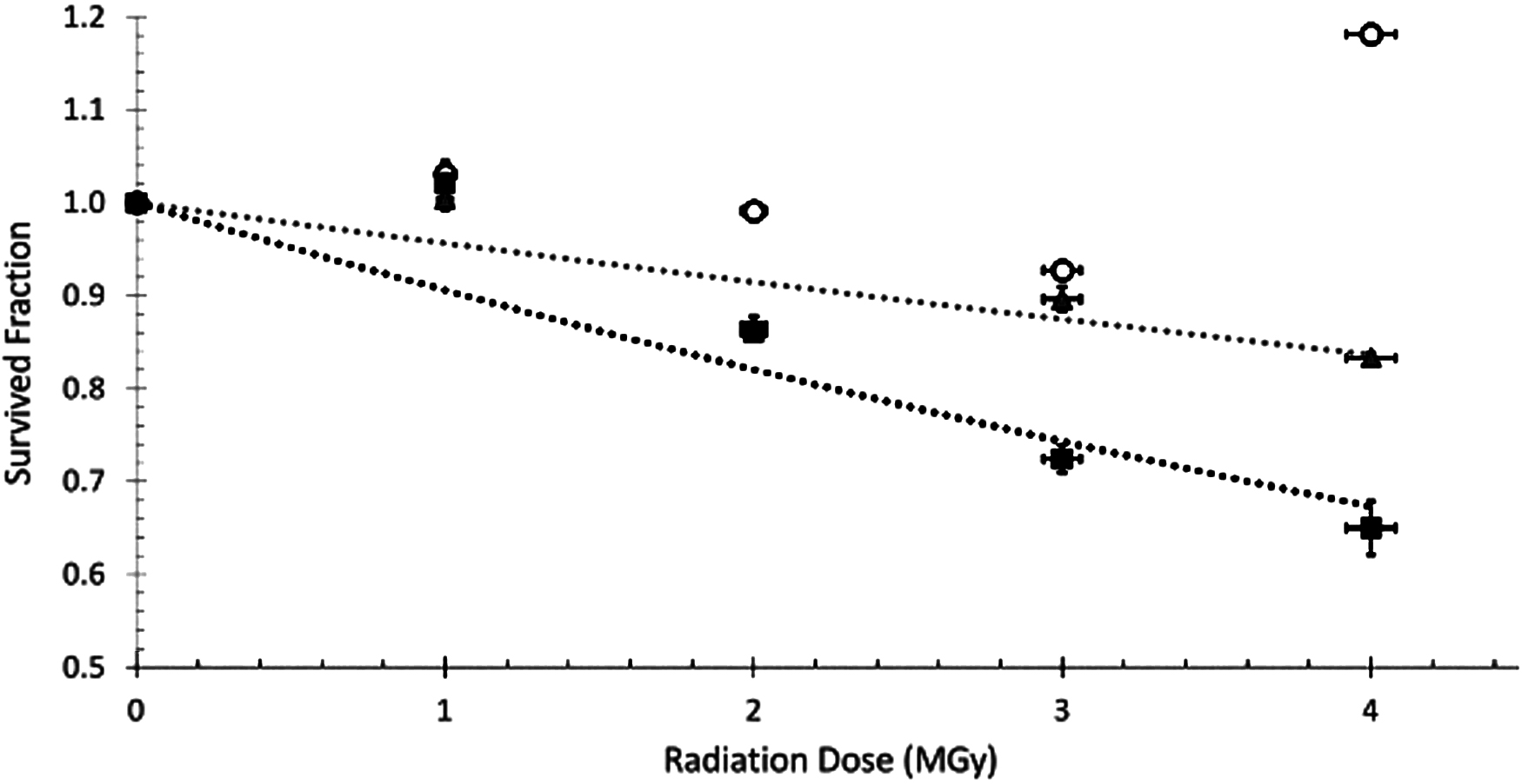
Irradiation of pure dry amino acids at room temperature. Glycine (circles), alanine (triangles, gray fitted line), and isovaline (squares, black fitted line). Small radiation dose errors (horizontal bars on data points) are due to ±2% uncertainty in radiation dose rate in the radiation source. Survived fraction errors are derived from the standard deviation of three injections. All reported survived fraction values in all graphs are blank subtracted. The data point for glycine at 4 MGy could be due to contamination or sample workup efficiency. Errors of 5–10% have been observed based on sample workup efficiency. Note that our control (unirradiated in the gamma source) samples were still exposed to Earth's radiation background of ∼2 × 10−3 Gy/year during preparation and storage.
The samples were analyzed on an Acquity H-Class Xevo G2 XS time-of-flight mass spectrometer (Xevo G2 XS) and/or an Acquity Classic LCT Premier time-of-flight mass spectrometer (LCTP) both equipped with an electrospray ionization source in a positive mode. Separation of amino acids was accomplished by injecting 10 μL of the derivatized sample onto an Acquity UPLC BEH C18, 50 × 2.1 mm column (1.7 μm particle size) with an Acquity UPLC BEH Phenyl, 150 × 2.1 mm column (1.7 μm particle size) maintained at 25°C.
Chromatographic separation was achieved using 50 mM of ammonium formate buffer (8% methanol, pH 8) as eluant A and 100% methanol as eluant B. Analytes were eluted using a flow rate of 150 μL/min and the following gradients: 0.0 min (0% B), 35.0 min (55% B), 45.0 min (100% B), 50.0 min (100% B), 50.1 min (0% B), and 55.0 min (0% B). The Waters Acquity UHPLC was equipped with a fluorescence detector set to λexcitation = 340 nm and λemission = 450 nm.
The OPA/NAC measurements were conducted using the Xevo G2 XS with an electrospray capillary voltage set to 1.2 kV, sample cone voltage set to 30 V, source temperature set to 120°C, cone gas flow set to 50 L/h, desolvation temperature set to 500°C, and desolvation gas flow set to 800 L/h. The ToF-MS analyzer was operated in a V-optics mode, which used a reflectron to achieve a full-width at half-maximum resolution of 22,000 based on the mass-to-charge ratio (m/z) value of leucine enkephalin. The m/z range over which data were collected was 100–600. A mass tolerance of 10 ppm was implemented for accurate mass identification. For Ultra High Performance Liquid Chromatography analysis, a 100 μL syringe and 15 μL needle were used.
