
Abstract
Cryopreservation by freezing is usually employed for storage of allogeneic valves destined for clinical use; however, disruption of leaflet extracellular matrices by ice may occur. This study was performed to determine the effects of cryoprotectants employed in VS55 for ice-free, vitreous cryopreservation upon adherent porcine heart valve leaflet-derived myofibroblasts in culture. Low-passage myofibroblasts exhibiting strong actin and myosin staining were employed for these experiments. Three cryoprotectants, dimethylsulfoxide, formamide, and propanediol, were tested individually and in combination. Exposure experiments demonstrated that the individual cryoprotectants (0–5 M) were generally cytotoxic in a dose-dependent manner with little if any loss of cells as determined by measuring metabolic activity and DNA content. Exposure to formamide resulted in the greatest loss of cells and reduction in viability. Combination of the three cryoprotectants demonstrated that the cytotoxic effects of each cryoprotectant were not cumulative. Cell viability and DNA content were equivalent to dimethylsulfoxide and propanediol and higher than formamide alone over most of the 0–5 M dose–response curve. After cryopreservation by slow-rate freezing, the benefits of the combination of cryoprotectants over individual cryoprotectants were demonstrated at the 4 M concentration range for both cell viability and cell retention. In conclusion, these studies demonstrated that, with the exception of the lower concentrations of propanediol, the combination of cryoprotectants employed in these studies result in equivalent or better cell viability and attachment than individual cryoprotectants.
Introduction
The principles that govern the cryopreservation of mammalian cells and tissues have been recently reviewed.23,24 Successful cryopreservation of tissues is not a simple matter of extrapolating the well-established principles of cell cryopreservation to more complex tissues, such as heart valves. 23 The reason is that tissues are much more than the aggregate sum of their component cells. They are invariably comprised of a variety of cell types intimately associated with basement membranes and ECMs, which may impede cryoprotectant permeation and may demand special consideration for successful cryopreservation. These differences are manifest in additional mechanisms of injury, which must be circumvented for successful preservation. 25 Ultimately, these differences in responses to freezing between individual cells and tissues are principally due to extracellular ice formation.
There has been a major focus during the last decade on the development of low-temperature preservation techniques that avoid ice crystallization by vitrification.23,24 Prevention of freezing by vitrification means that the water in a tissue remains unfrozen in a noncrystalline state during cooling. Vitrification is the solidification of a liquid without crystallization. As cooling proceeds, however, the molecular motions in the liquid permeating the tissue decrease, and eventually, an “arrested liquid” state known as a glass is achieved. It is this conversion of a liquid into a glass that is called vitrification (derived from vitri, the Greek word for glass). A glass is a liquid that is too cold or viscous to flow and is essentially in molecular stasis. Vitrification does not have any of the biologically damaging effects associated with freezing because no appreciable degradation occurs over time in living matter trapped within a vitreous matrix. Vitrification has been shown to provide effective preservation for a number of cells, including monocytes, ova and early embryos, and pancreatic islets.26–29 Cryopreservation employing ice-free vitrification has been applied to cardiovascular tissues.30–35 We have previously proposed that interstitial ice formation in allograft heart valves during cryopreservation and thawing may result in structural deterioration in vivo.32,33 This occurs in tissues preserved by commercial methods employing dimethyl sulfoxide (Me2SO) as a cryoprotectant. Although the current, widely used, clinical cryopreservation methods can preserve cell viability, major matrix disruption can occur in the presence of excellent cell survival because of extracellular ice formation.33,35–37 Mirabet et al. 38 and Gerson et al. 39 failed to see any ECM changes in frozen cryopreserved valves. Whether this was due to differences in experimental technique or design is not clear. However, a more recent publication clearly demonstrates that frozen cryopreserved allogeneic pulmonary heart valves perform poorly in a growing juvenile sheep implant model compared with valves that were cryopreserved employing an ice-free method. 40 Explant heart valve leaflet pathology also demonstrated statistically significant differences. If allograft failure can be prevented, it would considerably improve clinical outcomes, may ultimately reduce healthcare costs, and have significant benefits for the general public. Therefore, this study was performed to determine the effects of cryoprotectants employed to inhibit extracellular ice formation on an adherent in vitro cell model based upon porcine aortic heart valve leaflet-derived myofibroblasts prior to initiation of large animal heart valve studies.
