
Abstract
Background:
The microbial community in human milk is associated with many maternal and neonatal factors. This study aimed to investigate the effect of antibiotic exposure on the microbial community structure of colostrum.
Methods:
Twenty women with antibiotic treatment immediately after delivery and 10 age-matched control women were enrolled at the Guangdong Women and Children Hospital. Colostrum samples were collected within postpartum 30 hours. The V4 variable region of the bacterial 16S rRNA gene was sequenced to characterize the microbial profile using Illumina MiSeq platform.
Results:
Phyla Proteobacteria and Firmicutes were the predominant bacteria in colostrum samples. The core and abundant genera in colostrum included Streptococcus, Staphylococcus, and Pseudomonas. Compared with the control group, principal coordinate analysis based on the Bray-Curtis distance showed a significant difference in milk microbial community in women with antibiotic exposure, accompanied by a significantly lower alpha diversity and a different microbial ecological network. Furthermore, the relative abundances of genera Actinomyces, Anaerobacter, and Clostridium_sensu_stricto significantly decreased after antibiotic treatment.
Conclusions:
This study provided evidence of alterations in the colostrum microbial community with antibiotic exposure, improving our understanding of the effects of antibiotic treatment on the milk microbiome.
Introduction
Human milk provides nutrients (including oligosaccharides, hormones, antibodies, fatty acids) and microbes to the gastrointestinal tract that play an important role in helping seed and determine the infant's gut microbiota.1–3 If this process is disrupted, the infants may have a high risk of developing allergy, asthma, and obesity with a dysbiotic microbial community.1,4 As human milk contains a complex microbial community that influences the colonization of the infant's intestinal tract, more studies are needed on the community structure of human milk microbiome.3,4 Increasing evidence indicates that phyla Firmicutes and Proteobacteria, and genera Staphylococcus, Streptococcus, and Pseudomonas are the predominant microorganisms in human milk by using both culture-dependent and culture-independent methods.1,5 With the development of next-generation sequencing technology, some bacteria (Bacteroides, Clostridium, Faecalibacterium, Roseburia, etc.), which are difficult to culture, have also been identified in breast milk. 2
The milk microbiota composition and diversity are influenced by multiple maternal and environmental factors,1,2 including geographical location, maternal nutrient intake, delivery mode, time from birth, exposure to disinfection agents, and chemotherapy.
Maternal exposure to antibiotics at the time of delivery is used to prevent or reduce the risk of postpartum maternal infection, particularly when a cesarean section or episiotomy is performed.6,7 To date, there is little information about the effect of these prophylaxis regimens on the milk microbiota. Hence, we investigated this issue using 16S rRNA sequencing to provide evidence on the association between antibiotic exposure and colostrum microbiome.
Methods
Subjects, study design, and sample collection
This study was approved by the Ethics Committee of Guangdong Women and Children Hospital (No. 201901049), and was conducted according to the principles of the Declaration of Helsinki. After excluding women with a complicated singleton pregnancy, medical problems, adverse outcomes during pregnancy, known fetal anomalies or complications, and antibiotics or other antimicrobial therapy during pregnancy, 30 healthy and pregnant women with gestational age (GA) >37 weeks were recruited in this study. All the study subjects were healthy lactating women without clinical signs of breast disease upon examination by an experienced obstetrician. Among them, 20 women were treated with antibiotics (cefuroxime, intravenous infusion with a dose of 1.5 g each time, twice a day for 1–2 days) immediately after delivery (AT group) according to their clinical manifestations as judged by the experienced obstetrician, while 10 age-matched women without antibiotic use were recruited as the control group (NT group).
All participants provided written informed consent before recruitment. Clinical data, including basic demographics, pregnancy situation, and recent medications, were extracted from health records.
All colostrum samples were collected within postpartum 30 hours. After cleaning the breast with an iodine swab, colostrum was manually expressed into a sterile milk container by a trained nurse wearing sterile gloves, and the first few drops were discarded. The samples were immediately frozen at −20°C after collection and transferred to −80°C in 30 minutes until DNA extraction.