The OPA/NAC measurements were conducted using the LCT Premier with an electrospray capillary voltage set to 3800 V, a sample cone voltage of 30 V, a source temperature of 120°C, a cone gas flow of 70 L/h, a desolvation temperature of 250°C, and a desolvation gas flow of 700 L/h. The ToF-MS was calibrated over the 50–1200 m/z range. The ToF-MS analyzer was operated in the V-optics mode, with a maximum resolution of 5000. For UHPLC analysis, a 250 μL syringe, 50 μL loop, and 15 μL needle were used.
Samples were derivatized with AccQ•Tag reagents according to the Waters manufacturer's protocol (Boogers et al., 2008,). Briefly, 10 μL of either a standard or a sample extract mix solution was mixed with 70 μL of AccQ•Tag Ultra borate buffer, and 20 μL of AccQ•Tag reagent previously dissolved in 1.0 mL of AccQ•Tag Ultra reagent diluent was added. The reaction was allowed to proceed for 10 min at 55°C. The UHPLC solvents were prepared according to the Waters manufacturer's protocol.
The samples were analyzed via the commercial Waters AccQ•Tag protocol on the Xevo G2 XS and/or an LCTP described above. Separation of amino acids was accomplished by injecting 1 μL of the AccQ•Tag derivatized sample onto an Acquity AccQ•Tag Ultra C18, 150 × 2.1 mm column (1.7 μm particle size) maintained at 55°C. Chromatographic separation was achieved using 100 μL of the AccQ•Tag concentrate A with 900 μL of ultrapure water as eluant A and Waters AccQ•Tag B as eluant B. Analytes were eluted using a flow rate of 700 μL/min and the following gradients: 0.00 min (0.1% B), 0.54 min (0.1% B), 5.74 min (10.0% B), 7.74 min (21.2% B), 8.04 min (59.6% B), 8.64 min (59.6% B), 8.73 min (0.1% B), and 10.00 min (0.1% B). The Waters Acquity UHPLC was equipped with a fluorescence detector set to λexcitation = 266 nm and λemission = 473 nm.
AccQ•Tag measurements were conducted using a Xevo G2 XS with the electrospray capillary voltage set to 1.2 kV, the sample cone to 40 V, the source temperature to 120°C, the cone gas flow to 70 L/h, the desolvation temperature to 500°C, and the desolvation gas flow to 1000 L/h. The ToF-MS analyzer was operated in the V-optics mode, which used a reflectron to achieve a full-width at half-maximum resolution of 22,000 based on the m/z value of leucine enkephalin. The m/z range over which data were collected was 100–600. A mass tolerance of 10 ppm was implemented for accurate mass identification. For UHPLC analysis, a 100 μL syringe and 15 μL needle were used.
The AccQ•Tag measurements were conducted using the LCT Premier and an electrospray capillary voltage set to 3500 V, a sample cone voltage of 50 V, a source temperature of 120°C, a cone gas flow of 70 L/h, a desolvation temperature of 500°C, and a desolvation gas flow of 1000 L/h. The ToF-MS was calibrated over the 50–1200 m/z range. The ToF-MS analyzer was operated in the V-optics mode, with a maximum resolution of 5000. For UHPLC analysis, a 250 μL syringe, 50 μL loop, and 15 μL needle were used.
Selected ion traces were quantified. A linear least-square model was fit to each proteinogenic amino acid in the standard calibration set, and these calibration curves were used to quantify the analytes in the samples. For the nonproteinogenic amino acids, the relative instrument responses for these analytes compared with alanine were calculated by using calibration standards prepared and analyzed at a later date, and these responses were used together with the alanine calibration curve to quantify these compounds in the samples. A procedural blank (empty sealed glass tube) carried through the same analytical procedures as the samples was used to subtract procedural and laboratory amino acid background from the samples, which were all analyzed in triplicate.
The amino acid concentrations in the procedural blank varied from compound to compound and ranged from 0.001 to 0.1 μM for glycine, serine, and L-alanine (Fig. 9, blank, e.g.). The lower range is below the detection limit for the techniques used in this study. Most of the analyzed samples were spiked with amino acid concentrations in the mM range (>1000 × above background). For some of the samples that degraded when irradiated, the detected amino acid levels dropped to near-background levels, increasing the quantitation error as indicated.
Results
In this study, we first calculated the radiation accumulation rate depth profile (Fig. 1) at the Cumberland mudstone location using the GEANT4 numerical code similarly to its use by Pavlov et al. (2012) but assuming the rocks’ density and elemental composition from the work of Farley et al. (2014). GCR fluxes are modulated by the solar activity and vary during the solar cycle significantly. However, gradual radiolytic changes in the organic abundance in rocks over millions of years are dependent on the average GCR flux rather than instantaneous. Therefore, in the present study, we followed the approach of Pavlov et al. (2014). We assumed a rather conservative integrated GCR flux of 0.77 nucleons/(cm2·s) as the GCR flux in the middle of the solar cycle, year 2006 (Maiorov et al., 2011 see their figure 2) at the Earth's orbit.
The GCR spectrum was taken from the work of Adriani et al. (2011) as it corresponds closely to the mean GCR spectrum and value over the solar cycle, when the PAMELA (Payload for Antimatter Matter Exploration and Light-nuclei Astrophysics) mission was taking measurements. We considered only hydrogen and helium as the primary GCR particles. We did not take into account heavy ions. The energy range for primary particle calculations was from 20 MeV to 10,000 GeV (Adriani et al., 2011). The GCR flux was assumed to be isotropic. To calculate ionization losses, we used GEANT4's BIC (Binary Cascade) model for both protons and helium particles. SCR particles were excluded from these calculations because their contribution is significant only in the top 2 cm of rock as shown by Pavlov et al. (2012).
Based on our calculations, the average radiation accumulation on the million-year timescale in the top 10 cm of the Cumberland mudstone is about 0.05 Gy/year. This rate is about 30% lower than the rate observed by Hassler et al. (2014) at the martian surface during the MSL mission. Given 0.05 Gy/year as an average radiation accumulation rate, the exposure to cosmic rays of 80 Myr at the Cumberland drill location determined by cosmogenic nuclides (Farley et al., 2014) corresponds to ∼4 MGy of gamma ray exposure. Therefore, in all our experiments we kept the maximum sample exposure dosage at 4 MGy.