Materials and Methods
Isolation and culture
Cultures of porcine heart valve leaflet myofibroblasts were established by placing small pieces of minced leaflet (<1.0 mm3) in 24-well culture plates (Falcon) coated with 0.2% gelatin (Fisher). Enough Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS) was added to cover the bottom of the wells without submerging the pieces of tissue. The plate was covered and left at 37°C with 5% CO2 in air until visible outgrowth occurred, about 3–5 days. At that time, more media were added to the wells, ∼1.0 mL, and outgrowth was allowed to continue until the well was confluent. Upon reaching confluence, the cells were washed with phosphate-buffered saline (PBS) and trypsinized before being transferred to T25 cell culture flasks (Falcon). The cultures were expanded and maintained in DMEM (10% FCS) at 5% CO2 until they were ready to be used. The cells between passages 2 and 20 were used for experiments.
Immunohistochemistry
Characterization of the cells was performed using immunohistochemical staining for actin and myosin (Fig 1). Cells were allowed to adhere to glass slides (Superfrost-Fisher) before being fixed with methanol. After washing with PBS, the slides were blocked for 1 h at 37°C in PBS containing 0.1% Triton X-100 with 10% serum. Staining for actin was accomplished using a monoclonal antibody to α-actin (1:250) (Sigma) conjugated with phalloidin–flourescein isothiocyanate. Myosin staining was done using a monoclonal antibody (Sigma; 1:500) followed by a goat anti-mouse secondary antibody (1:100) conjugated with Texas Red. Incubation with the primary antibodies was allowed overnight at 4°C, whereas secondary antibody incubation was for 1 h at room temperature. After washing with PBS + 0.1% Triton X-100, the cells were left in 4% paraformaldehyde for 10 min before being mounted and stored in the dark at 4°C.
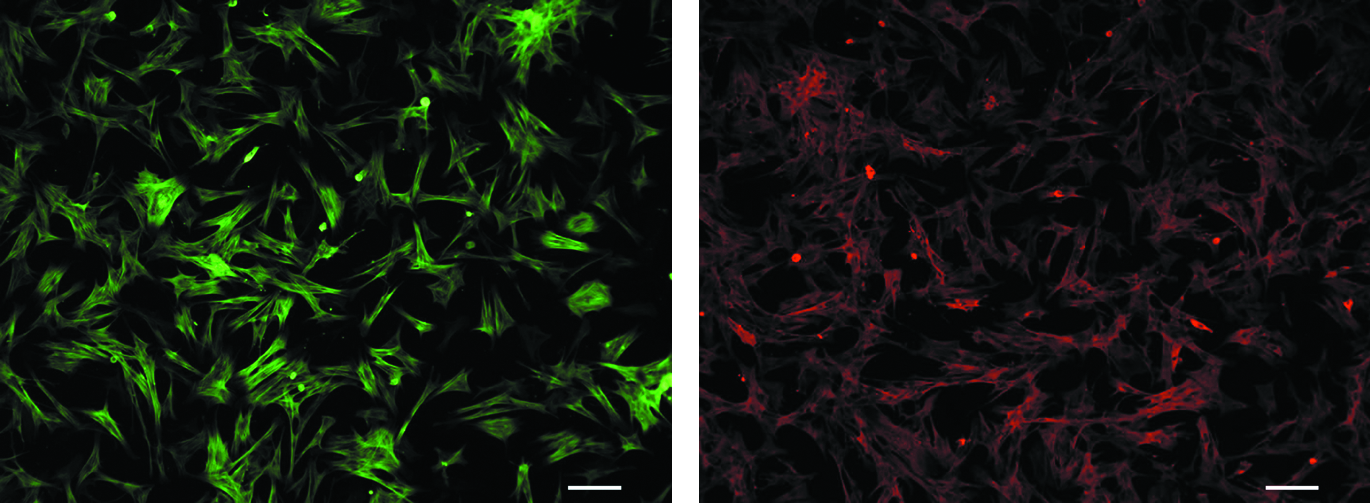
Primary myofibroblast staining. Primary myofibroblasts isolated from porcine heart valve leaflet were stained with actin (green) and myosin (red) to confirm their origin. Pictures were taken under 10 × magnification using Qcapture software.
