
Abstract

In view of the exponential increase of the number of DNA samples to be stored, classical conservation in freezers appears cumbersome and costly and may be subjected to the risks of natural disasters. Room temperature storage of dehydrated DNA emerges as an alternative solution because
• the main degradation pathways (depurination, base deamination, and base or sugar oxidation) of DNA involve water, • solid state is known to generally slow down chemical processes because of reduction of molecular mobility, and • there are multiple examples of retrieval of DNA aged at room temperature.
However, DNA, even thoroughly dried, if left exposed to air, regains a fair amount of water and still undergoes strand breaking and oxidation. Moreover, the presence of the atmospheric pollutant ozone, very reactive toward DNA, should not be neglected. Thus, it appears that long-term storage of DNA at room temperature may require an absolute protection from the atmosphere. However, this protection cannot be provided by plastic containers because they are known to be permeable to moisture vapor. We therefore developed a new procedure for room temperature storage of DNA wherein DNA samples are stored under an anoxic and anhydrous atmosphere in small glass vials fitted in stainless-steel, laser-sealed capsules. Under these conditions, at room temperature or 70°C, no DNA degradation was detected after 8-month or 1-week storage, respectively, whereas samples kept in air in closed plastic tubes, either naked or in the presence of matrices, underwent clear or strong degradation and/or aggregation. This study demonstrates the necessity of protecting DNA from air to preserve its integrity for room temperature storage.
Introduction
However, before seriously considering room temperature for DNA storage, some important points should be taken into account. First, it is known that reactive oxygen species are still active in the solid state.7–10 Second, even thoroughly dried, DNA, if left exposed to air, regains a fair amount of water: about 10 molecules per nucleotide at 50% relative humidity. With purified nucleic acids or their components, this has been shown to be sufficient to promote depurination, base deamination, and oxidation.2,11
Third, ancient DNA can be recovered from archaeological remains only if the surrounding environment has been reasonably dry and anoxic. Even under those conditions, DNA appears heavily broken and bears signs of oxidation, cytosine deamination, and depurination. 12
An interesting observation is that, once bones have been unearthed, it seems that the DNA degradation rate increases sharply. 5 One can speculate that this is due to air exposure.
In summary, these observations seem to indicate that hydrolysis and oxidation still take place in the solid-state DNA and can be avoided only by protecting it from atmospheric water and oxygen. A last parameter to consider is ozone pollution because, on one hand, ozone has been shown to rapidly react with solid DNA and, on the other hand, a correlation has been established between ozone pollution, DNA adducts formation in cells, and lung cancer. 13
Hence, many evidences presume that DNA cannot be reliably stored at room temperature without protecting it from the atmosphere. Nature has found only a limited answer with anhydrobiotes such as tardigrades, as their maximum life expectancy, when dried, is about 5 years. 14 Spores or seeds use a much better approach, as some of them have been brought back to life after almost 2000 years of storage. 15 But, for practical reasons, man has to find a solution on his own. Moreover, polymers are known not to be really airtight, 16 and in a previous work, we confirmed this to be true for all the plastic tubes we tested. 2 The same is true for glass vials because of the rubber stoppers. Another problem with plastic tubes is the fact that they often release chemicals able to interfere with DNA or protein analysis and biological assays.17–19 For all these reasons, we developed a procedure for DNA room temperature storage based on improved containers.
Material and Methods
Plasmid DNA was extracted as previously described. 2 Genomic DNA from horse blood was prepared by classical phenol–chloroform extraction or using the Puregene kit (Qiagen). Plant genomic DNA was extracted using the plant DNeasy Qiagen extraction kit.
