
Abstract
Thiophene and two derivatives (2-methylthiophene and 3-methylthiophene) have been detected on the surface of Mars with the Sample Analysis at Mars instrument suite onboard NASA’s Curiosity rover. Thiophene could serve as a secondary chemical biosignature since the secondary biosynthesis of thiophene is considered an important production pathway. However, it is critical to understand the abiotic formation and destruction of thiophene and its derivatives since these pathways could affect the molecules’ stabilities on planetary surfaces over geological timescales. Here, we present the radiolytic destruction kinetics of thiophene, 2-methylthiophene, and 3-methylthiophene as single-component ices and when diluted in water ice at low temperatures. Using infrared spectroscopy, we determined the destruction rate constants and extrapolated our radiolytic half-lives to the surface of Mars, assuming the measured and modeled surface dose rates. We found that our rate constants strongly depend on temperature and presence of water ice. Based on our determined radiolytic half-life for thiophene under conditions most similar to those of thiophene groups in Martian macromolecules, we expect thiophene to be stable on the surface for significantly longer than the Martian surface exposure age of sites in Gale crater where thiophenes have been detected.
Introduction
The Martian surface and subsurface contain organic molecules trapped in macromolecular structures embedded in clay-bearing sediments. These organic molecules, in addition to the geological features that suggest the presence of surface and subsurface liquid water, provide mounting evidence of a potentially habitable environment in Mars’ distant past. The identities and chemical pathways (e.g., production and destruction) of these organic molecules directly probe the astrobiological potential of the ancient wet Mars.
Instruments onboard the Mars Science Laboratory’s Curiosity rover identified a variety of organic molecules at various sites in Gale crater, a location selected for its evidence of ancient aqueous activity and potential for astrobiology (Wray, 2013; Vasavada, 2022). Data from the pyrolysis evolved gas analysis (EGA) and gas chromatography-mass spectrometry (GC-MS) instrument suite onboard Curiosity rover revealed the presence of likely indigenous chlorobenzene (Freissinet et al., 2015), which is consistent with a reanalysis of data from the Viking landers (Guzman et al., 2018). More recent efforts using the pyrolysis EGA and GC-MS instruments detected several sulfur-bearing organic molecules, including thiophene and alkylthiophenes (2-methylthiophene and 3-methylthiophene) in samples from lacustrine mudstones in Gale crater (Eigenbrode et al., 2018; Millan et al., 2022).
The structures for thiophene (C4H4S), 2-methylthiophene (C5H6S), and 3-methylthiophene (C5H6S) are shown in Figure 1. Both thiophene and alkylthiophenes are likely indigenous to the Martian surface as components of refractory organic macromolecules, which was inferred from the pyrolysis temperatures required for decomposition of the macromolecules and subsequent thiophene detection. Most interesting, these previous studies suggested that thiophene and alkylthiophenes could serve as preserved secondary chemical biosignatures on Mars due to their importance in the preservation of organic material on Earth. Terrestrially, thiophenes form through reactions between reduced sulfur (e.g., H2S) and various organic materials during the diagenesis of sedimentary material, and both of these “reactants” can be biotic or abiotic in origin (see report by Heinz and Schulze-Makuch, 2020, and references therein). Li et al. (2023) demonstrated that collisions between gas–phase sulfur atoms and conjugated dienes, including 1,3-butadiene and isoprene, might provide additional abiotic pathways to form thiophenes, and these pathways may be important for planetary and interstellar environments.
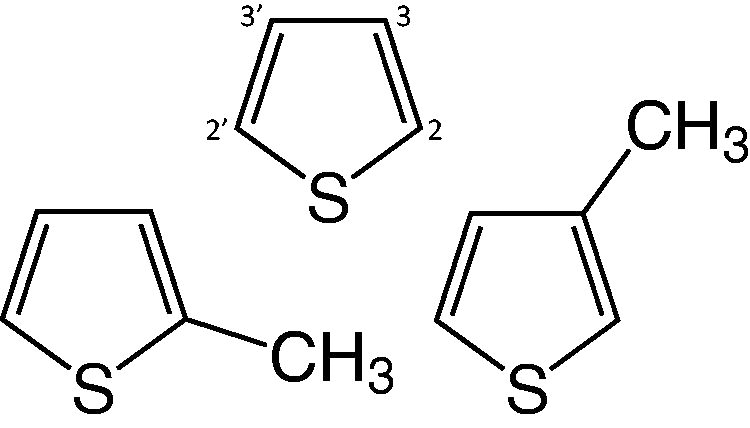
Molecular structures of thiophene (top), 2-methylthiophene (left), and 3-methylthiophene (right).