Cytotoxicity assay
The cells were plated in 96-well microtiter plates (Falcon) at 104 cells per culture well. The next day, the cytotoxicity assay was carried out as outlined in Table 2. The cryoprotectants solutions (Table 1) were prepared in EuroCollins solution (194 mM dextrose, 15 mM KH2PO4, 42 mM K2HPO4, 15 mM KCl, 10 mM NaHCO3) and both the microtiter plate and the solutions were kept on ice (4°C ± 1°C) for the duration of the assay. Each solution was added and then removed prior to the addition of the next step as shown in Table 2. For some steps, the solutions were added on top of the previously added solution, as indicated in the table. The cells were allowed to incubate at each step for 5 min except for step 7. Here the cells were left in the cryoprotective agents (CPAs) for 10 min before beginning the removal of the CPAs. The mannitol concentrations that were used to load the cells with CPAs did not exceed the final CPA concentration for a given set of cells. For example, cells that were incubated in a final CPA concentration of 1.0 M were first preincubated with 0.5 M mannitol. The first 2 steps during the removal of the CPAs simply dilute the CPA present in the well. After step 8, the CPA concentration was ½ of the final concentration added in step 7, and after step 9, the CPA concentration was ⅓ of the final concentration added in step 7. Once the CPAs have been removed, the cells were washed with cell culture medium (DMEM with 10% FCS) and then allowed to recover in fresh culture medium for 1 h at 37°C before viability assessment.
Me2SO (3.1 M), formamide (3.1 M), and 1,2-propanediol (2.2 M).
The number of addition steps is based on the final CPA concentration.
At these steps, a volume equal to step 7 is added.
Cryopreservation assay
The cells were plated at 2 × 104 cells per culture well. The protocol employed for the cytotoxicity assay was used with minor modifications (Table 2). After step 7 when the final concentration of CPA was added, the plates were equilibrated at 4°C in a controlled-rate freezer (Planar) for 10 min before cryopreservation at a controlled rate of −1.0°C/min to −80°C. Under the slow-rate cooling conditions employed, all experimental groups should freeze, except for those containing the highest concentration (8.4 M) of the combined cryoprotectant formulation, which is free of ice. The plates were then transferred to the vapor phase of liquid nitrogen for storage overnight. The next day, the plates were placed in a −20°C freezer for 30 min followed by rapid warming at 37°C. 41 During the brief incubation (1–2 min) at 37°C, an equal volume of 0.5 M mannitol in DMEM (10% FCS), warmed to 37°C, was added to each well. Each plate was then placed on ice for 5 min and removal of the CPAs was performed as shown in Table 2.
Assessment of cell viability
Cell viability was determined using the noninvasive metabolic indicator alamarBlue™ (Trek Diagnostics). AlamarBlue measures the oxidation/reduction reactions within cells and can be read using fluorescence or absorbance. A volume of 20 μL of alamarBlue was added to cells left in 200 μL of DMEM (10% FCS) after the cytotoxicity and cryopreservation assays described above. After incubation at 37°C for 3 h, the plate was read using a fluorescent microplate reader (Molecular Devices) at an excitation wavelength of 544 nm and an emission wavelength of 590 nm. Figure 2 demonstrates the applicability of alamarBlue for measuring metabolic activity over a range of cell densities. For the cytotoxicity experiments, the data were normalized against cells that were treated with Euro-Collins alone (0.0 M), whereas the data were normalized against an untreated control for the cryopreservation experiments. The values are presented as the means (±standard error of the mean) of 16 replicates.