Degradation kinetics were conducted on vacuum dried plasmid or genomic DNA. DNA samples prepared for air exposure were deposited at the bottom of 0.3-mL cylindrical glass inserts and those treated with the commercial matrix were placed in Eppendorf-type 1.5-mL plastic tubes, as indicated by the supplier. These inserts were then fitted into stainless-steel capsules and vacuum dried so that kinetics tests could be run in the absence of air and moisture. The capsules were transferred into a vacuum (0.2 mbar) chamber, which was subsequently filled with anhydrous and anoxic argon (<1 and 0.5 ppm of water and oxygen, respectively) and samples were equilibrated overnight. Then the chamber was opened in a glove box maintaining in the same atmosphere. A stainless-steel top was fitted on the capsules, which were hermetically closed by laser welding.
After storage, DNA samples were rehydrated by adding water. Genomic DNA samples were generally denatured either with NaOH (pH>12) or by heating to 95°C for 5 min or both. All the denaturation procedures gave the same results as discussed later. Native or denatured genomic DNA samples and plasmid DNA samples were applied to nondenaturing agarose gel electrophoresis (0.8% agarose in 20 mM Tris acetate and 0.5 mM EDTA electrophoresis buffer). Gels were stained with ethidium bromide or Sybrgreen I and then photographed with a digital imaging device. The images were analyzed with the Kodak 1D or Bio1D (Vilbert-Lourmat) image analysis software.
Plasmid degradation was measured as follows: for ethidium bromide- and Sybrgreen I-stained gels, the image analysis software provided a fluorescence intensity for open circular (OC) and supercoiled (SC) forms of DNA: FOC,sample and FSC,sample, respectively. For highly degraded samples, the linear form (L) fluorescence intensity was added to that of the OC form. Then, the remaining percentage of SC (%SC left) was calculated according to the following formula:
Normalization by the control sample was used to correct for the slight variations between gels. Moreover, the classical α=1.4 value was factored into the equation to account for the weaker fluorescent signal for the SC form than the OC form. This value was determined using dilution series of the SC form, OC form, and their mixtures. To calculate the degradation rate, data points were fitted to a first-order kinetic: %SC left=100×e−nkt where t is in seconds. The fit gave n×k value in s−1; n is the plasmid total nucleotide number (nt). From that, we could calculate k in s−1 nt−1, a value independent from the size of the plasmid.
Results and Conclusions
The effect of air exposure on the stability of DNA was first studied by room temperature kinetics conducted in different conditions.
In the first experiment, plasmid DNA samples were placed in closed Eppendorf-type tubes containing air. In that case (Fig. 1, closed circles), degradation could not be assessed beyond 1 month because, after that time, only miniscule amounts of DNA could be recovered. It is likely the samples underwent aggregation and adsorption on the container walls as is commonly observed with dried DNA stored in the presence of moisture. 20 This phenomenon has been shown to be efficiently prevented by using trehalose or other sugars. 21 So we ran a similar kinetic in the presence of trehalose (triangles) or a commercial matrix (diamonds). It can be seen that aggregation was prevented, but degradation still occurred. At last, when DNA samples were kept in sealed capsules (squares), no degradation was detected at the end of the 8-month period of the experiment. The yield of DNA recovery was close to 100%.