Radiolytic degradation of pure amino acids
Figure 2 shows the results from the irradiation of pure dry amino acids with gamma rays up to 4 MGy at room temperature. The data in Fig. 2 indicate that there is a relatively minor radiolytic destruction of pure amino acids under these conditions. Glycine did not show any degradation even after 4 MGy exposure. L-alanine showed losses of only up to ∼16.5% over 4 MGy, while L-isovaline had losses of up to 32.7%. The observed degradation rate of dry pure amino acids in our study is not as high as that reported by Kminek and Bada (2006). Specifically, calculations using their radiolysis constants produce expected losses of glycine, alanine, and isovaline of 28%, 36%, and 43%, respectively, after 4 MGy of gamma irradiation. The differences between our observations and those calculations could be due to reactions occurring between the amino acids and air present during the Kminek and Bada irradiation. Kminek and Bada used a nitrogen flow to remove air in the sample but had to remove the nitrogen flow and expose the samples to atmospheric air while sealing the tubes. In contrast, the samples in our study were sealed under vacuum (∼20 mTorr), which eliminated exposure to reactive species present in atmospheric air. Kminek's nitrogen flow would also have been unable to remove water attached to the inner walls of the test tubes, whereas in our sample preparation, any residual water in a test tube was removed during sealing under vacuum (∼20 mTorr). As we demonstrate later in this paper, the presence of small amounts of water can significantly enhance the amino acid degradation rate under gamma irradiation.
We report radiolysis experiment results using the standard exponential equation:
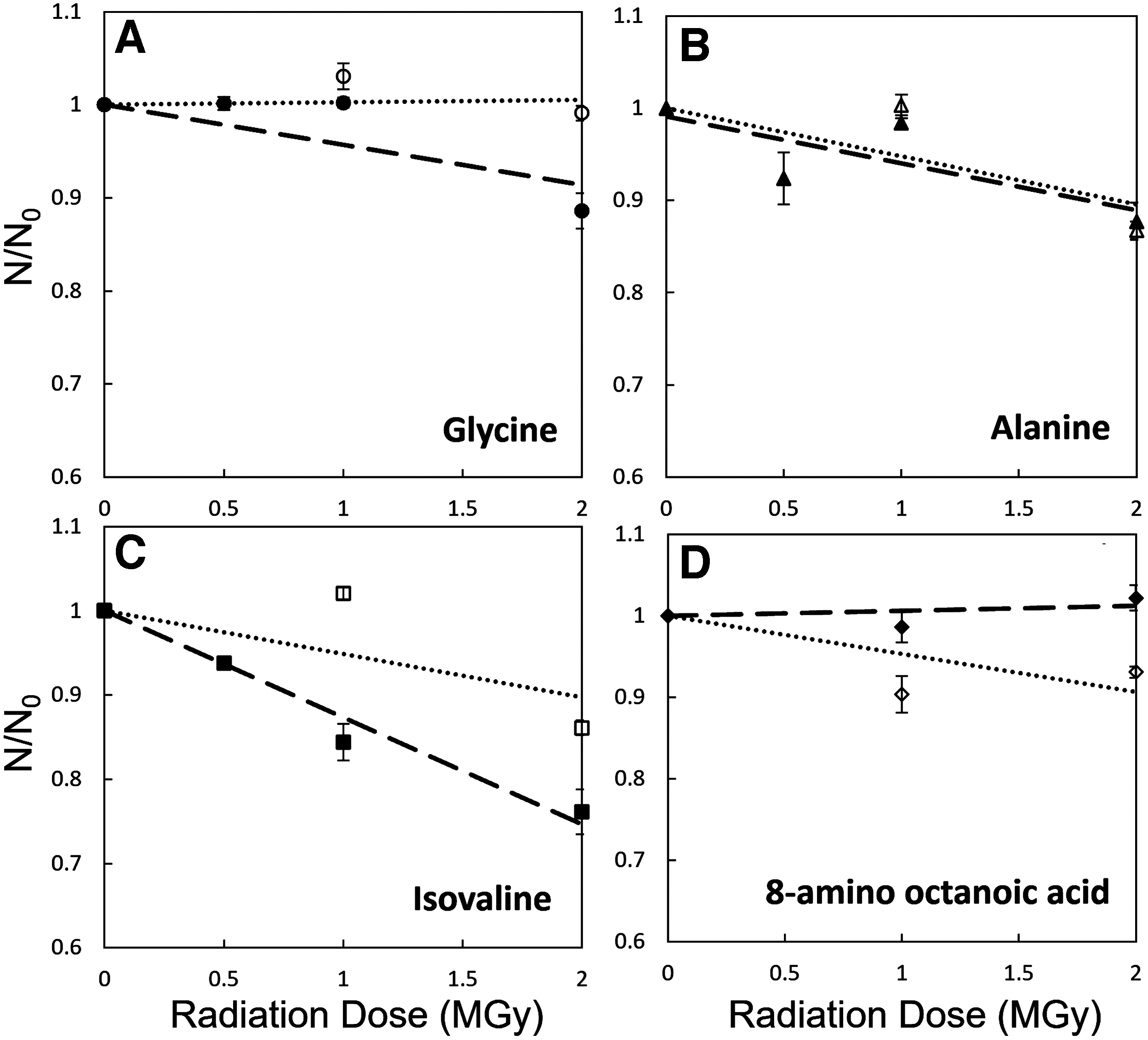
Comparison of degradation of the amino acids in an amino acid mixture versus amino acids irradiated individually. The filled shapes (long dash lines) are individually irradiated amino acids and the nonfilled shapes (dotted line) are the amino acids that were in a mixture. A, B, C, D subpanels - correspond to various amino acids.
Although Fig. 2 shows little degradation of individual amino acids under gamma radiation (even slower than previous studies), it is unrealistic to expect clumps of individual amino acids in any natural samples. Organic matter in both biological samples and particularly in meteorites contains a wide variety of amino acids. Therefore, it is important to determine how individual amino acids would degrade in the presence of other amino acids. The analysis of oligopeptides is beyond the scope of this study and will be discussed elsewhere.
Figure 3 illustrates that the degradation of amino acids when in a mixture (containing glycine, serine, alanine, serine, α-aminobutyric acid, isovaline, and 8-amino octanoic acid) is significantly faster than the degradation of pure individual amino acids. After 2.0 MGy of exposure, isovaline degraded by up to 30% in the amino acid mixture versus ∼18% when isovaline was irradiated in a pure form. Glycine, which did not show any degradation under gamma radiation in the pure form even at 4.0 MGy, did exhibit a loss of ∼15% in the amino acid mixture after only 2.0 MGy of exposure. One possible explanation to such a phenomenon is a partial reformation of amino acids during irradiation. Once cleaved by gamma radiation, an individual amino acid (such as any organic molecule after radiolysis) has some probability to spontaneously reform itself from radiolysis products and radicals. Hence, we report the net loss of amino acids during irradiation: Fnet (degradation) = Fdegradation − Fproduction. If an unirradiated sample is composed of the same molecules (e.g., glycine), then the number of types of radiolysis products would be smaller compared with the number of products from samples with other amino acids present in a mixture.