Unlike Earth, Mars lacks a global magnetic field capable of shielding its surface from radiation, which allows galactic cosmic rays (GCRs) and solar energetic particles to penetrate through the thin CO2 atmosphere (Acuña et al., 1998). Given enough incident energy, these particles penetrate into the regolith, depositing energy in the surface and creating secondary energetic particles that induce chemistry. GCRs provide the dominant source of radiation to the top three meters of the Martian regolith (Hassler et al., 2014). This radiation-driven chemistry decomposes or degrades organic molecules, providing a destruction pathway for molecules that serve as potential biosignatures. Previous laboratory work has demonstrated that biologically important molecules, including amino acids (Gerakines et al., 2012; Gerakines and Hudson, 2013, 2015; da Costa et al., 2021; Mejía et al., 2022) and nucleobases (Materese et al., 2020; Gerakines et al., 2022; Vignoli Muniz et al., 2022; Mejía et al., 2023), decompose when exposed to ionizing radiation (Tolbert and Lemmon, 1955). Thus, to effectively use thiophene or a thiophene derivative as a secondary biosignature, it is critical to understand the effects of radiation on thiophene groups within larger solid macromolecules. Early works examined the effects of radiation on liquid thiophene or aqueous solutions of thiophene and identified many of the radical intermediate species produced (Saunders et al., 1978; Saunders, 1978). However, the rate of radiation-driven destruction of thiophene or of its derivatives in the solid phase, as would be expected on Mars, remains undetermined. These rates provide additional astrochemical context to the recent detections of thiophene and alkylthiophenes on the surface of Mars.
Here, we report the results of laboratory experiments to measure the radiation-induced destruction of thiophene and thiophene derivatives as single-component ices and when mixed with water ice at 20 K and 125 K. We identify the likely radiolytic products and quantify the rates of destruction with infrared (IR) spectroscopy. Finally, we describe the implications of our destruction rates for the Martian surface.
We performed all experiments in a high-vacuum chamber with a base pressure of ∼1 × 10−7 Torr at the lowest temperature studied (Hudson and Ferrante, 2020; Qasim et al., 2022; Materese, 2022). Briefly, we vapor-deposited samples onto a precooled KBr or ZnSe substrate in thermal contact with a helium cryostat. Note that the choice of substrate did not alter the acquired spectra. We produced single-component ices or ice mixtures at 20 K and 125 K. Although these temperatures are considerably lower than the average Martian surface temperature (∼220 K near the equatorial latitudes; Vasavada et al., 2017), thiophene and its derivatives begin to substantially sublimate above 125 K in our vacuum chamber, which limits our experimental temperature. All compounds were degassed by three freeze-pump-thaw cycles with liquid nitrogen to remove excess dissolved gases from the samples prior to vapor deposition. We determined the ice thickness by using the interference fringes of a 670 nm laser near perpendicular to the substrate. For mixtures, we codeposited each compound from independent vapor lines using precalibrated leak valves to achieve a relative ratio of water to thiophene or thiophene derivative of 100:1 (i.e., 100 water molecules for every 1 thiophene or thiophene derivative molecule). This mixture ratio was chosen to isolate the thiophene or thiophene derivative molecules within the matrix and limit interactions between organic molecules during radiolysis while maintaining sufficient IR signal-to-noise to quantify the radiolytic destruction.
We determined the deposition rate of the organic molecules required to produce the desired relative ice composition using
Density and Index of Refraction at 670 nm, and the Calculated Total Stopping Power and Projected Range of 0.9 MeV p+ in Thiophene, 2-Methylthiophene, 3-Methylthiophene, and When Diluted in Water
The density and index of refraction were determined at 18 K.
H2O HPLC Grade, Fisher Chemical.
Narten et al. (1976); Compact Amorphous H2O Ice, 77 K.
Weast et al. (1984).
Values for 100:1 mixtures of water and thiophene and thiophene derivatives, respectively.