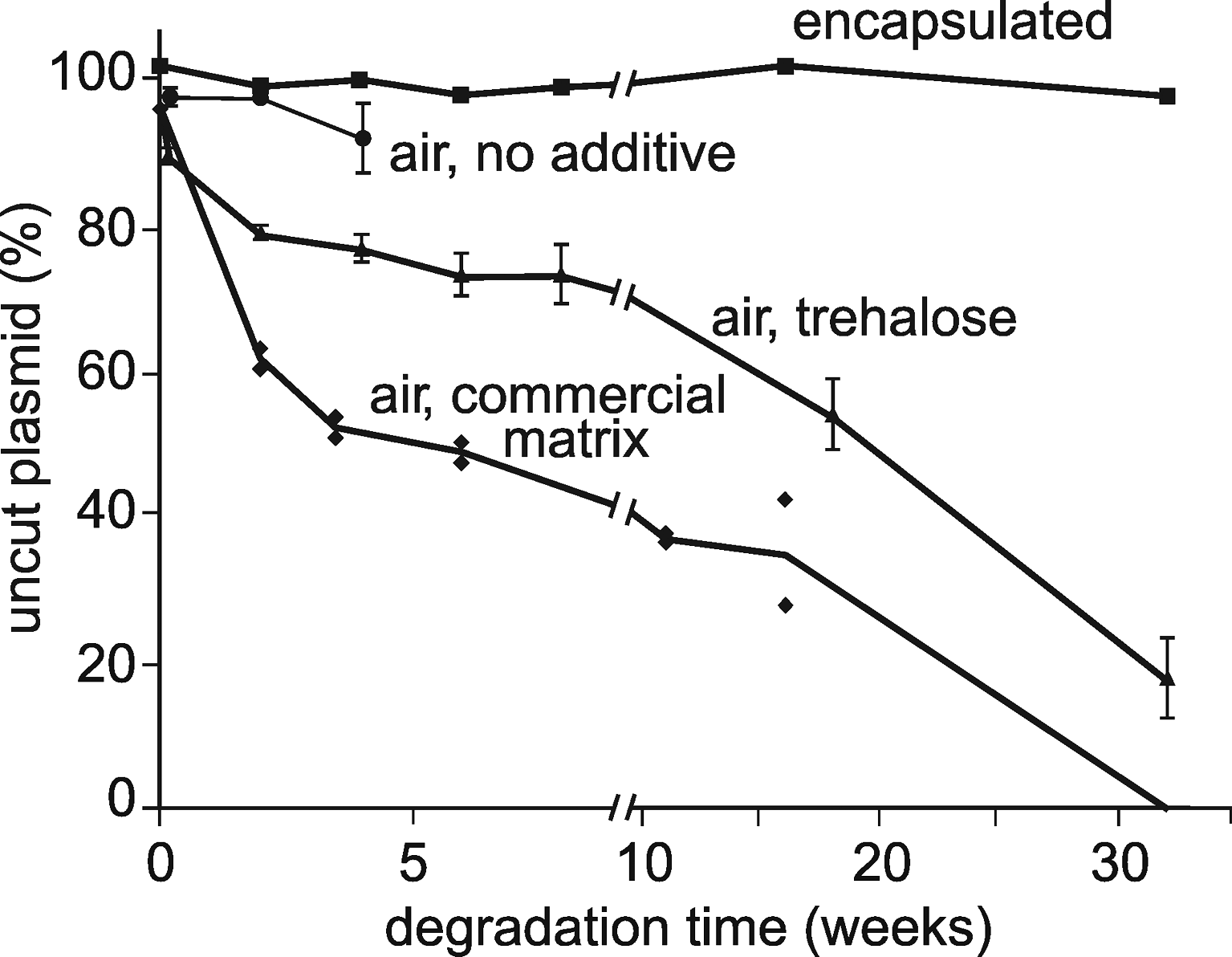
Influence of air and matrices on plasmid DNA degradation at room temperature. Dehydrated plasmid DNA (200–250 ng) was incubated at room temperature under the following conditions: encapsulated (squares), that is, protected from moisture and oxygen (see Material and Methods section); unprotected, without additive, in glass inserts inside closed plastic tubes (circles); unprotected, embedded in a trehalose matrix, in glass inserts inside closed plastic tubes (triangles); unprotected, embedded in a commercial matrix, inside closed plastic tubes (diamonds). Supercoiled plasmid proportion was estimated as described in the Material and Methods section and normalized by comparison to the same plasmid stored at −20°C. Triangles and circles are the average of 3 measurements; diamonds and squares were single measurements.