The larger the number of radiolysis products, the lower the probability to reform a specific initial amino acid molecule in a sample—Fproduction is low. In contrast, Fdegradation likely remains essentially the same—samples have similar total organic content, mass, and no additional oxidants. Thus, the net loss of an amino acid [Fnet (degradation)] for the same radiation dosage should be larger in samples with amino acid mixtures compared with the individual amino acid samples.
It is possible that some amino acids are reformed from amino acid decomposition products generated during the radiation experiments. If so, amino acid racemization would be expected. Figure 9 shows the absence detectable of racemization in the case of fused silica/amino acid samples, and thus, an insignificant amount of amino acid reformation. However, in the case of pure amino acid samples, the destruction and synthesis reaction may be possible, but were not investigated.
Kminek and Bada (2006) determined that the radiolysis constant is linearly proportional to the amino acid weight—the more massive amino acids would have a larger cross section for radiolysis and, thus, will degrade at a faster rate. Our results (Supplementary Fig. S2) suggest that the dependence of radiolysis on the molecular mass is more complicated. Although we did observe that the light amino acids (e.g., glycine [75 Da] and L-alanine [89 Da]) are destroyed at a slower rate than the slightly heavier L-isovaline (105 Da), we also discovered that the heaviest amino acid in the amino acid mixture—8-amino octanoic acid (159 Da)—did not show any degradation (Fig. 3). This inconsistency in degradation based on molecular weight hints that there are other factors to the degradation of individual amino acids more related to structure than just molecular weight. A systematic physical chemistry analysis would be necessary to fully understand these observations.
To determine the effect of an inert silicate surface on the degradation of amino acids, we irradiated fused silica (∼1 g per sample) spiked with trace amounts of amino acid mixture (∼100 ppm) (see Section 2.1). Based on the reported organic molecule abundances on Mars (Freissinet et al., 2015; Eigenbrode et al., 2019; Szopa et al., 2020), silicate samples with ppm levels of organic molecules would be much more representative of martian rocks and regolith than pure amino acids or amino acid mixtures. It was our objective to determine whether the rate of amino acid radiolysis changes in the presence of silica in Mars-like conditions.
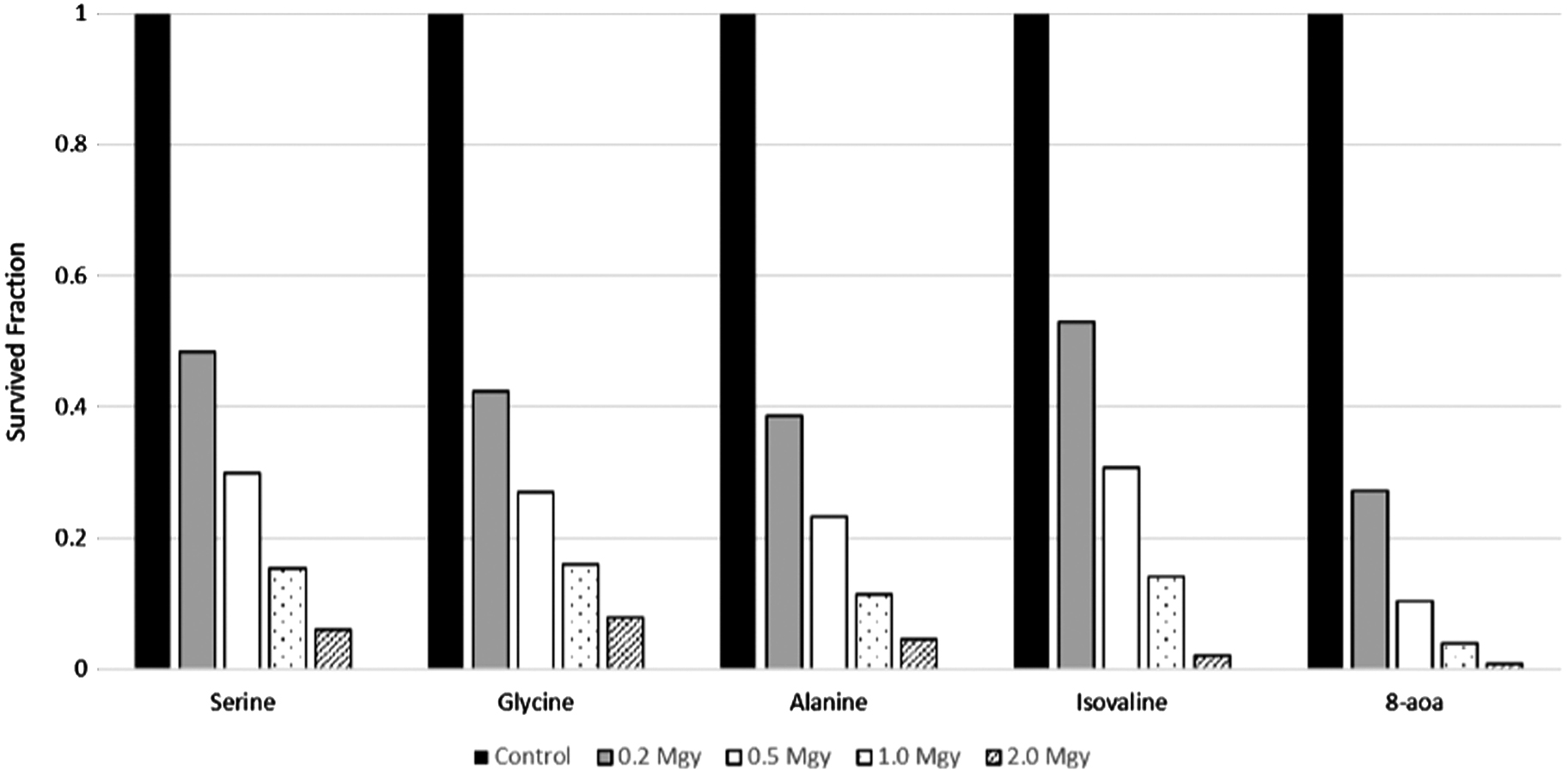
We observed that when amino acids are present in trace amounts in a dry fused silica, the rate of their radiolytic degradation increased dramatically (Fig. 4) compared with the pure amino acid samples. For example, glycine levels in fused silica decreased by 80% after 2 MGy irradiation under −50°C to −55°C temperatures, while glycine in the amino acid mixture under the same temperature conditions decreased only by ∼10% after 2 MGy exposure (Fig. 3).