After deposition, we irradiated the ice with ∼0.9 MeV protons generated by a Van de Graff accelerator and directed perpendicular to the substrate surface (Loeffler and Hudson, 2018) at a current of
Radiolysis of thiophene and methyl-substituted thiophenes
Figure 2 contains the IR spectrum of a thiophene ice grown at 125 K and the spectrum after each irradiation step. The initial spectrum is consistent with previous low-temperature (130 K and 12 K) spectra of solid thiophene (Quigley et al., 1996; Cesaro et al., 1999). Spectral assignments and drawings of the fundamental vibrational modes for thiophene can be found in the works of Rico et al. (1965) and Christensen et al. (1988), respectively. Note that the initial thiophene features decrease as the radiation dose increases, which indicates the radiation-driven destruction of thiophene. Additionally, we note the emergence of several absorption features as the thiophene was irradiated. Specifically, distinct features appear at 3267, 3225, and 1514 cm−1, as well as a shoulder at 3067 cm−1 (Fig. 2). The features at 3067 and 1514 cm−1 are consistent with the formation of various bithiophenes, including 2,2′-bithiophene, 3,3′-bithiophene, 2,3′-bithiophene, and a-thienothiophene, and polybithiophenes (Wynberg and Bantjes, 1959; Rasch and Vielstich, 1994), however we cannot identify the specific isomer(s) produced. The 3067 cm−1 feature may, in part, also be due to the destruction of the thiophene crystalline lattice at 125 K. The broader features centered at 3267 cm−1 and 3225 cm−1 suggest the formation of alkyne groups (H-C≡C-R). This assignment is consistent with several studies that examined the radiation- and photochemistry of benzene, which is structurally comparable to thiophene since both are fully conjugated planar aromatic molecules. Strazzulla and Baratta (1991) noted the emergence of spectral features at 3288 cm−1 and 3232 cm−1 in irradiated benzene ices and attributed these bands to monosubstituted acetylenes and acetylene, respectively. Ruiterkamp et al. (2005) confirmed these spectral features in irradiated benzene ices and noted that the 3278 cm−1 feature might be due to acetylene aggregates. Qi et al. (1999) exposed collimated molecular beams containing thiophene to a 193 ArF excimer laser and identified several photodissociation pathways that included the formation of thioketene and ethyne. More recently, Kim et al. (2006) examined photolyzed 2,5-diiodothiophene using matrix-isolation spectroscopy and found evidence of ethynylthioketene, a ring-opening product of thiophene that contains an alkyne group. The presence of this pair of alkyne-group features in our irradiated thiophene ices suggests that the thiophene ring might open during radiolysis. Radiation product spectral features at ∼3270, ∼3230, and ∼1515 cm−1 are present for irradiated thiophene and methyl-substituted thiophene samples at both temperatures studied.
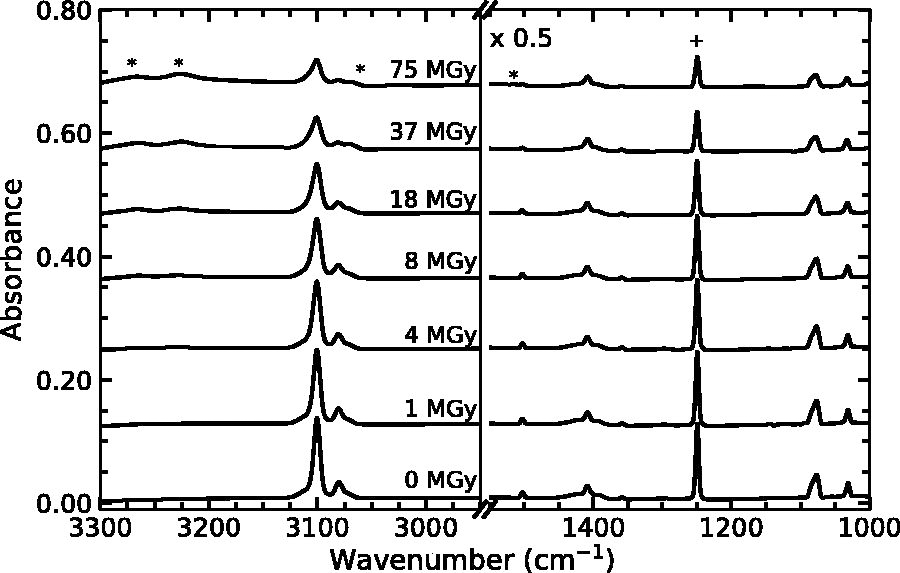
Infrared spectra of thiophene irradiated at 125 K. From top to top, spectra correspond to radiation doses of 0, 1, 4, 8, 18, 37, and 75 MGy. The plus sign and asterisk correspond to the fundamental thiophene band of interest and a radiolytic product, respectively. Spectra are vertically offset for clarity, and the lower wavenumber range spectra (right panel) have been scaled by 0.5 for clarity.