In the second experiment, we used genomic DNA. Samples were kept at room temperature and degradation was assessed by gel electrophoresis before and after DNA denaturation. It can be seen (Fig. 2, left gel) that, in the absence of air, no degradation was detectable in the 8-month storage period as expected from our previous estimations. 2 On the contrary, in the samples unprotected from air, DNA degradation clearly occurred, and the average single strand size was estimated as low as 14 kb. Assuming a constant chain-breaking rate, this average size corresponds to a degradation rate of about 4×10–12 nt−1 s−1. This value is in agreement with the plasmid breaking rate (7×10–12 nt−1 s−1) estimated from the kinetics of Fig. 1 and with our previous estimates of the chain-breaking rate at room temperature of solid-state DNA exposed to air (about 1.6×10–12 to 10×10–12 nt−1 s−1). 2

Influence of air and matrices on genomic DNA degradation at room temperature. After storage in the conditions indicated above the gel, 500 ng aliquots, either denatured (left) or nondenatured (right), were run on a nondenaturing gel (see Material and Methods section for details). M1 is the “1 kb” λ phage marker (denatured). M2 is the “HindIII” λ phage marker (nondenatured). The marker lane (M1) was darkened to make bands more visible. The DNA used for the 2 different kinetics had the same origin (horse blood) but were prepared differently: for the capsules, gDNA1 was phenol–chloroform extracted, and for the commercial matrix, gDNA2 was extracted using the Puregene kit (Qiagen). Both techniques gave DNA with equivalent quality.
These results are not particularly in contradiction with those presented for validating commercial matrixes. The differences can be explained on mere technical ground. First, a frequently used technique for showing a lack of degradation of DNA is polymerase chain reaction (PCR) because any cut in the target sequence will prevent its amplification. However, on one hand, end-point PCR will show a decrease only when there is almost no intact copy left, and on the other hand, real-time quantitative PCR is generally used with short amplicons (100–300 bp) and is thus quite insensitive to DNA degradation. Second, a common way to assess DNA degradation is to use nondenaturing agarose gel electrophoresis of genomic DNA but, as shown in Fig. 2, this considerably underestimates the degradation state of DNA. Third, the technique using plasmid relaxation is very sensitive: for instance, a 50% decrease of SC corresponds for the plasmid we used to an average of one cut per 20,000 nucleotides. This level of degradation could have been easily overlooked by the less-sensitive techniques generally used.
To confirm these results, we conducted accelerated aging studies. We heated DNA samples at 70°C at 50% relative humidity for 7 days. The experiment was done on 7 different horse or plant DNA preparations. All of them gave the same results. A representative gel analysis is given in Fig. 3. The following conclusions can be drawn: (1) encapsulated (fully protected) DNA in the presence of trehalose matrix exhibited no detectable degradation, but in the absence of trehalose a part of the DNA was lost probably through aggregation/adsorption; (2) when exposed to moist air in the presence of the trehalose matrix, DNA samples were clearly degraded; and (3) when exposed to moist air in the presence of the commercial matrix or in the absence of any matrix, the DNA was completely lost by degradation and/or aggregation/adsorption.

Influence of air and matrices on genomic DNA degradation at 70°C. Horse genomic DNA was heated for 7 days at 70°C, in the conditions indicated above the gel. DNA samples (500 ng), either denatured (right) or nondenatured (left), were run on a nondenaturing gel (see Material and Methods section for details). M1 is the “1 kb” λ phage size marker. On the left gel, samples were run in duplicates.
In conclusion, this study demonstrates the necessity of protecting DNA from air to preserve its integrity at room temperature. These findings further confirm our previous accelerated degradation studies 2 that DNA can be preserved at room temperature for periods that must meet the most demanding storage requirements.
Footnotes
Acknowledgments
This work was supported by the “Conseil Régional d'Aquitaine,” the “Pôle Aquitaine Santé,” and “Oséo Innovation.” The authors thank Christian Oste for helpful suggestions and manuscript editing.
Author Disclosure Statement
The authors may have competing interests in relation to this work: J. Bonnet is a shareholder and a consultant for Imagene; M. Colotte and D. Coudy are employees of Imagene; and S. Tuffet is CEO and shareholder of Imagene.