
Abstract
Access to biospecimens and their derivatives, that is, human biological materials (HBM), for translational research (TR) is considered a major bottleneck hindering successful bench to bedside translation. Clinical trials offer a unique opportunity to collect HBM in a specialized setting that allows prospectively designed, high-quality TR that would be difficult to fulfill from community- or population-based HBM collections alone. Increasingly, as the field advances toward personalized treatment of cancer patients, access to HBM is becoming a necessity for patient enrollment in a new generation of clinical studies that are designed and driven by molecular hypotheses. The European Organization for Research and Treatment of Cancer (EORTC) is one of the largest networks for clinical trials in oncology. The EORTC is re-focusing its strategy, building on experiences and expertise gained over the years from specific initiatives such as EORTC Group activities and the EORTC Virtual Tumour Bank, by developing new mechanisms to support investigators with the practical aspects of HBM collection as part of EORTC clinical studies. Due to the complex, multidisciplinary nature of HBM collection and TR, integration of HBM collection into clinical trials warrants careful upfront planning and input from a range of expertise. To simplify HBM collection in clinical studies, the EORTC has developed a simple checklist containing the key elements of HBM collection setup and combines these into a simple tool for practical use. Through identifying and managing key risk areas, this can maximize the HBM collection success while achieving efficient clinical trial development. This article focuses on the key elements of HBM collection and the approaches of the EORTC for efficiently integrating this collection into clinical trial development.
Introduction
HBM collection in population-/community-based biobanking has much value for hypothesis generation, assay development, and analytical validation (the ability of the test to measure accurately and reliably the genotype or phenotype of interest) and assessment of biomarker frequency in a population-based setting. However, numerous findings from TR performed using such heterogeneous sets of convenience samples have often not been confirmed in subsequent studies. 1
Recently, many methodological issues regarding TR study design and the lack of reproducibility and reliability have been addressed via guidelines relating to the design and conduct of biomarker studies. Examples include reporting criteria for prognostic and predictive biomarker studies and the Level of Evidence (LOE) scale for evaluation of the strength of evidence of a biomarker and whether to recommend its use in clinical practice.2,3 Clearly, clinical trials offer a unique opportunity to implement these guidelines for TR with a high degree of rigor. Most importantly, the treatments and the follow-up of patients in clinical trials are standardized, whereas this is less the case outside of clinical trials. As a result, many important elements determining robustness and reliability of TR results such as patient selection and data collection, standardized HBM collection, and statistical design of TR projects can be controlled to a greater degree. Increasingly, as the field advances toward personalized treatment of cancer patients, access to HBM is becoming a necessity for patient enrollment in a new generation of clinical studies designed and driven by molecular hypotheses. Therefore, HBM collection in clinical trials is of increasing importance to increase the rate of adoption of the use of biomarkers in clinical practice.
Due to the multidisciplinary nature of HBM collection and TR, integration of HBM collection into clinical trials warrants careful upfront planning and requires input from a range of different expertise. As much as possible the HBM collection should be integrated into standard clinical practices that are used in the participating hospitals and should be made as simple as possible to facilitate the collection.
The European Organization for Research and Treatment of Cancer (EORTC) is one of the largest networks for clinical trials in oncology. The EORTC groups have a long-standing expertise in collecting HBM in many early clinical studies to evaluate drug pharmacokinetics and pharmacodynamics. 4 These studies require careful sampling according to strict protocols, as required by registration agencies such as the U.S. Food and Drug Administration and the European Medicines Agency. Many of this HBM has subsequently been used for discovery and evaluation of other pharmacodynamics and pharmacogenetic biomarkers. This led to the association of toxicity of both cytotoxic and targeted drugs with certain genetic polymorphisms, as well as associations with outcome of therapy. However, the various requirements for specific drugs revealed limitations for retrospective discovery of other potential biomarkers, in particular with novel methodologies such as genomics and proteomics. Additionally, collection of tumor biopsy specimens was not routinely implemented in these studies.