Irradiation of a mixture of amino acids with fused silica at room temperature.
Figure 4 also demonstrates that the dependence of amino acid radiolysis in silica on the amino acid mass was rather weak. There was no significant increase in the survival of “light” amino acids (e.g., glycine) compared with “heavy” amino acids (e.g., L-isovaline). Instead, under room temperature conditions, <10% of amino acids survived 2 MGy of irradiation regardless of the amino acid molecular weight. We did not include the data from α-aminoisobutyric acid in Fig. 4, since the degradation rates were similar to isovaline.
We examined the effects of temperature on degradation of amino acids when mixed with silica. As Fig. 5 shows, for all dosages, samples irradiated at −50°C to −55°C exhibited less amino acid degradation than samples irradiated at room temperature.

Comparison of degradation of glycine in fused silica mixtures at −50°C to −55°C (black) and room temperature (gray) up to 2 MGy.
Fused silica is an incomplete analog of the sedimentary rocks on Mars and martian regolith. Martian rocks can be hydrated and contain salts. Therefore, we explored the effects of water and perchlorate salts added to fused silica on the rate of amino acid degradation. Figure 6 shows the effect of 10 wt % H2O (by mass) added to fused silica previously spiked with amino acids. This experiment was conducted at −50°C to −55°C to ensure that the added water remained in the frozen state during irradiation. Despite the cold temperature, the rate of amino acid degradation increased significantly with no amino acids (above the detection limit) surviving 1 MGy of exposure. The amino acid mixture with fused silica and water shows rapid and significant degradation. The degradation rates are faster than those observed in mixtures of fused silica and amino acids (Figs. 4 and 5).

Irradiation of amino acids in a mixture with fused silica and water at −50°C to −55°C. Values are blank subtracted.
Similarly, we observed a significant increase in amino acid degradation if, instead of 10 wt % water, we added 2 wt % of dry magnesium perchlorate to fused silica samples (Fig. 7). We found that almost all amino acids do not survive 1 MGy of gamma exposure in fused silica samples with perchlorate. The only amino acid that did survive at ∼10% level of 1 MGy exposure was L-serine (Fig. 7).

Irradiation of amino acids in a mixture with fused silica and perchlorate at room temperature. Values are blank subtracted.
Figure 8 demonstrates the relative efficiency of various factors on the destruction rate of L-isovaline by gamma radiation. Note that L-isovaline was taken as an example—all amino acids studied in our experiment follow the same pattern. Specifically, pure amino acids degrade the least efficiently. An individual amino acid exposed as a part of an amino acid mixture (dry “cell-like” analog) degrades somewhat faster than the pure individual amino acids. A dramatic increase in degradation rate occurs when amino acids in trace concentration are mixed with dry silica. Addition of perchlorate or water accelerates the destruction rate even further.

Isovaline degradation under different irradiation conditions at room temperature: alone (circles), in an amino acid mixture (diamonds), with fused silica (triangles), with fused silica and perchlorates (squares).
The krad values for isovaline for the pure individual amino acids are 0.099 ± 0.012 MGy−1, for the amino acid mixtures are 0.21 ± 0.02 MGy−1, the amino acids and fused silica are 2.08 ± 0.14 MGy−1, and for amino acids and fused silica plus perchlorate are 3.48 ± 0.25 MGy−1. The drastic shifts in krad values indicate that the addition of silica and silica plus perchlorates dramatically increases the rate of radiolytic amino acid degradation under Mars-like temperatures.
Amino acid abundances and enantiomeric ratios were calculated in both the unirradiated controls and the irradiated samples. Figure 9 shows that the amino acid concentration decreases with increased radiation, indicating degradation. The amino acid standard mix contains racemic amino acids. Small amounts of L-serine and L-alanine were observed in the procedural blank. In the 0 MGy (control) amino acid standard mixture LC-FD/ToF-MS selected ion chromatograms (Fig. 9), L-serine, L-alanine, and L-isovaline were observed along with small amounts of D-serine and D-alanine.
The D/L ratios for isovaline could not be determined since a D-isovaline peak was not detected in any of the measurements (Fig. 9), while for serine, the D/L ratios were calculated to be 0.15 ± 0.01 for the 0 MGy (control), 0.16 ± 0.01 for 1 MGy, and 0.11 ± 0.15 for 2 MGy, and the D/L ratios for alanine were 0.06 ± 0.03 for the 0 MGy (control), 0.20 ± 0.05 for 1 MGy, and 0.15 ± 0.22 for 2 MGy. Since D-isovaline was not detected after 1 and 2 MGy gamma ray exposure of the standard, there is no evidence for any racemization of L-isovaline in these experiments. Both the L- and D-alanine and L- and D-serine abundances decreased with increased gamma radiation exposure.
The D/L serine ratio measured in the 1 MGy exposure was identical within error to the serine D/L ratio in the nonirradiated control sample, and therefore, no significant serine racemization occurred during irradiation. However, we did measure a higher D/L alanine ratio outside of error in the 1 MGy exposed standard relative to the control, indicating that some radioracemization of L-alanine to D-alanine may have occurred. However, the racemization rate for alanine is much lower than the observed degradation rate, as has been observed in photodestruction (Ehrenfreund et al., 2001) experiments.
Discussion
We emphasize that our results on amino acid degradation due to exposure to ionizing radiation on Mars should not be confused or misinterpreted with studies of organic matter destruction by ionizing radiation. In the present study, we estimated how long a primordial amino acid would remain as an amino acid under ionizing radiation in the surface rocks on Mars. We did not study the products of amino acid radiolysis. We did not identify a fraction of carbon that is converted by radiation from amino acids to “inorganic” CO and CO2 versus a fraction of carbon that would be transformed from amino acids into some other organic molecules. Therefore, any timescales of degradation reported in this study should not be used to justify the presence or absence of organic molecules on Mars in general.
Hence, there is no controversy that various S- and Cl-containing organic molecules, including chlorinated alkanes, chlorobenzenes, methanethiol, dimethyl sulfide, thiophene, and methyl thiophene, have been detected in Gale crater sedimentary rocks at concentrations ranging from ∼1 to 300 ppb by weight (Freissinet et al., 2015; Eigenbrode et al., 2018; Szopa et al., 2020). It is possible that the observed organic molecules are not primordial but rather products of radiolysis of some more complex refractory organic matter. The irreversible destruction of organic matter by gamma radiation and its conversion to CO2 on Mars have been estimated in a recent article by Vivas et al. (2021).