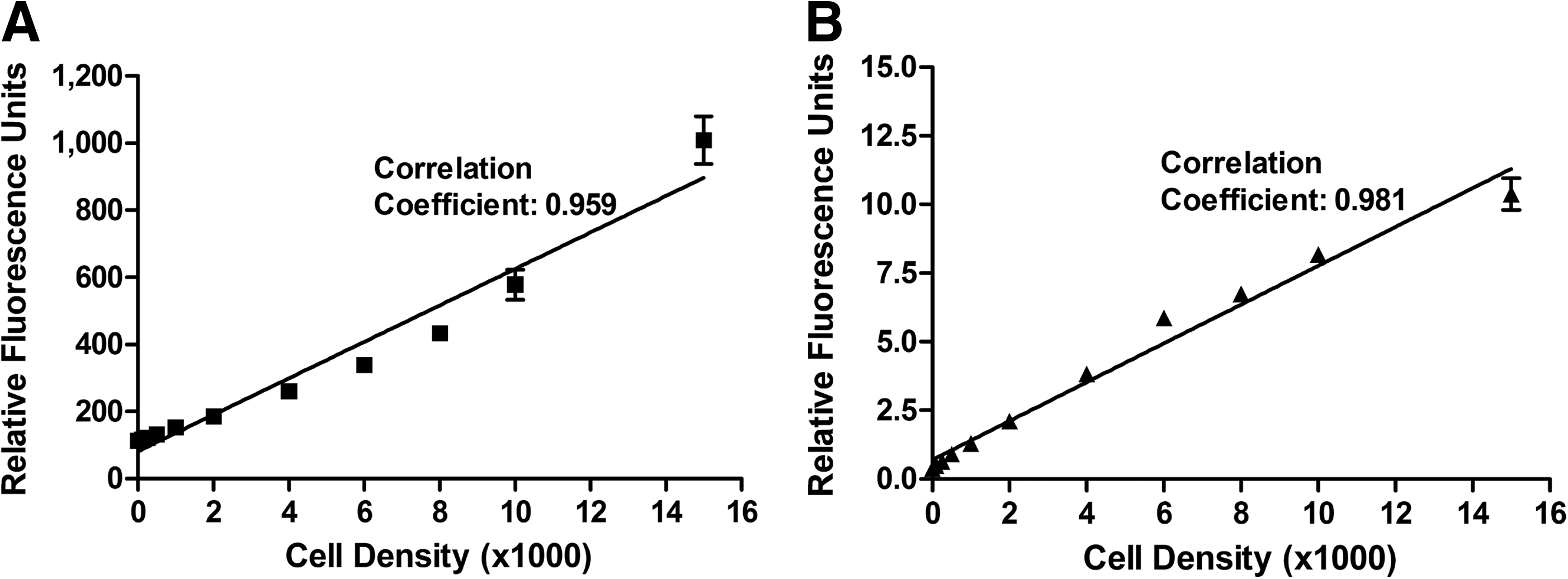
AlamarBlue™ and Cyquant assay standard curves for myofibroblasts. Myofibroblasts were plated at various cell densities and assayed for
Assessment of DNA content
The proportion of cells remaining in the well of the microtiter plate after cytotoxicity screening and cryopreservation was assessed by measuring the DNA content of the cells within each well. The DNA content was used as an indicator of cell number. The Cyquant assay (Molecular Probes) uses a fluorescent dye to label nucleic acids, which can then be measured using a fluorescent microplate reader with an excitation wavelength of 485 nm and an emission wavelength of 538 nm. The assay included a step using RNAse A (Sigma) to eliminate the variable amount of RNA within individual cells and thereby provide a direct measure of the DNA content. There was a linear relationship between cell number and DNA content (Fig. 2). The plate was read and the data were normalized against untreated control cells. The data are presented as the mean (±standard error of the mean) of 16 replicates.
Analysis of ice formation in solutions
Characterization of ice crystal formation and growth was assessed in bulk samples (75 mL) of 2 cryoprotectant (CPA) solutions cooled to −100°C under slow cooling conditions (1°C–2°C/min) in an insulated glove box. Digital photographs were taken at various temperatures and quantified. The number of ice crystals was counted and the relative area occupied by ice in the bulk sample was calculated as a percentage (Table 3). The 2 CPA solutions were the vitrification solution (VS55 containing 55% CPA consisting of 3.1 M Me2SO, 3.1 M formamide, and 2.2 M 1,2-propanediol) and a diluted version of the same CPA formulation containing 49% CPA (VS49).
The number of experiments performed is given within parentheses.
ME2SO (2.75 M), formamide (2.75 M), and 1,2-propanediol (2.0 M).