In addition to the radiolytic products mentioned above, we found evidence for radiation-driven amorphization of our ices at 125 K. Previous laboratory and computational studies have demonstrated that liquid thiophene undergoes many stable and metastable crystalline phase transitions between 25 K and 125 K when cooled from room temperature (Miyazaki et al., 2021, and references therein), and differentiation between phases with IR spectroscopy requires higher spectral resolution than 4 cm−1 (Migliorini et al., 1974). Regardless of the identity of the specific crystalline structure, our 125 K samples are crystalline. We expect that our vapor-deposited thiophene and methyl-substituted thiophene samples at 20 K are amorphous, based on observations of ices composed of other aromatic heterocycles (e.g., pyridine; Hudson and Yarnall, 2022).
Figure 3 contains the spectrum of 2-methylthiophene deposited at 125 K (crystalline) before and after irradiation (to a dose of 37 MGy), and the spectra of amorphous unirradiated 2-methylthiophene and 3-methylthiophene deposited at 20 K for comparison. Although most spectral features decrease after a dose of 37 MGy, several features appear (1077, 1044, and 1035 cm−1) or increase (1451 and 1440 cm−1) with dose. These changes are consistent with the appearance of amorphous 2-methylthiophene (Fig. 3; third from the top) and suggest that our high-temperature ices undergo partial amorphization, which is likely incomplete as evident from the residual sharpness of the 3098 cm−1 and 3083 cm−1 bands. We note that these spectral changes are greatest near a dose of 37 MGy and gradually decrease up to 75 MGy (data not shown). Additionally, these changes did not occur in our ices that were deposited and irradiated at 20 K.

Infrared spectra of 2-methylthiophene (2MT) irradiated at 125 K, and unirradiated 2MT and 3-methylthiophene (3MT) for comparison. From the top to the top, spectra correspond to 2MT grown at 125 K, 2MT grown at 125 K and irradiated to a dose of 37 MGy, 2MT grown at 20 K, and 3MT grown at 20 K. Spectra are vertically offset for clarity. Vertical dotted lines indicate spectral features that increase with dose and correspond to reference spectral features.
Figure 4 contains the spectrum of 3-methylthiophene deposited at 125 K (crystalline) before and after irradiation (37 MGy), and the spectra of amorphous unirradiated 3-methylthiophene and 2-methylthiophene deposited at 20 K for comparison. Similar to that described above, the spectrum of 3-methylthiophene deposited at 125 K and irradiated to 37 MGy (second from the top; Fig. 4) resembles the spectrum of 3-methylthiophene deposited at 20 K (third from the top; Fig. 4), which suggests that the sample undergoes radiation-driven amorphization. This process is likely incomplete based on the shape of the 1451 cm−1 band and residual structure near ∼1030 cm−1.

Infrared spectra of 3-methylthiophene (3MT) irradiated at 125 K, and unirradiated 3MT and 2-methylthiophene (2MT) for comparison. From the top to the top, spectra correspond to 3MT grown at 125 K, 3MT grown at 125 K and irradiated to a dose of 37 MGy, 3MT grown at 20 K, and 2MT grown at 20 K. Spectra are vertically offset for clarity. Vertical dotted lines indicate spectral features that increase with dose and correspond to reference spectral features.
In both Figures 3 and 4, we note that several of the changes might in part be due to radiolytic rearrangement, specifically the migration of the methyl group between the second and third carbon on the thiophene ring (Fig. 1). Early studies of aqueous samples containing alkylthiophenes demonstrated the photoisomerization of 2-methylthiophene to 3-methylthiophene (Kellogg et al., 1970). Photoisomerization is also seen for the oxygen-bearing analog alkylfuran in the gas phase (Hiraoka and Srinivasan, 1968). However, for our ices, this isomerization does not seem to be significant since the contribution of the strongest bands of amorphous 2-methylthiophene (1239 cm−1) and 3-methylthiophene (1451 cm−1) in the corresponding irradiated crystalline spectra is minimal (Figs. 3 and 4).
Most important, the amorphization of our 125 K ices impacts the determination of the radiation-driven destruction rate constants since the band strengths of amorphous materials are typically different for the same bands of crystalline material. To estimate the impact of any radiation-induced amorphization, we grew thiophene or methyl-substituted thiophene ices at 20 K, warmed them to 125 K at a rate of 2K min−1, and found that the band of interest (∼1250 cm−1 for thiophene and 2-methylthiophene; ∼1150 cm−1 for 3-methylthiophene) increased in area by ∼15% for thiophene and 3-methylthiophene, and ∼35% for 2-methylthiophene when the ice crystallized. This increase in band area suggests that we might be overestimating the amount of radiolytic destruction for a fraction of material at 125 K that undergoes radiation-induced amorphization (discussed further in Section 4.1).