DNA extraction and sequencing
As previously reported, 5 DNA was extracted from the breast milk samples using the QIAamp DNA Stool Mini Kit (Qiagen, United Kingdom) according to the manufacturer's instructions. The concentration and purity of extracted DNA were measured using the NanoDrop One (Thermo Fisher Scientific, MA). Thereafter, the V4 region of the 16S rRNA gene was amplified using 515F (5′-CCTACGGGAGGCAGCAG-3′) and 806R (5′-CCGTCAATTCMTTTRAGT-3′) primers. The length and concentration of the polymerase chain reaction (PCR) products were analyzed using 1% agarose gel electrophoresis, and samples with bright main strips between 290 and 310 bp were used for further experiments. PCR products were mixed equally and sequenced on an Illumina MiSeq platform with 250 bp paired-end reads. The sequence data were deposited in the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA BioProject ID PRJNA760237).
Sequence data processing
The sequencing reads were first assigned to each sample according to the barcode using custom Perl scripts. Paired-end reads were then assembled using FLASH v1.2.118 with a minimum overlap of 10 bp, and a maximum allowable error ratio of 0.1 in the overlap region. Mothur v1.39.5 software 9 was used for further processing of assembled sequences. Taxonomic assignments for each sequence were analyzed using the Ribosomal Database Project Naïve Bayesian Classifier 10 against RDP 16S rRNA gene training set (version 16) at an 80% pseudo-bootstrap confidence score. Functional annotation analysis was performed using PIRCRUSt2.0 software, 11 and the differences between the AT and NT groups were investigated using STAMP software. 12
Alpha diversity was represented using the Shannon and Simpson indexes, and beta diversity was represented using the Bray-Curtis dissimilarity index. Principal coordinate analysis (PCoA) based on the Bray-Curtis distance was performed, accompanied by permutational multivariate analysis of variance (PERMANOVA, 999 permutations) using R software. The microbial ecological network was constructed using SPIEC-EASI package 13 in R software and visualized with Gephi software.
Statistical analysis
The continuous demographic variables and categorical variables were presented as the mean ± standard deviation and numbers (percentages, %), respectively. Comparisons were made using t-tests and chi-square tests. Statistical significance was accepted as p < 0.05. Significantly different microbes between the AT and NT groups were determined using the generalized linear model (GLM) in R software with p < 0.05 to adjust maternal and neonatal factors.
Results
Characteristics of study subjects
Thirty pregnant women were enrolled in this study. All the subjects were of Han ethnicity with age range of 20–38 years. The subjects lived in Guangzhou City, and their diet was mainly Cantonese cuisine. All women gave birth between the 37th and 42nd gestational week. The colostrum samples were collected from postpartum 16–30 hours. There were no differences in age, GA, prepregnancy body mass index (BMI), BMI during pregnancy, neonatal birth weight, delivery mode, and sampling time between the AT and NT groups (Table 1). Among the women with cesarean section, 10 had uterine scarring, 3 had fetal distress, 1 had placental abruption, 1 had pelvic outlet, and the remaining 10 were voluntary.
Characteristics of the Women Included in This Study
AT, after delivery; BMI, body mass index; NT, control group; SD, standard deviation.
Alterations of the microbial community in women after antibiotic exposure
Sequencing of colostrum samples generated 28,876,936 reads for 30 samples, with a mean read length of 292 bp. An average of 707,802 and 665,846 reads per sample were obtained for the AT and NT groups, respectively.
The Shannon and Simpson indexes were used to estimate microbial alpha diversity in this study, which considers both microbial richness and evenness. After adjustment for maternal age, mode of delivery, prepregnancy BMI, neonatal gender, and so on, a significantly higher alpha diversity was observed in the NT group compared with the AT group (Fig. 1a), with a greater value of Shannon index (3.44 ± 0.45 versus 3.02 ± 0.41, p = 0.026) and a smaller value of Simpson index (0.13 ± 0.08 versus 0.21 ± 0.07, p = 0.015). Furthermore, PCoA based on Bray-Curtis distance showed a significant difference between the two groups (PERMANOVA, R2 = 0.12, p = 0.001) after adjusting for maternal and neonatal factors (Fig. 1b), indicating alterations of colostrum microbial community after antibiotic treatment.

Comparisons of alpha and beta diversities between the AT and NT groups.