Me2SO (3.1 M), formamide (3.1 M), and 1,2-propanediol (2.2 M).
Data are expressed as the mean +1 standard error of the mean.
CPA, cryoprotective agent.
Statistical analysis
All experiments were repeated at least 4 times with 4 replicates in each experiment. Statistical differences were assessed by 2-way analysis of variance. P values <0.05 were regarded as significant.
Results
Cryoprotectants and their concentrations were chosen based on the composition of VS55 (Table 2). VS55 exhibited a small amount of ice formation at the slow cooling rates (1.5°C/min) employed in these studies. Relatively few ice crystals formed (∼46 in 75 mL of solution) in VS55 and they occupied ∼1% of the solution (Table 3). In contrast, reduction of the total cryoprotectant concentration to 49% (VS49 solution) exhibited extensive ice formation occupying ∼100% of the solution at slow cooling rates (2.4°C/min). These experiments indicate that all experimental cryopreservation groups, except the highest concentration groups exposed to full-strength VS55, were cryopreserved by freezing but not vitrification.
The cells used in this study were isolated from porcine heart valve leaflet explants. The myofibroblasts migrated from the tissue onto the plastic culture substrate and were used in the experiments. Immunohistochemistry verified their smooth muscle origin (Fig. 1). The cells expressed both actin (green) and myosin (red), indicative of smooth muscle cells.
All the assays for cytotoxicity and preservation experiments were performed in 96-well microtiter plates. One plate was not treated and was considered the fresh control for cells in other plates. To account for variation between plates, several experiments were performed. The first series of experiments evaluated cryoprotectant cytotoxicity. There was a marked tendency for the fresh untreated control plate in each of the cytotoxicity studies to result in normalized experimental data slightly higher than 100% (Fig. 3). The data can also be calculated using the zero CPA exposure data as the control, in which case the results are all below 100% (data not shown). Both the individual CPAs at a concentration range of 0–5.0 M and the complete vitrification solution