To standardize HBM collection in clinical trials (not only plasma, serum, or urine, but also other specimens such as whole blood and frozen, formalin-fixed, paraffin-embedded tumor tissue [FFPE]), the EORTC is developing strategies to support investigators with the practical aspects of collection, storage, and access to HBM, which builds on the existing experiences of the EORTC groups. This article focuses on the considerations required for such collections and approaches taken by the EORTC to integrate HBM collection in clinical trial protocols.
Prospective rather than retrospective HBM collection
As access to HBM as part of clinical studies is becoming critical for the development of the field, 3 the EORTC is identifying processes and mechanisms to streamline HBM collection. An identified difficult area is where HBM collection is not foreseen in the clinical trial protocol and investigators wish to retrospectively retrieve HBM from multiple locations after trial closure. Here, many factors can lead to a poor retrieval of HBM suitable for research, including filing and obtaining positive votes from ethics committees (EC) in different centers and countries, loss of HBM due to varying local site policies for retention, and release of HBM and variable quality (and quantity) of HBM.
The adopted strategy to best secure the HBM is through prospective planning of the collection in the protocol. In newer trials, HBM collections are expressly written (type of material, amount/volume, time point, etc). Prospective HBM collection allows harmonization of the collection and handling techniques, and planning for logistical and financial support, and allows integration with other aspects of the protocol, for example, sampling at the same moment as imaging studies. By including HBM collection in clinical trials, the amount of information collected from each patient's participation is increased, making the best possible use of the donation of each patient to further research efforts toward better cancer care.
Integrated vs. correlative TR
HBM collected within the framework of trials can be used in an enormous range of different TR studies, ranging from biomarker discovery and validation to assessment of clinical validity and clinical utility. To facilitate the integration of TR in clinical studies at the EORTC at an early stage in development, TR is dichotomized into one of two types: correlative or integrated TR.
Correlative TR is where the biomarker question being addressed is accommodated within a trial that is designed and powered to address another hypothesis. The TR does not form part of the trial design itself and can be considered as a side-study. An example of a correlative TR study was the use of archived HBM from the EORTC 26981/22981, a randomized trial comparing radiotherapy alone with radiotherapy combined with concomitant and adjuvant treatment of glioblastoma patients with temozolomide. In this study, correlative TR generated strong evidence that patients whose tumor contained a hypermethylated MGMT gene had a greater benefit from temozolomide than patients whose tumor did not have MGMT hypermethylation. 5
At the EORTC the term “integrated TR” is used to describe the situation where a biomarker assay forms part of the clinical study design. This integrated TR drives the trial design; for example, it could be a stratification factor, a randomization factor, or even a surrogate endpoint. An example of integrated TR is the gene expression signature used in MINDACT (Microarray In Node-negative and 1 to 3 positive lymph node Disease may Avoid Chemo Therapy, EORTC Protocol 10041-BIG 3-04). MINDACT is a randomized study in which a 70-gene expression signature (MammaPrint) is combined with the risk by classical pathobiological criteria to place patients into 1 of 2 risk groups, a “consistent” risk group (where the gene signature risk and classical pathobiological risk agree, and the chemotherapy decision is taken according to these agreeing risk assessments) and a discordant risk group in which patients are randomized to have chemotherapy or no chemotherapy. 6 For integrated TR, all patients must have available HBM of sufficient quality to enable the biomarker assessment to take place and hence integrated TR can also lead to eligibility/noneligibility for the study. Guidelines for HBM collection in similar breast cancer studies have been published recently. 7
A checklist for HBM collection setup
Establishing an HBM collection within the context of a clinical study is complex and requires a range of expertise. As many different factors influence the success of HBM collection, these must be considered at an early stage to ensure that the collection is feasible, making upfront planning of any collection indispensible. Many of the elements influencing the feasibility of an HBM collection are inter-dependent; hence, changing one factor such as the type of HBM being collected or the frequency of sampling can dramatically impact on the final planning and cost of an HBM collection (Fig. 1).