Figure 5 contains the IR spectrum of thiophene diluted in a water–ice matrix at a ratio of 100:1 grown at 125 K and irradiated with 0.9 MeV p+. Broad absorption features near 1600 cm−1 and 2200 cm−1 dominate the spectrum and correspond to the H2O bending mode and the second overtone of the lattice vibration, respectively (Hardin and Harvey, 1973; Hagen et al., 1981). Two much sharper features at 1407 cm−1 and 1250 cm−1 indicate the presence of thiophene in the ice mixture. These features decrease as the radiation dose increases, which indicates radiation-driven destruction of the thiophene. At high doses, we note the emergence of a band at 2340 cm−1, which we attribute to the formation of CO2. This is consistent with previous laboratory studies that demonstrated that the irradiation of mixtures of H2O and various hydrocarbons (including CH4 and C2H2) produces CO2 (e.g., Moore and Hudson, 1998; Palumbo et al., 1998), and similar hydrocarbons (H-C≡C-R) are expected when thiophene or its derivatives are irradiated as described in Section 3.1. In addition to CO2, a weak band at 2853 cm−1 appears, which indicates a small amount of H2O2. Also, our ice contained a small D2O/HOD contamination, evident from the absorption band at 2422 cm−1 (Hornig et al., 1958); however, D2O/HOD does not influence the observed radiation chemistry.
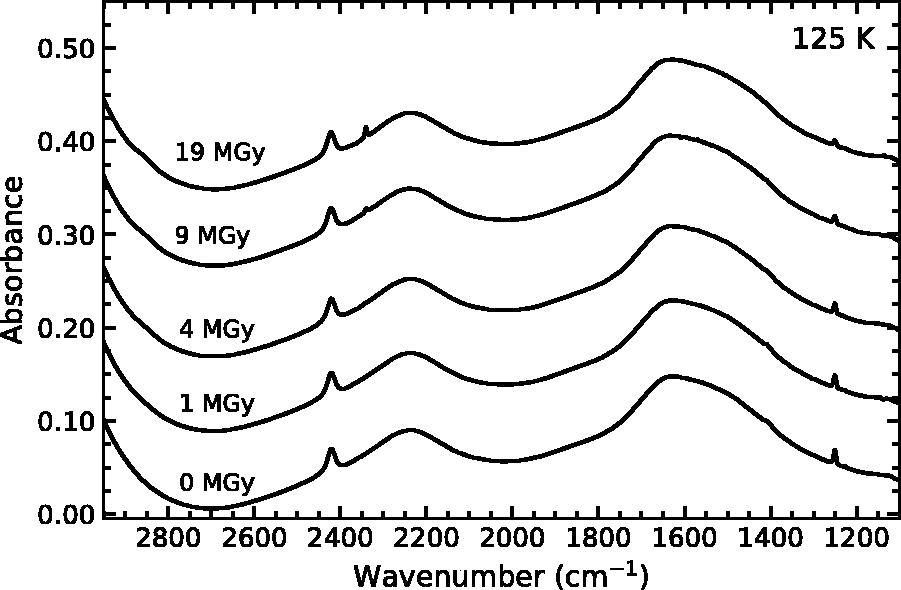
Infrared spectra of a mixture of water ice and thiophene (100:1) irradiated at 125 K. From top to top, spectra correspond to radiation doses of 0, 1, 4, 9, and 19 MGy. Spectra are vertically offset for clarity.
Although each of the thiophene-derivative mixtures produces similar radiolytic products, there were several differences when the initial ice mixture was deposited and irradiated at 20 K. Figure 6 contains the IR spectrum of thiophene diluted in a water–ice matrix at a ratio of 100:1 as deposited at 20 K and after irradiation. In addition to CO2, which is also produced at 125 K, a spectral feature appears at 2137 cm−1, which we attribute to CO, a known radiolytic and photoproduct of CO2 (e.g., Moll et al., 1966; Moore et al., 1991). Any CO formed in the ices at 125 K would be expected to sublimate from the ice. The greater abundance of H2O2 observed in low-temperature H2O ices is consistent with the generally accepted formation mechanism of H2O2 (i.e., the limited diffusivity of OH radicals below 80 K).