Different microbes between women with and without antibiotic exposure
The dominant phyla in colostrum microbiome were Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria (Fig. 2a). Compared with the NT group, the average relative abundances of Bacteroidetes (4.05% versus 2.88%) and Firmicutes (48.08% versus 41.59%) increased, whereas Actinobacteria (3.47% versus 4.99%) and Proteobacteria (43.57% versus 49.71%) decreased in the AT group. A least average relative abundance >1% was selected to show genus in Figure 2b. The abundant genera in milk samples were Acinetobacter, Chryseobacterium, Clostridium_sensu_stricto, Corynebacterium, Escherichia/Shigella, Gemella, Neisseria, Pseudomonas, Rothia, Sphingomonas, Staphylococcus, Stenotrophomonas, and Streptococcus.
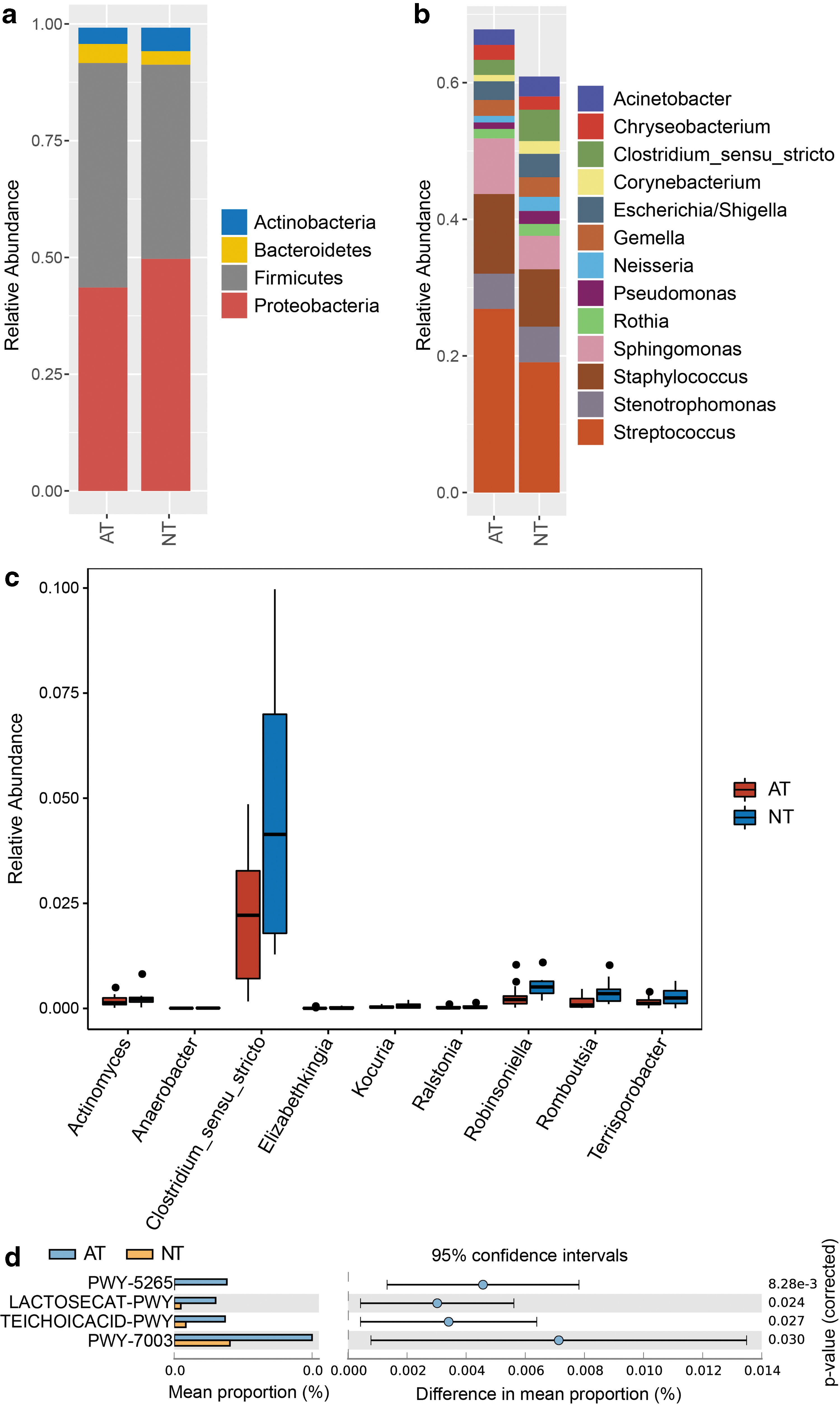
Relative abundance of microbiota at the phylum and genus levels in human colostrum.