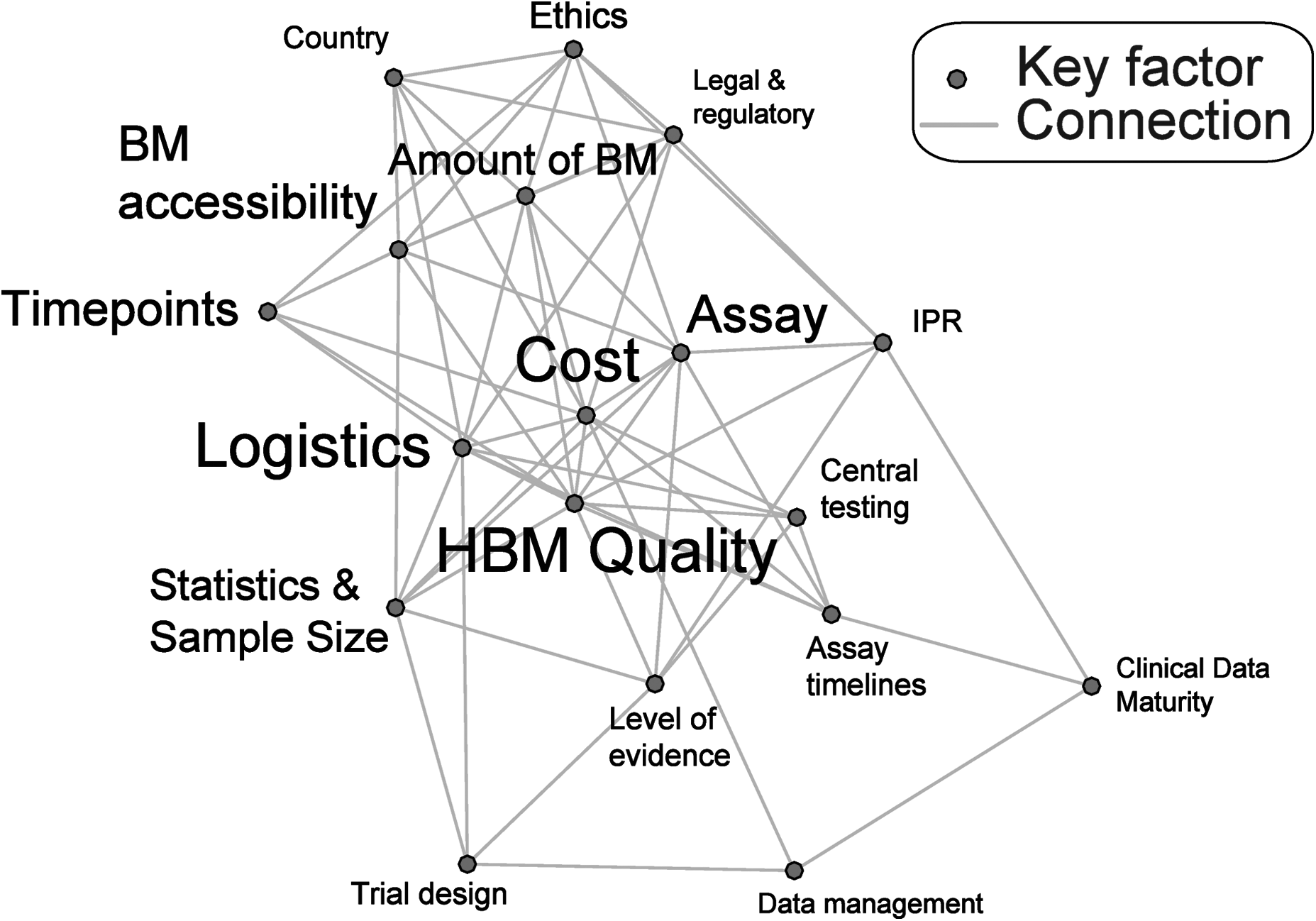
A network diagram illustrating the inter-relation between the key factors involved in an HBM collection setup. Factors occupying a central position on the graph have the highest number of connections (and hence the most inter-dependency); this property is also reflected by the size of the text. IPR, intellectual property rights. HBM, human biological materials.