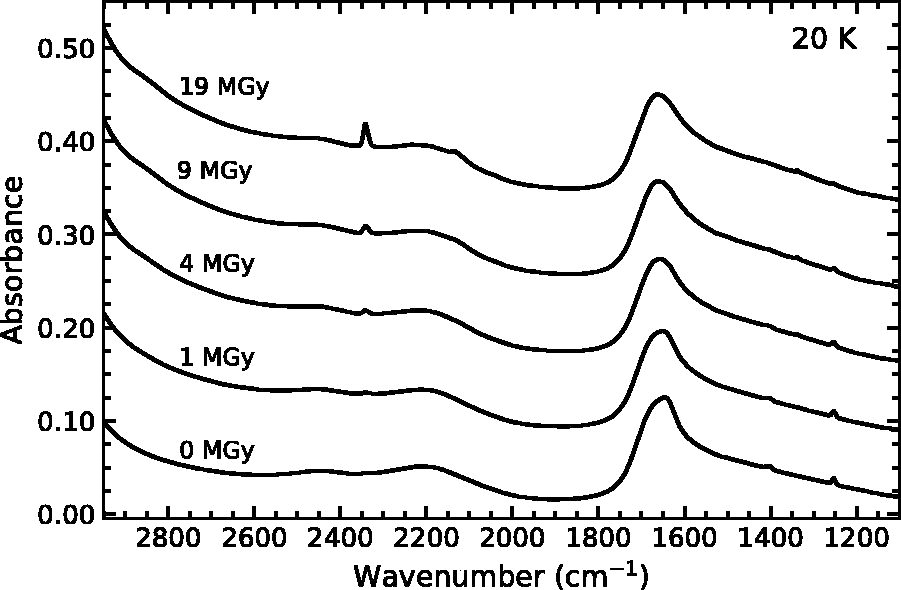
Infrared spectra of a mixture of water ice and thiophene (100:1) irradiated at 20 K. From top to top, spectra correspond to radiation doses of 0, 1, 4, 9, and 19 MGy. Spectra are vertically offset for clarity.
To quantify the destruction kinetics of thiophene and its derivatives, we assume that the radiation-driven destruction follows reversible pseudo first-order kinetics where
Although this is undoubtedly a simplification, first-order kinetics provide reasonable fits to the data, despite the partial amorphization described in Section 3.1. Our group has successfully applied this analysis technique to ices that contain amino acids (Gerakines et al., 2012; Gerakines and Hudson, 2013, 2015) and nucleobases (Materese et al., 2020; Gerakines et al., 2022). Given the forward and reverse reactions, the differential rate law for the destruction of thiophene or thiophene derivative is written as
We fit this function to our normalized band areas

Normalized thiophene abundance (
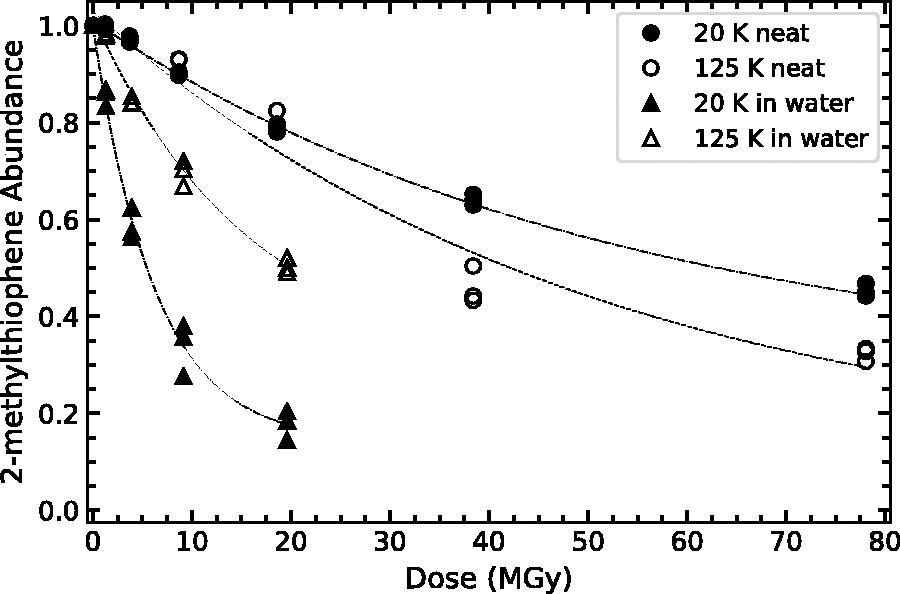
Normalized 2-methylthiophene abundance (
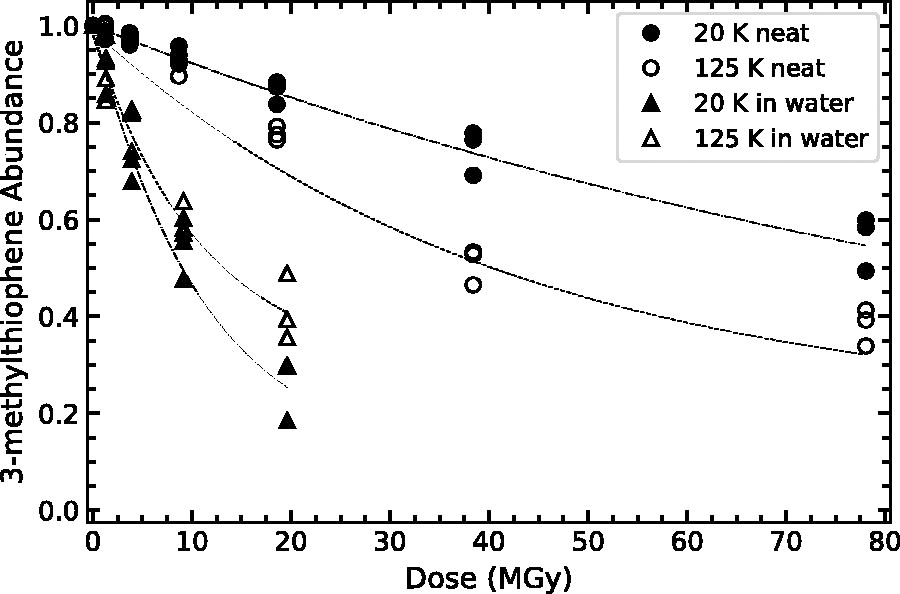
Normalized 3-methylthiophene abundance (
Curve Fit Parameters for the Radiolytic Destruction of Thiophene and Thiophene Derivatives
All mixtures with water have a relative abundance of 100:1 (water:organic).
Rate Constants and Radiolytic Half-Lives for Thiophene and Thiophene Derivatives
All mixtures with water have a relative abundance of 100:1 (water:organic).
k1 was calculated using fitted parameters a and b.
Note that for two samples (low-temperature thiophene and 3-methylthiophene), only a lower limit could be established for the radiation half-life (see Section 3.3).
Radiolytic destruction rate constants
As shown in Table 3 and Figures 7–9, the destruction rate constant of thiophene and thiophene derivative is larger in a water–ice matrix as compared to the single-component ices. For example, the destruction rate constant of thiophene is a factor of 7.3 higher when irradiated in a water–ice matrix as compared to a single-component ice at 20 K (0.08 vs. 0.011). This behavior resembles that of other categories of organic molecules, including simple aromatics (benzene; Ruiterkamp et al., 