using a range of increasing total CPA concentrations (0–8.4 M) were assessed for potential cytotoxicity to primary myofibroblasts (Fig. 3A, C). The choice of CPA and the concentration were found to be significant (P < 0.001). Me2SO and propanediol demonstrated cell viabilities of ∼75% at concentrations of 2–3 M, but formamide clearly demonstrated decreased viability at concentrations >1 M. Cell viabilities of ∼75% were observed using a combination of the 3 cryoprotectants up to a 4 M concentration before a decrease in cell viability was detected relative to control cells without CPA (0 M). The cryoprotectant 1,2-propanediol demonstrated the highest viability and statistical significance compared with formamide (P < 0.001). Formamide showed the greatest cytotoxicity of the CPAs tested either individually or combined.
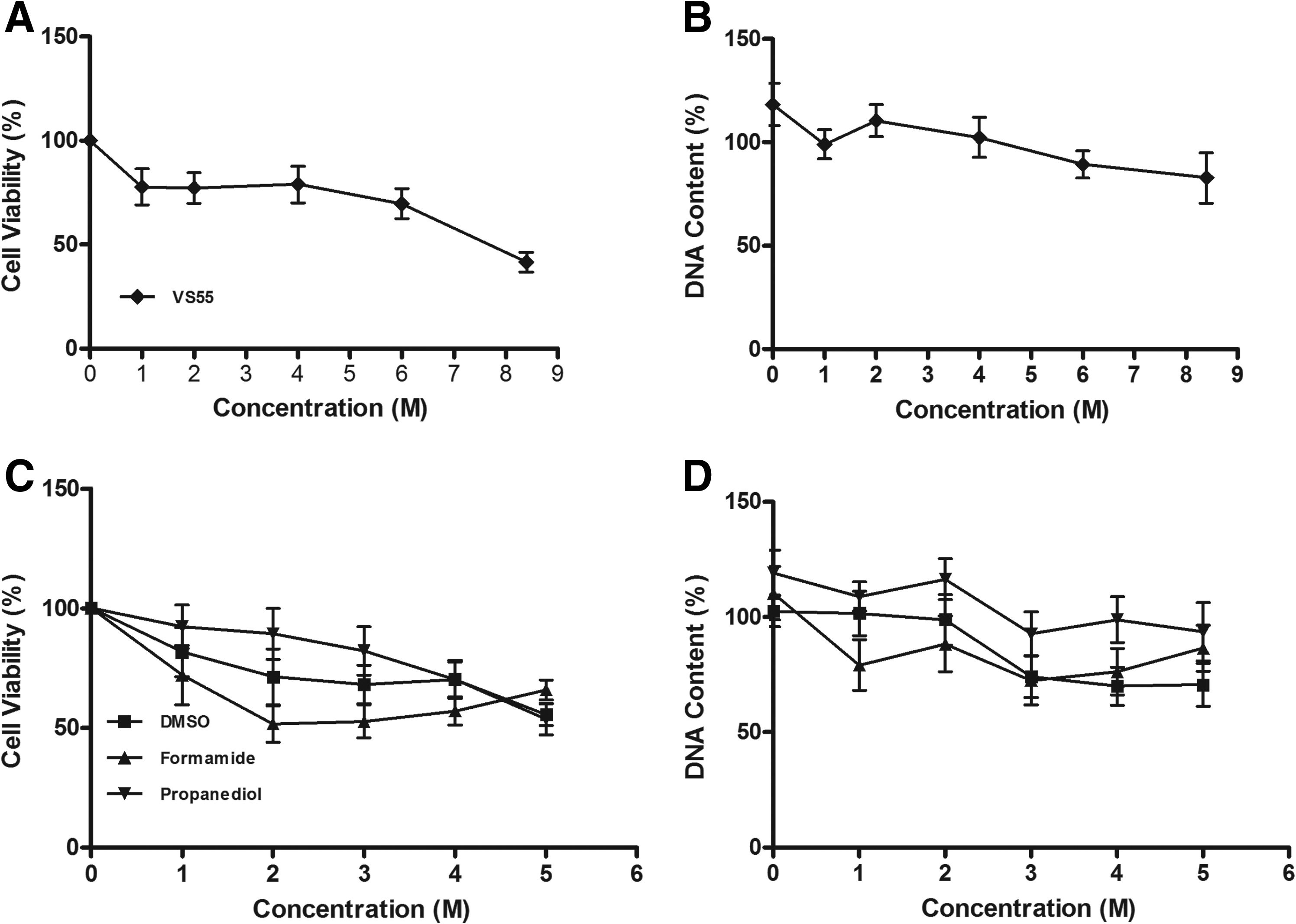
Cytotoxicity of cryoprotectants. Cytotoxicity and cell loss during exposure to Me2SO, 1,2-propanediol, formamide, and the cryoprotectant combination (VS55). Myofibroblast viability
In addition, the ability of the cells to remain attached to the microtiter plate was assessed by measuring DNA content (Fig. 3B, D). Again the choice of cryoprotectant and its concentration were significant factors (P < 0.001). Each of the CPAs tested demonstrated DNA content that was at least 75% of untreated controls. Formamide demonstrated the greatest loss of cells. DNA content remained high even when the combination of all 3 CPAs was used.
In the next series of experiments, the myofibroblasts were subjected to cryopreservation and rewarming using the same CPAs and concentration ranges that were used for the cytotoxicity studies (Fig. 4A, C). The choice of cryoprotectant and its concentration were statistically significant (P < 0.001). Cells cryopreserved in the presence of Me2SO, 1,2-propanediol, or the dilutions of the cryoprotectant combination (VS55) demonstrated a “classical” dependence on cryoprotectant concentration with optimum survival in the range of 1–2 M (P < 0.001). In contrast, cells frozen with formamide alone showed a reduced level of recovery over the entire range of concentrations, indicating that formamide alone is a poor cryoprotectant.