Integrating HBM collection into the timelines of the setup of a clinical trial adds additional challenges and warrants a careful coordination to ensure the input of various areas of expertise at different moments in protocol development. To simplify planning and integration, a checklist containing the key components for HBM collection setup has been developed by the EORTC Translational Research Unit (TRU) in collaboration with other units of the EORTC headquarters, the EORTC groups with experience in HBM collection and biobanking, including the EORTC PathoBiology Group (PBG) and the Pharmacology and Molecular Mechanisms (PAMM) Group of the Translational Research Division (TRD) (Fig. 2). The key components of this checklist will be addressed in greater detail.

A practical checklist tool for HBM collection setup. The checklist contains key elements for any HBM collection as part of a clinical trial.
Key Components for HBM Collection
Rationale and concept of the TR, the trial design, and the LOE of the biomarker
Obviously, for the conduct of any type of TR project, it is fundamentally important that the rationale and goal should be clear. Equally, the role that the biomarker assay will play in the clinical trial should be clear and the rationale for its use should be scientifically sound.
Further, the design of the trial within which the HBM collection is being accommodated is a crucial element influencing the range and type of integrated and correlative TR. Clearly, different questions may be more applicable in early phase I/II trials, with fewer patients and where the goal may be to better understand drug activity, for example, using omics approaches that may necessitate the collection and use of high-quality frozen tissue samples. In contrast, a large phase III trial with a treatment and control arm will be an ideal situation to demonstrate the utility of a known marker for predicting response to therapy, for which standard clinically available HBM such as FFPE tissue specimens could be used.
In addition, the LOE of which the TR study is intended to achieve could lead to the implementation of different requirements for HBM collection. Recently an update of the LOE scale was suggested by Simon and colleagues. 8 This LOE scale is based on the key elements that influence the robustness of a TR study including aspects relating to HBM collection. For example, a higher LOE for a biomarker can be achieved through studies with prospectively planned HBM collection where specimens are analyzed in real time for the biomarker of interest but also through prospective collection of HBM into biobanks using generic standard operating procedures (SOPs). HBM may then be analyzed later, perhaps after completion of the trial, with appropriate preplanning of the statistical design. In contrast, a lower LOE would be obtained from biomarker studies using HBM that was collected, processed, and archived without SOPs.
Therefore, to perform a TR study with a high LOE, clinical trials offer a unique context for prospective collection of HBM from a specific preselected patient population using defined and standardized protocols for HBM collection; this is quite the contrast from performing a retrospective analysis of a biomarker on a set of convenience samples taken from patients with heterogeneous treatment regimens and follow-up.
In the circumstances where the final goal of the TR is not yet known at the time of writing the clinical study protocol, HBM can be collected wherever possible and archived in a biobank for future use, under appropriate conditions.
Assay
The selection of the assay (that is dictated by the rationale of the research) will drive the HBM collection requirements of the study. Variability in specimen collection, storage, processing, and analysis represent major potential sources of bias in TR and could produce artifacts resulting in misinterpretation of test results. For this reason, each biomarker study, whether correlative or integrated, requires robust, validated assays that are fit for the intended purpose. Most importantly, the type of biomarker to be investigated will determine the type of assay platform, for example, a correlative TR biomarker discovery project using gene expression profiling or other genomic profiling techniques will have different requirements than protein profiling in a proteomics setting. This type of work often requires gold-standard frozen tissue. Subsequently, these validated, robust clinically applicable assays can be used in a biomarker qualification/validation study, which are often performed in a surrogate tissue such as serum.
Less robust assays may be more sensitive to subtle variations in the HBM collection and preservation methods, making the need for tightly controlled, standardized collection protocols important. The degree of analytical validity required for the assay must be established, both for integrated and correlative TR. In this context, quality assurance programs should be activated to establish consistency in assay methodologies and reagents employed to determine inter- and intra-assay variability and to minimize errors that could affect assay results. Quality assurance and quality control programs can be implemented at different levels, that is, to check preanalytical conditions (since the quality of HBM markedly affects the quality of TR), to assess assay performance and reproducibility, and to analyze, interpret, and report assay results.
Particularly for integrated TR, consideration of the expected failure rate of the assay is important for assessing the feasibility of the implementation of the biomarker. Assay failure rate can impact on the number of patients who need to be screened to enter the trial, and early consideration should be given on how to follow-up and treat patients that are not assessable for the status of the biomarker.