2005), amino acids (glycine; Gerakines et al., 2012; Gerakines and Hudson, 2013), and nucleobases (thymine and uracil; Materese et al., 2020; Gerakines et al., 2022). This enhanced decomposition is likely due to the additional reaction pathways provided by the water molecules, ionized products, and neutral radicals. Studies of aqueous thiophene have demonstrated the importance of H2O radiolysis products, especially OH, H, and e-, on the production of thiophene radicals (Saunders et al., 1978; Saunders, 1978). This higher reactivity in water–ice mixtures is also observed in our lower-temperature ice mixtures containing 2- and 3-methylthiophene. At low temperatures, OH does not readily diffuse within an ice (Siegel et al., 1961), and this may result in faster organic decomposition, possibly due to reactions between OH radicals and neighboring thiophene-derivative molecules. Bierbach et al. (1992) demonstrated that rate coefficients for gas-phase reactions between methylfuran and OH radicals were higher than for reactions between furan and OH radicals. That study suggested that the substituted methyl group provided an inductive effect (i.e., additional electron density on the heterocyclic ring, attracting the strong electrophile OH; Marusawa et al., 2002) since methyl groups are less electron-withdrawing than a hydrogen atom. We note that H2O2 (a strong, low-temperature oxidant; Loeffler and Hudson, 2013) present in our low-temperature ices likely does not contribute to any thermally driven chemistry of thiophene or thiophene derivatives at these temperatures, since oxidation reactions between these species typically require catalysts at room temperature (Brown and Espenson, 1996). Materese et al. (2020) suggested that the water ice matrix structure (e.g., crystalline or amorphous) might affect the destruction of organics (thymine) at low temperatures due to segregation of organics in more crystalline ices. Although we cannot rule out a structural effect, the enhanced reactivity highlighted here occurs for only the methylated thiophenes, and our organics are effectively matrix isolated (100:1), which will suppress any rate enhancement due to matrix structure and resultant organic segregation (see Fig. 7 from Materese et al., 2020).