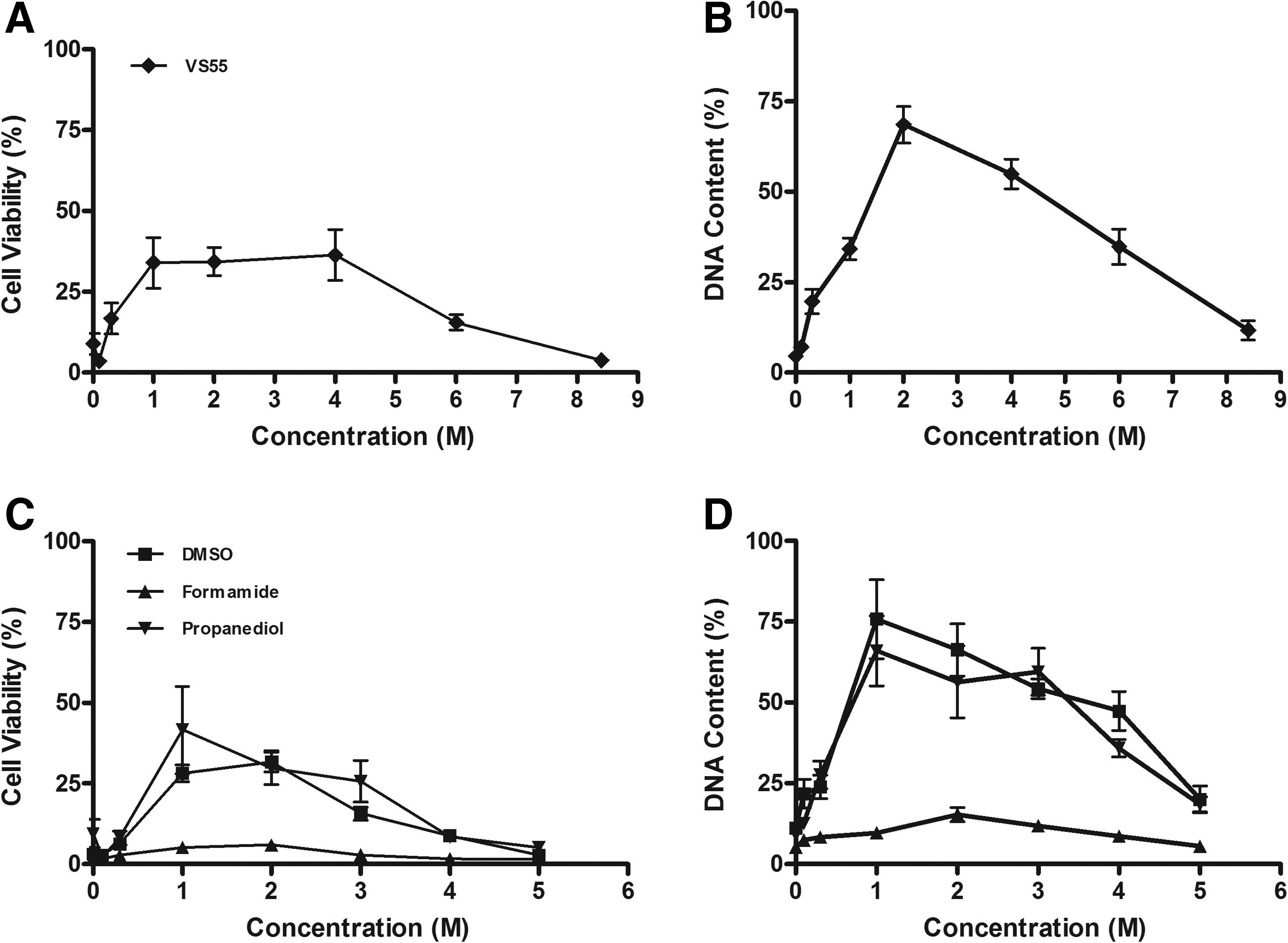
Effect of cryopreservation on cell survival. Cells were exposed to cryoprotectants (Me2SO, 1,2-propanediol, formamide, and the cryoprotectant combination, VS55) and cryopreserved. Viability
Cell attachment after cryopreservation was assessed using the same experimental design (Fig. 4B, D). Cell attachment of ∼75% was achieved using a concentration of 1–2 M of Me2SO or 1,2-propanediol and a concentration of 2.0 M of the combined cryoprotectants, whereas significantly less attachment was observed at all formamide concentrations tested (P < 0.001).