Lastly, the selected assay may determine the final scoring system and cut-point of the biomarker; that is, is it a quantitative, semi-quantitative, or categorical variable? This is an important issue if the biomarker is to be used for treatment decisions and subsequently impacts on the statistical design and power calculations.
Statistical considerations and sample size for the design of the TR project
The field of biomarker research has suffered from poor study design resulting in inconsistent conclusions or results that are in direct contradiction to one another.3,9 Common methodological pitfalls include inadequate sample sizes and failure to account for multiple hypothesis testing.9,10 Hence, once the hypothesis and type of data generated by the assay are clearly defined, statistical considerations can be addressed. For correlative TR, it is important that the TR project is robustly designed and that the statistical analysis methods are clarified and described in sufficient detail to allow up-front consideration of the power and sample size. Even if the TR project is exploratory in nature, some rationale for sample size should be given of why that number of specimens should be collected or better still the TR study should be designed to detect a specified effect size, for example, a specified hazard or odds ratio. This sample size should be matched with the context of the trial design itself to see if the two are compatible and the correlative TR is accommodated by the trial design. A key parameter that impacts on the sample size estimation is the frequency of occurrence of the biomarker (eg, an infrequent biomarker requires a large sample size to achieve the same power), which can greatly influence the feasibility of accessing the required number of specimens to perform the work, particularly for integrated TR.
Amount or volume of HBM required for analysis, the accessibility of HBM, and time points for collection
The selected assay may impose necessities on the amount of HBM required, for example, to allow sufficient amounts of DNA to be extracted, or nondegraded proteins to be isolated. The availability of the right amount of HBM specimen is a critical feasibility factor for any TR project. This assessment must take into account the normal treatment of the patient, the physical location of the specimen, and how amenable to biopsy or collection it is, the risk of intervention in that particular clinical setting, and the time-points for collection. It is important to assess whether residual HBM from healthcare samples (eg, diagnostic tissue samples or FFPE blocks) can be used or whether additional HBM should be taken to allow the TR to take place. If the HBM accessibility is not compatible with the requirements of the selected assay, further work for developing a more robust assay may need to take place, or a reconsideration of the selected biomarker or assay in question. These steps may take several rounds of iteration before the right balance is achieved.
Participating clinical sites, timelines for assay processing, and centralized vs. decentralized biomarker testing
As the details and requirements of the clinical trial itself become clear, the countries and centers that will be contributing to the HBM collection will be clarified. Additionally, in the case of integrated TR, the ability and willingness of a site to perform the HBM collection required for the trial to take place can be taken into account in the selection of the participating sites. In relation to HBM collection, different countries may have different regulations, regarding handling and shipping. For example, whether the countries are within the European Union or not can impact on costs and timelines of shipment across borders and through customs. In addition, specific considerations will need to be made on a country by country basis for the implementation and content of the patient information sheet and informed consent (PIS/IC) to ensure compliance with local legislation. In an integrated TR study, careful consideration of the total time from shipment to analysis and obtaining the biomarker assay result is needed, since the timing of the patient treatment may be dependent on the outcome of the biomarker assessment and the patient must receive treatment within an acceptable period of time. These feasibility elements combined with the maturity of the technology, any implemented quality assurance/quality control program and the level of robustness of the assay contribute to the decision of whether a centralized testing of an integrated TR biomarker should be implemented or whether the testing should be done locally.
Writing the PIS/IC document
HBM can only be collected, stored, and used with the appropriate consent of the study participant and/or EC, as applicable. A PIS/IC document relating to HBM collection and its use in research is presented to the patient at the time of asking consent for the patients participation in the clinical study. For storage of HBM in a biobank for future research not known at the time of protocol writing, the patient can be asked a broad consent (eg, for “cancer research”) for the HBM to be stored and used in new research projects not yet known at this time, subject to approval of new projects by an EC. For the use of HBM in research where no specific consent for nonpredefined research had been obtained (eg, older clinical studies), EC (and/or other regulatory bodies if applicable) should be approached for advice, review, and approval before commencement of the project.