Through GLM analysis, the average relative abundances of genera Actinomyces (0.17% versus 0.25%, p = 0.03), Anaerobacter (0.006% versus 0.01%, p = 0.02), Clostridium_sensu_stricto (2.25% versus 4.59%, p = 0.04), Elizabethkingia (0.009% versus 0.02%, p = 0.04), Kocuria (0.04% versus 0.07%, p = 0.02), Ralstonia (0.03% versus 0.04%, p = 0.04), Robinsoniella (0.27% versus 0.56%, p = 0.03), Romboutsia (0.14% versus 0.4%, p = 0.002), and Terrisporobacter (0.14% versus 0.27%, p = 0.005) significantly decreased in the AT group, compared with the NT group (Fig. 2c).
Core genera of human milk were defined as at least 95% of individuals with a minimum 1% mean relative abundance in this study. Overall, 13 core genera were identified, which comprised Streptococcus, Staphylococcus, Sphingomonas, Stenotrophomonas, Clostridium_sensu_stricto, Escherichia/Shigella, Gemella, Acinetobacter, Chryseobacterium, Rothia, Neisseria, Pseudomonas, and Corynebacterium. These genera represented 67.80% and 60.90% of the average relative abundance in the AT and NT groups, respectively.
Functional analysis against the MetaCyc database showed that pathways PWY-5265 (peptidoglycan biosynthesis II), LACTOSECAT-PWY (lactose and galactose degradation I), TEICHOICACID-PWY (poly (glycerol phosphate) wall teichoic acid biosynthesis), and PWY-7003 (glycerol degradation to butanol) significantly increased in the AT group with STAMP software analysis (Fig. 2d).
Different microbial ecological networks between the AT and NT groups
SPIEC-EASI software was used to study the ecological network of colostrum microbiome at the genus level. A more complex and different network was observed in the AT group (Fig. 3a), with 167 nodes and 581 edges. Whereas the same number of nodes was present in the NT group, only 361 edges were observed (Fig. 3b). In addition, PageRank algorithm indicated that the top 3 important genera in the AT group were Cohnella, Alloprevotella, and Corynebacterium, which were different from Mycobacterium, Clostridium_IV, and Olsenella in the NT group. Furthermore, modularity analysis indicated 10 modularity classes in the NT group, which was reduced to 9 in the AT group.

Microbial ecological network at the genus level.
Discussion
This study demonstrated that the dominant phyla in milk microbiota were Firmicutes and Proteobacteria, accounting for an average of >90%, which was in accordance with previously published results. 1 The core genera defined in this study included Streptococcus, Staphylococcus, Sphingomonas, Stenotrophomonas, Clostridium_sensu_stricto, Escherichia/Shigella, Gemella, Acinetobacter, Chryseobacterium, Rothia, Neisseria, Pseudomonas, and Corynebacterium. Among them, Streptococcus and Staphylococcus have been reported as common constituents of the human milk microbiota by culture-dependent and culture-independent methods.1,14,15 Similarly, Pseudomonas is known to account for a high relative abundance in human milk samples from postpartum 1st to 12nd week, 5 and Acinetobacter has a high relative abundance in a community type of human milk microbiome.14,15 Stenotrophomonas, Escherichia/Shigella, Rothia, Corynebacterium, and Sphingomonas are commonly found in human milk.1,5,15,16
The dominant genera identified in this study were in accordance with previously published results. In addition, a high proportion of Rothia and a low abundance of Pseudomonas have been found in neonatal oral microbiome. 14 Clostridium_sensu_stricto and Escherichia/Shigella are common in gut microbiome.17,18 It was in agreement with the hypothesis of human milk microbiome origin, mainly from entero-mammary translocation of maternal gut and retrograde inoculation of infant's oral microbiome. 1
A distinct bacterial community structure was observed in the AT group with PCoA based on the Bray-Curtis distance. The genera Actinomyces, Anaerobacter, Clostridium_sensu_stricto, Elizabethkingia, Kocuria, Ralstonia, Robinsoniella, Romboutsia, and Terrisporobacter were significantly higher in the NT than in the AT group. Actinomyces can cause breast infection, 19 and Ralstonia is associated with subacute mastitis and mastitis. 20 In addition to maternal health, these bacteria also play key roles in neonatal health. The members of genus Clostridium_sensu_stricto can produce acetate, butyrate, lactate, and so on.21,22 Its relative abundance is closely related to infants with IgE-mediated food allergy 23 and necrotizing enterocolitis. 24 Anaerobacter plays an important role in IgE-mediated food allergy. 23 Furthermore, these significantly different genera in the AT group formed a more complex ecological network with more links. The decrease in these genera in the milk microbial community after antibiotic treatment might be transferred to neonates by breastfeeding, affecting neonatal health.