In addition to the apparent matrix effects (i.e., single component or within a water–ice matrix), we find that the inclusion of a methyl functional group may subtly increase the destruction rate constant of the thiophene derivatives as compared to thiophene. This effect is most evident for the single-component ices at 125 K and in water–ice mixtures at 20 K. However, this is not the case for 3-methylthiophene as a single-component ice at low temperatures, and 2- and 3-methylthiophene diluted in water at high temperatures. This discrepancy for 3-methylthiophene at low temperatures is possibly due to the considerably smaller intrinsic band strength resulting in higher relative uncertainty in the fitted and computed parameters. For water–ice mixtures at high temperatures, we hypothesize that this difference might be at least partially due to the increased diffusivity of OH radicals that results in fewer reactions between OH and neighboring methyl-substituted thiophenes.
Finally, we note the appearance of a slight temperature dependence for our single-component ice samples, where the destruction rate constant for a single composition is higher at higher temperatures. This dependence could be due to a simple Arrhenius-like behavior (i.e.,
Implications for astrobiology
To best apply our laboratory data to the conditions of the Martian surface, we computed the radiation half-life for thiophene at 125 K as a single-component ice and for thiophene diluted in water ice at various depths (Table 4). We used the dose rates determined from the data collected by the Radiation Assessment Detector onboard the Mars Science Laboratory’s Curiosity rover (Hassler et al., 2014). As expected, the half-life for thiophene is significantly shorter when diluted in a water–ice matrix as compared to a single-component ice. Based on previous work on the radiolytic destruction of glycine in water and in carbon dioxide ice (Gerakines and Hudson, 2013, 2015), we expect that thiophene may also be more susceptible to radiation-driven destruction in carbon dioxide than in water. This higher reactivity in carbon dioxide ice may be important for the Martian surface, where diurnal cycling of CO2 frost occurs even at low- and mid-latitudes (Piqueux et al., 2016). However, a detailed compositional study is outside the scope of the present work and left for the future.
Dose Rates at and Under the Martian Surface, and Calculated Thiophene (T), 2-Methylthiophene (2MT), and 3-Methylthiophene (3MT) Half-Lives (t1/2) at 125 K
Dose Rates at and Under the Martian Surface, and Calculated Thiophene (T), 2-Methylthiophene (2MT), and 3-Methylthiophene (3MT) Half-Lives (t1/2) at 125 K
Hassler et al. (2014).
Half of the initial thiophene remains after approximately 130 and 530 million years (in water ice and as a single-component ice, respectively) at the depths probed by the Curiosity rover drill (∼0.06 m; Millan et al., 2022). This stability increases substantially beyond 1 m into the surface. We emphasize that the data in Table 4 do not take into account all possible effects such as surface heterogeneity in composition (i.e., matrix), regolith porosity, and temperature at a given depth, which will all affect both the dose rates and the thiophene half-lives. Moreover, macromolecules in which thiophene groups are embedded might provide additional radiation resistance, which is not explored here. With these caveats in mind, we have demonstrated that thiophene and mixtures of thiophene and water are degraded via radiolysis; however, much of the thiophene at the Martian surface could remain for hundreds of millions of years and significantly longer at depths greater than 1 m. Furthermore, the surface exposure age of the floor of Gale crater (where thiophene was detected) is estimated to be 80 Myr (Farley et al., 2014), well within the radiation half-life of thiophene listed in Table 4. Consequently, thiophene, produced from abiotic or biotic sulfur or organic molecules, could be stable on and near the Martian surface for significant periods of time (∼600 Myr) without the need of an active replenishing source. The radiation stability of thiophene and, to a lesser extent, its derivatives might explain their presence on the Martian surface.
We measured the radiation-driven destruction of thiophene, 2-methylthiophene, and 3-methylthiophene at 20 K and 125 K as single-component ice
Footnotes
Acknowledgments
P.D.T. acknowledges Steve Brown and Eugene Gerashchenko for operating and maintaining the Van de Graff Accelerator in the Radiation Effects Facility at NASA GSFC, and Joseph Nuth for fruitful discussions. Data from this publication are available in the following Zenodo repository: 10.5281/zenodo.13850669.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work is supported by NASA's Planetary Science Division Internal Scientist Funding Program, through the Fundamental Laboratory Research (FLaRe) work package at the NASA Goddard Space Flight Center and the NASA Astrobiology Institute through Award 13-13NAI7-0032 to the Goddard Center for Astrobiology (GCA). This material is also based upon work supported by NASA under award number 80GSFC21M0002.
Associate Editor: Christopher P. McKay