As expected, it takes a much longer period of time to degrade organic carbon completely rather than just alter amino acid molecules. Specifically, Vivas’ radiolysis constant for total organic destruction was 0.3 MGy−1, which is ∼7 times lower than the radiolysis of amino acids in pure fused silica in our experiments (∼2 MGy−1) (Fig. 8).
Our experimental results suggest that amino acids are destroyed or at least transformed by cosmic rays in the martian surface rocks at a much faster rate than was previously thought. To put things in perspective, we estimated the survived fraction of amino acids at various rock depths in Cumberland mudstone-like ancient rock on Mars (Fig. 10). Based on our experimental estimates of the radiolysis rates and GEANT4 simulations of the radiation accumulation dosage (Fig. 1), pure isovaline and other amino acids would have mostly survived (∼68%) even at the shallowest rock depth of 1 mm to 5 cm depths after 80 million years of exposure. However, the presence of solid pure amino acids is not a realistic Mars analog.

OPA/NAC selected ion chromatograms of gamma-irradiated amino acid mixture plus fused silica samples. The increase in baseline in the L-isovaline 2 MGy chromatogram after elution of the L-isovaline due to an unknown instrument artifact did not impact accurate quantification of this amino acid. OPA/NAC, o-phthaldialdehyde/N-acetyl-L-cysteine.
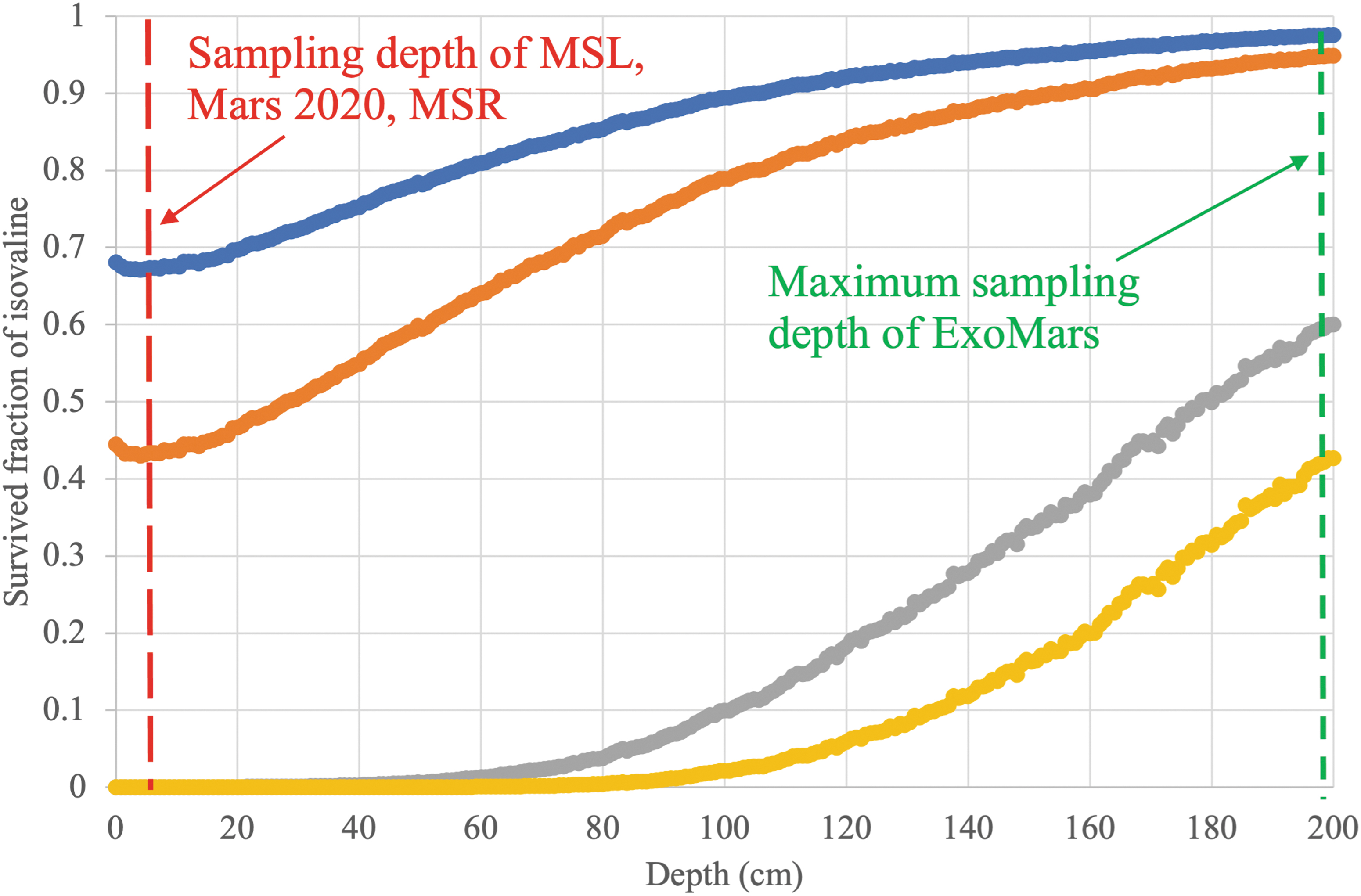
Calculated surviving fraction of isovaline amino acid at various depths in Cumberland-like rock after 80 million years of exposure to GCRs on Mars: alone (blue), in an amino acid mixture (orange), with fused silica (gray), with fused silica and perchlorates (yellow). Ionization rate was taken from Fig. 1 and the radiolysis constants were determined from this study (Fig. 8). Survived fractions of amino acids in the silicate or silicate+perchlorate mixtures are extremely small at depths of MSL, Mars 2020, and MSR sampling (5 cm). In contrast, ExoMars might be able to sample a significant fraction of the ancient amino acids at 2 m depth if the exposure age is 80 Myr. MSR, Mars Sample Return.
A mixture of pure amino acids, which can be expected in an organically rich sample, would have survived as well after 80 million years of exposure (∼45%). However, organic-rich ancient rocks containing amino acids have not yet been found on Mars (Millan et al., 2020). In fact, any reported reduced carbon molecule abundance in martian rocks is in the sub-ppm range. Therefore, we believe that a more realistic analog of organic molecules in martian surface rocks would be fused silica samples (with salts or without) spiked with trace amounts of amino acids. As reported in Section 3, the radiolysis constants for amino acids in such samples are at least an order of magnitude higher than the radiolysis constants of pure amino acids (Fig. 8).