Key items that should be mentioned in the PIS/IC include what additional intervention or burden the patient may experience by participating in the HBM collection (eg, the number of extra blood samples), if HBM may be shared with third parties for scientific research collaborations, and that HBM may be transferred out of the patients' own country. Patients may withdraw their consent for the use of their HBM in research at any time, and it should be clearly explained that patients waive any claim to intellectual property rights resulting from research on the patients' HBM.
Logistics
Establishment of the logistical planning may come late in the process of the HBM collection setup, as many of the earlier factors must be clarified before a clear view of the logistics can be envisaged. Considerations for the frequency and timing of shipments of HBM, such as batched shipments or real-time sending, the conditions (eg, frozen or ambient), the countries and sites involved, and the requirements of the biomarker assay turn-around time should all be considered. HBM should also be traceable, that is, the location of the HBM at any time should be known. Logistics and handling protocols for HBM collection should be developed and delivered prospectively to the sites participating in HBM collection. As much as possible, the HBM collection should be integrated into standard clinical practices and must be made as simple as possible for those on site performing the collection to facilitate the success of the collection. To this purpose, personnel involved in all the preclinical phases of TR projects should attend training courses and updating of the procedures and their competency should be checked on a regular basis.
Data collection
As well as collecting the HBM itself, it is of paramount importance to collect information regarding the HBM. Key variables such as deviations from collection SOPs, data on HBM handling, surgical techniques used to obtain the specimen, the time of processing, and storage of HBM can subsequently influence the later use of the HBM. Also, additional information on the patient (eg, diet and health background etc.) may be important to collect within the context of a given TR project. The specific data to be collected can vary between different HBM types and the type of TR proposed. Critically important is the collection of pathology data such as histology and grade, and follow-up information such as survival times and the treatments received. These additional variables should be considered side by side with the case report forms of the trial itself to ensure that the correct minimum data set is captured. At the EORTC clinical information can be collected via a remote data capture system using case report forms and new developments for a new tool for tracking the locations and times of HBM shipment as well as quality information of the HBM are underway.
Budget for the HBM collection
At the end stages of the checklist, a final cost of the HBM collection can be established. The feasibility of the HBM collection or even the trial itself (as in the case of integrated TR) may be heavily influenced by this final budget estimation. Several re-iterative steps may be required to readjust decisions on each element of the checklist to reach a feasible and implementable budget for the HBM collection.
Integrating the checklist into clinical trial protocol development
To integrate HBM collections in clinical trial protocols, the concept of the TR project(s) must be clarified early during the protocol development process. For that purpose, two submission forms have been implemented at the EORTC, one for integrated and one for correlative TR. These submission forms contain questions addressing the key components of the clinical trial but also the associated HBM collection and have to be completed by the investigators.
The EORTC translational research advisory committee (TRAC) is a committee that provides expert scientific and practical advice on TR projects conducted within the EORTC. Using the submission forms, the TRAC makes an early review of the clinical study concept and proposed HBM collection to stimulate ideas for TR, to clarify rationale, and to give a feasibility assessment. The recommendations of the TRAC are discussed and coordinated with the TRU at the EORTC headquarters and are disseminated to the protocol writing team. Later in clinical trial development, a more detailed protocol outline is developed and more practical aspects of HBM collection are addressed, for example, participating sites and countries, setting up logistics, drafting the PIS/IC, and defining the data to be collected. SOPs for the collection of HBM are developed prospectively and distributed to the involved centres prior to HBM collection (Fig. 3).