Discussion
The only methods currently known to be of use for storage and stabilization of cells and tissues involves the application of cryobiology, that is, the study of the effects of temperatures lower than normal physiologic ranges upon biologic systems.42–44 Sub-zero storage was founded upon the discovery of glycerol 45 and Me2SO 46 in the middle of the last century. Since the discovery of these CPAs, the field of cryoprotection during freezing and vitrification of biological materials has become established and many other cryoprotectants have been identified.
Although many types of isolated cells and small aggregates of cells can be frozen simply by following published procedures, obtaining reproducible results for more complex tissues, such as heart valves, requires an understanding of the major variables involved in tissue cryopreservation. The extent of freezing damage depends upon the amount of free water in the system and the ability of that water to crystallize during freezing. Cryoprotectant selection for heart valve cryopreservation by freezing has been usually limited to Me2SO and occasionally glycerol. Both Me2SO and glycerol are intracellular cryoprotectants with low molecular weights, which reduce intracellular water by cell permeation. On occasion, combinations of 2 cryoprotectants, which may or may not permeate the cells, result in additive or synergistic enhancement of cell survival.47,48 Intracellular cryoprotectants, such as glycerol and Me2SO at concentrations from 0.5 to 3.0 M, are effective in minimizing cell damage in slowly frozen biological systems.
In this study, we have investigated the effects of individual CPAs up to 5.0 M (Me2SO, formamide, and 1,2-propanediol) and combinations of the 3 CPAs up to 8.4 M in the VS55 formulation upon heart valve leaflet-derived myofibroblasts to evaluate the CPA effects upon cell viability and cell adhesion or retention on their culture plastic substrate. Cells attached to microtiter plates cryopreserved by freezing in the presence of Me2SO demonstrated optimum survival at 1–2 M, similar to most reports on nonadherent cells in suspension in the literature. Propanediol and the cryoprotectant combination (VS55) were equally effective during freezing and both appeared to be effective at wide concentration ranges (Fig. 4A, C). In contrast, formamide was not an effective cryoprotectant under the experimental conditions employed in these studies. Formamide in combination with Me2SO and propanediol demonstrated higher cell viability at the 4 M total CPA concentration than any of the CPAs individually at 4 M. This observation supports a previous report that formamide in combination with other cryoprotectants may act to reduce the overall cytotoxicity of the cryoprotectant solution, allowing higher CPA concentrations to be used. 49
Lower DNA content is explained by the cells becoming detached from their substrate, particularly after cryopreservation (Fig. 4B, D). The extent to which cell adhesion mechanisms may be compromised during cryoprotectant exposure and cryopreservation has not been extensively studied. However, if cells are becoming detached during cryopreservation and/or rewarming are they dead or inevitably committed to cell death? The high concentrations of CPAs required for effective vitrification were not well tolerated in this cell culture model. However, tissues are more complex than cell monolayers and it is well established that tissues can tolerate the higher concentrations of CPAs required for vitrification with ∼80% viability and function after preservation.31,34,35,42,50 Further studies of combinations of the 3 CPAs are needed to better understand the mechanism(s) by which combinations of CPAs result in higher viability than similar concentrations of individual CPAs in both cell culture and tissue models. In addition, is it possible that CPA exposure affects cell adhesion mechanisms in tissues but the cells cannot detach because they are embedded in ECMs and that many of them subsequently recover after removal of CPAs under physiological conditions?
Conclusions
Cryopreservation by freezing is usually employed for storage of allogeneic valves for clinical use; however, disruption of leaflet ECMs by ice frequently occurs. This study was performed to determine the effects of cryoprotectants employed for ice-free, vitreous cryopreservation upon adherent porcine heart valve leaflet-derived myofibroblasts after exposure to cryoprotectants and after cryopreservation. Exposure experiments demonstrated that the individual cryoprotectants (0–5 M) or in combination were cytotoxic in a dose-dependent manner. After cryopreservation, the benefits of the combination of cryoprotectants over individual cryoprotectants were clearly demonstrated at 4 M (P < 0.001).
Footnotes
Acknowledgments
This work was supported in part by a cooperative agreement (No. 97-07-0039) between the U.S. Department of Commerce, National Institute of Standards and Technology—Advanced Technology Program, and Organ Recovery Systems, Inc., and by a grant (No. HL5973) from the National Heart, Lung, Blood Institute of the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.