In the context of the Cumberland mudstone, it would mean that the primordial amino acids would not have survived at 0–40 cm depths after 80 million years of exposure at detectable levels (Fig. 10). At 2 m depths, the maximum expected drill depth of the future ExoMars mission, some amount of amino acids will survive, up to 40% if perchlorates are present in silicates and 60% if perchlorates are absent. However, if the exposure at the place of the future ExoMars drill is several times longer than 80 million years (e.g., about 500 Myr), then even a 1.5 m depth would not be sufficient to shield ancient molecules from a very significant radiolytic degradation and transformation. Specifically, in a rock at 2 m depth and the exposure age of 500 million years, the original (primordial) abundance of amino acids would be decreased by a factor 103 if a rock is a silicate and perchlorate-free and by a factor of 105 if a silicate rock at that depth contained 1% or perchlorate.
A useful exposure limit on samples in a future MSR mission may be estimated from amino acid abundances detected in the Robert Massif (RBT) 04262 martian meteorite. This shergottite discovered in Antarctica is ∼225 Myr old (Usui et al., 2010), but the exposure age is only ∼2 Myr (Nishiizumi and Caffee, 2010) determined by cosmogenic nuclides. It means that the meteorite was ejected from martian depths below the penetration of the cosmic rays. Several achiral, straight-chained amino acids of martian origin were identified in RBT 04262 at concentrations ranging from 4 to 130 ppb (Callahan et al., 2013). Thus, 100 ppb might be a reasonable estimate of amino acids in unirradiated igneous martian subsurface rocks.
In the best-case scenario for preservation—when martian silicate rocks are perchlorate-free and have a very low water content, the primordial “unirradiated” 100 ppb of amino acids would be diminished to <0.1 ppb in just 70 million years of exposure at the MSR sampling depths. If amino acid levels drop below 0.1 ppb, then it would be below the current detection limits of amino acids in laboratories on Earth (Callahan et al., 2013). We can only speculate on the possible primordial abundance of amino acids (biological or abiotic) in the martian sedimentary rocks. Hypothetically, if “unirradiated” original levels of amino acids in the preserved martian sediments would have been in the ppms range, then it would still take only 100 million years of exposure to bring the amino acid levels below 0.1 ppb by radiolysis.
Therefore, the search for amino acids in samples at MSL and Mars 2020 sampling depths with >100 Myr exposure may not be a useful chemical indicator of ancient martian life.
Our results have serious implications for the current MSL, Mars 2020, and future MSR, ExoMars missions. If one of the primary objectives for these missions is to detect unaltered ancient organic molecules, then the knowledge of the exposure age at the drill locations is critical before sampling at shallow rock depths to avoid highly radiologically processed material. The Perseverance rover does not have the capability to measure the exposure age of the collected samples in situ. The Curiosity rover had the remarkable capability to measure multiple cosmogenic nuclides in situ (e.g., Farley et al., 2014; Martin et al., 2017; Cohen et al., 2019) and estimated the exposure ages for two different mudstones in Gale crater.
However, due to problems with MSL's drill, the precise calculation of a drilled sample's mass is no longer possible, precluding any reasonable estimates of the cosmic ray exposure age of drilled samples. It would be therefore pivotal to identify “fresh” outcrops (0–50 Myr of exposure) for future MSR samples.
Another interesting implication of our experiments is the effect of hydration of silicates on the radiolysis rates under martian temperatures. It is currently accepted that one of the best types of rocks to look for the ancient organic biomolecules are clays and other phyllosilicates (Bishop et al., 2013; Gil-Lozano et al., 2020). Phyllosilicates are sedimentary rocks that, on Earth, can effectively absorb biological organic molecules (e.g., amino acids) due to their sheet mineral structure. However, phyllosilicates also absorb water and may contain 7–9% of H2O on Mars. Specifically, phyllosilicate-rich regions in Mawrth Vallis and Nili Fossae are observed to have ∼7–9 wt % water, which is an increase of ∼2–4 wt % water compared with the surrounding areas (Milliken et al., 2007).
Our irradiation experiments of the hydrated silica samples (10 wt % water) spiked with amino acids under martian-like temperatures (Fig. 6) indicate that the high content of water in a silicate rock would be worse for organic molecule preservation. More than 90% of the original amino acids would be destroyed in just 10 million years of exposure or less. Therefore, it would be critical to sample essentially unirradiated hydrated phyllosilicates to have a chance to detect primordial biomolecules in them.
Our study has several simplifications, assumptions, and limitations, which should be addressed in future studies.
First, in our experiments, we used gamma ray irradiation as a proxy of the irradiation by cosmic rays. This is a standard approximation, which was used in several previous studies on this subject (Bonner et al., 1985; Kminek and Bada, 2006; Quinn et al., 2013). Incident GCRs and SCRs bombarding the martian surface are mainly energetic protons and He ions (alpha particles). While passing through the atmosphere and a material such as rock, these protons and alpha particles gradually lose their initial energies due to ionization of the target material and generation of secondary protons, neutrons, electrons, gamma rays, and so on with lower energies. The composition of the ionizing radiation (e.g., relative proportions of protons/neutrons/alpha particles/gamma rays and their energy spectrum) changes with rock depth (see figure 6 in Dartnell et al., 2007b). Therefore, organic molecules at various rock depths would experience irradiation by different compositions of energetic particles.
On the contrary, the 60Co gamma source that was used in this study generates exclusively gamma rays with energies ∼1.3 MeV. Gamma irradiation is cheaper than proton and neutron irradiation and does not produce radioactive isotopes in the irradiated material. Although both cosmic rays and gamma rays are energetic, can break any chemical bond in their path, and have a large penetration depth into the rock, it should be emphasized that no comparative studies on the destruction efficiency of various types of organic molecules by various types of ionizing radiation (e.g., gamma, protons, neutrons) have been conducted to date.
We do know that for biological samples and specifically for DNA damage, protons, neutrons, and particularly alpha particles are much more detrimental than gamma. To quantify the effects of various ionizing radiations for biological studies, the concept of relative biological effectiveness (RBE) is introduced (Hendry, 2014). RBE is a relative damage to cells for the same radiation dosage absorbed. RBE of gamma radiation is assigned as a reference to be 1, while proton's RBE is ∼2, and alpha particles’ RBE is ∼20. It means that at least for microorganisms, cosmic rays would produce more damage or produce a pattern of radiation damage more injurious to cell survival (e.g., more double-strand breaks) than gamma irradiation for the same exposure dosage.