As recommended by the World Health Organization, breastfeeding is the best way to provide the ideal nutrition for neonatal health. The microbial community and nutrients of human milk constitute an important source for neonatal growth,25,26 and they are vertically transferred from mother to infant through breastfeeding.5,27,28 Alterations of human milk microbial community with antibiotic use observed in this study might play a key role in maternal health and neonatal growth. However, this could not be verified due to the lack of neonatal growth and maternal health data. In the future, longitudinal data on maternal and neonatal health need to be examined.
Breast milk contains numerous nutrients, such as milk oligosaccharides, milk fatty acids, hormones, immune cells, antibodies, carbohydrates, fats, and proteins, which may help to develop the infant gut microbiome and are important for infant growth. 29 Notably, lactose, lipids, and oligosaccharides are abundant bioactive components in human milk. 29 In this study, the metabolic functional pathway “lactose and galactose degradation I” significantly changed after antibiotic exposure, which might be related to the metabolism of lactose and human milk oligosaccharides. Moreover, metabolic pathways “peptidoglycan biosynthesis II” and “poly (glycerol phosphate) wall teichoic acid biosynthesis” are involved in the synthesis of bacterial cell walls. 30 Their significant changes after antibiotic exposure might affect the proliferation of some microbes.
However, due to the lack of human milk nutrient data, the associations between the colostrum microbial alterations after antibiotic use and milk nutrients could not be evaluated. Moreover, the impact on the neonatal gut microbiome colonization could not be studied. In the future, the milk nutrients and neonatal gut microbiome data should be studied.
This study had some limitations, which should be addressed in future studies. First, the sample size should be increased in the future to confirm the observations. As ethnicity is associated with the milk microbial composition, 31 subjects from different ethnicities should be recruited in future studies. Notably, data on human milk nutrients and neonatal gut microbiome should be collected in the future. Metagenome sequencing should be considered to identify the taxonomy at the species level. Moreover, the milk microbial community is dynamic, 14 and only one time point with the same antibiotic use was examined in this study. Hence, it would be interesting to investigate different antibiotics, different lengths of exposure, and the longitudinal effect of antibiotic exposure in future studies.
In summary, the colostrum microbial community structure changed after antibiotic treatment, which was accompanied by a lower alpha diversity and a more complex microbial ecological network. The observations in this study improve our understanding of the effects of antibiotic use on the colostrum microbiome.
Footnotes
Authors' Contributions
Y.C. and Y.W. conceived of the study and planned the experiments. J.W., J.Z., and C.Z. conducted the experiments. X.Y., H.Y., and D.Y. designed the computational framework and analyzed the data. Y.W., J.W., X.Y., and Y.C. wrote the article. All authors discussed the results and contributed to the final article.
Ethics Approval and Consent to Participate
This study was approved by the Ethics Committee of Guangdong Women and Children Hospital (No. 201901049) and complied with the Helsinki Declaration. Informed consent was obtained from all participants in this study. All experiments were performed in accordance with the approved guidelines.
Availability of Data and Materials
The data set supporting the conclusion of this article is available in the NCBI Sequence Read Archive database (SRA BioProject ID PRJNA760237).
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by the Science and Technology Projects in Guangzhou (202102080378).