Although Blanco et al. (2018) conducted a comparative study of organic degradation by gamma versus electrons in air, we are not aware of such studies for protons versus gamma under vacuum. It makes sense qualitatively that the higher RBE seen for protons compared with gamma rays would apply to organic molecules in general. Gamma rays cleave electrons from the organic molecules, and the organic molecules are degraded during recombination. Protons (unlike gamma rays) have mass and, therefore, besides stripping electrons, can also transfer momentum to the target molecule directly and break it. Therefore, it is plausible that 4.0 MGy of gamma exposure in our experiments may produce less damage to amino acids in our samples than the damage to organic molecules that would be produced by a 4.0 MGy exposure to cosmic rays in martian rocks. In other words, we are likely underestimating the radiolysis constants for Mars cosmic ray exposure.
A future experiment to test the relative efficiency of organic degradation under proton radiation versus gamma radiation is scheduled.
Second, our estimates of the exposure dosage (Fig. 1) are based on a conservative estimate of the radiation accumulation rate. Specifically, Fig. 1 was generated assuming the current GCR flux and the atmospheric column of 7 mbar. As was demonstrated by Pavlov et al. (2012), less energetic SCRs can become the dominant source of radiation dosage in the top 2 cm of martian rocks due to periods of low obliquity when the atmosphere collapses (Armstrong et al., 2004).
Third, our experiments were designed to simulate the radiolytic destruction of the amino acids in shallow martian rocks. We deliberately conducted irradiation of all samples under a very small (∼20 mTorr) atmospheric pressure to avoid formation of extra oxidants from the atmospheric gases during irradiation. Additional oxidants would have added to the amino acid destruction. In other words, our approach represents destruction of amino acids in the subsurface rocks, which are mostly isolated from the martian atmosphere. In contrast, if the irradiation of mineral/organic mixtures occurs in the presence of CO2 (as would be the case in the loose martian regolith), then the rates of organic molecules destruction can be significantly faster than reported here (Ertem et al., 2021).
It is important to note that the gamma radiation exposure rate of ∼1.8 Gy/s (∼57 MGy/year) used in our experiments is significantly higher than the radiation accumulation rate of 0.05 Gy/year in the upper 10 cm of the martian surface. After a passage of a gamma ray through the target material, some molecules in the target will be in excited states. Conceivably, at some high radiation accumulation rate, excited molecules in the sample can become so abundant that they will have a chance to interact with each other before de-excitation.
In such a hypothetical case, the rate of degradation of amino acids can be artificially altered compared with the natural conditions on Mars. However, the radiation rate of 1.8 Gy/s is still orders of magnitude lower than that required for the scenario above. Specifically, by definition, 1.8 Gy/s means that about 1018 ion pairs are produced in a kg of target material per second. A typical lifetime of a molecule in an excited state in a solid body is 10−7–10−8 s or even shorter. Therefore, at any given moment ∼1.8 Gy/s radiation produces ∼1010–1011 excited molecules in a kg of target material. The relative abundance of the excited molecules in an irradiated sample would be ∼10−14 (0.1 ppt). Hence, the probability of any interaction between the excited molecules would be infinitesimal, and the radiation rate of our source is unlikely to artificially increase organic degradation.
Further research is needed to identify the exact mechanism by which the rate of amino acid degradation in dry fused silica increases dramatically compared with the irradiation of pure amino acids. Qualitatively, the mechanism must include the production of oxidative radicals of some sort when amino acids are in silica. It is possible that gamma rays form SiO2 + ions in the immediate vicinity of amino acid, and then the amino acid is degraded during SiO2 + recombination. Alternatively, when a gamma ray particle passes through silica it produces O-radicals. Those radicals can diffuse and attack amino acids.
In both mechanisms, the incident gamma ray photon does not have to hit amino acid molecules directly to cause damage to the amino acid. Thus, the effective cross section of amino acid radiolysis is larger than the geometric size of amino acid, and the overall rate of radiolysis increases. The addition of perchlorate or water to fused silica increases the rate of oxidant production (it is easy to produce O -radicals from perchlorate and OH-radicals from H2O) and, therefore, increases the rate of amino acid degradation even further.
No evidence of significant racemization is observed in the amino acids studied here. This observation is consistent with that of Bonner et al. (1985) with regard to L-leucine radiolytic degradation and racemization. Even though their gamma radiation experiments were conducted under different conditions (e.g., terrestrial atmosphere), they also reported that the radiolysis constants were 10–20 times larger than the radioracemization constants. Slow or even lack of radioracemization can work in favor of life detection on Mars. For example, if the amounts of amino acids in martian rocks or icy permafrost are low due to radiation exposure, and amino acids are still not fully racemic, that by itself might be strong evidence for the biological origin of amino acids.
We show that one potential reason for the lack of detection of complex organic molecules in the martian surface is due to their rapid alteration and degradation by cosmic rays. We conducted gamma irradiation experiments on various amino acids in silicate and hydrated silicate mixtures at various temperatures representative of Mars. We discovered that a broad range of amino acids should be effectively destroyed in just 20 million years of exposure or faster in the martian surface rocks. Based on our experimental results and the measured cosmic ray exposure ages for two martian mudstones in the Gale crater, there should be no primordial amino acids in the top meter at the Cumberland and Mohave drill locations—all ancient amino acids should have been completely destroyed by exposure to cosmic rays millions of years ago.
In general, it would be extremely unlikely to find primordial amino acid molecules in the top 1 m of martian rocks due to their effective radiolysis by cosmic rays regardless of a rock type or location on Mars. Since the current trend of search for “extinct” life through the search of ancient organic molecules will continue in the future (e.g., Mars 2020, ExoMars, MSR), it would be critical either to provide missions with at least 2 m drilling capabilities or select near-surface sampling locations with freshly exposed rocks (ideally with exposure age <10 million years) or rocks that are otherwise protected from radiation (e.g., inside a deep valley). These could include sampling locations near rapidly eroding outcrops or samples from recent impact craters. The future ESA/Roscosmos ExoMars mission will collect samples from up to 2 m depth. ExoMars includes the Mars Organic Molecule Analyzer (MOMA) instrument, which has the capability to detect amino acids and other ancient organic biomarkers. ExoMars will have the chance to sample a significant fraction of unaltered organic material from 2 m depths if the exposure age of rocks is <<500 Myr.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The material is based upon work supported by NASA under award number 80GSFC21M0002 and NASA's Planetary Science Division Internal Scientist Funding Program through the Fundamental Laboratory Research (FLaRe) work package.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Abbreviations Used
Associate Editor: Lewis Dartnell
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.