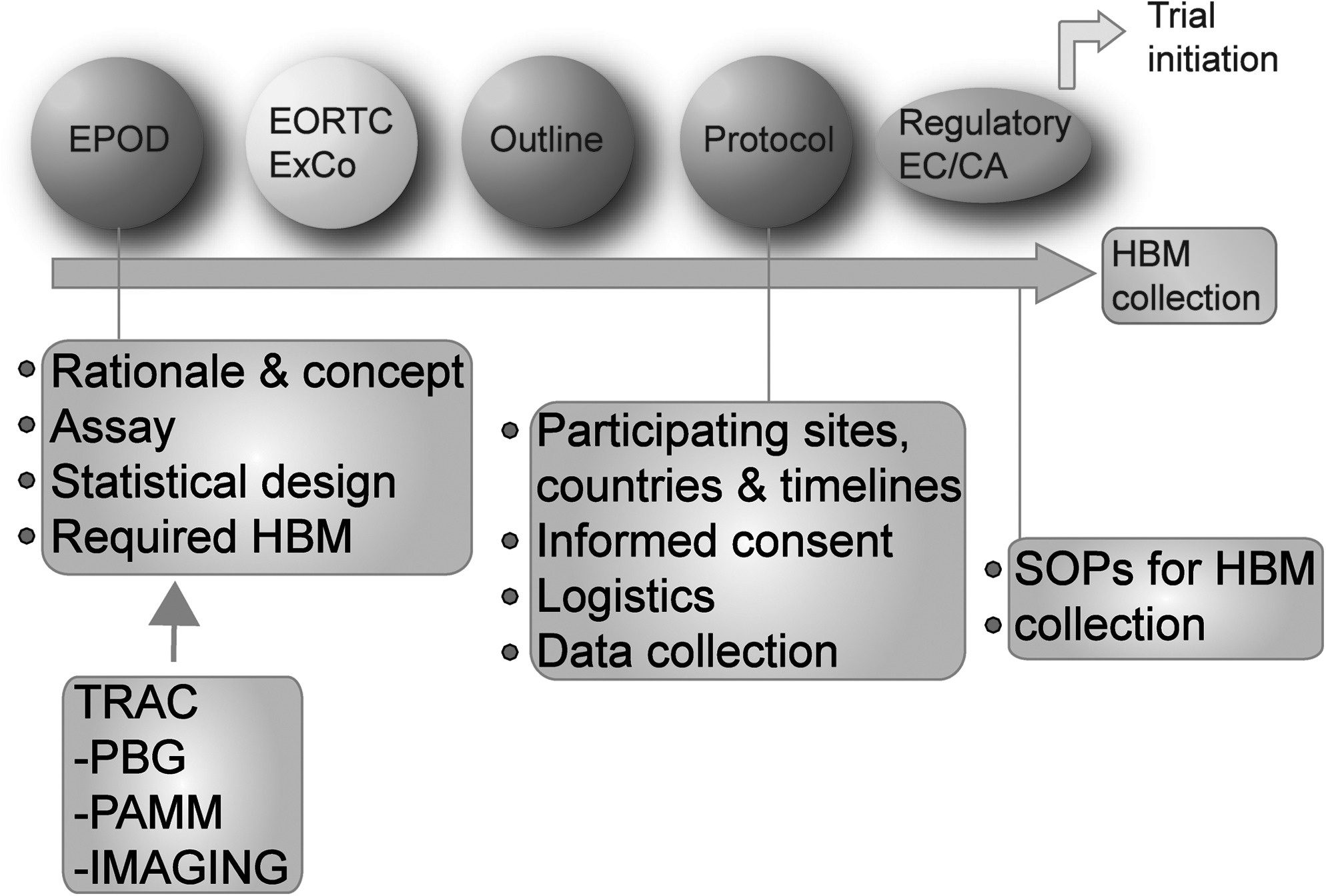
The integration of HBM collection setup into the process of clinical trial protocol development. New clinical trial concepts enter via the Early Project Optimization Department (EPOD) at the EORTC headquarters. The EPOD offers strategy advice, project support, and optimization. Clinical studies and associated TR are assessed by the EORTC Executive Committee (ExCo), and successful studies are developed into a more detailed protocol outline and subsequently a full protocol document within which the HBM collection is expressly written. After approval of the final protocol, SOPs for HBM collection are sent to sites before HBM is collected from patients. EORTC, European Organization for Research and Treatment of Cancer; SOP, standard operating procedure.
Conclusion
It is widely appreciated that HBM collection for TR in the context of prospective clinical trials is essential for further progress in oncology. However, it is also clear that HBM collection is a complex process with many potential factors endangering the ultimate success of the TR study.
By establishing the crucial elements for HBM collection, we aim to provide a tool to navigate the complexity of HBM setup, bringing critical decision points to a manageable level, thus increasing the effectiveness of HBM collection in clinical trials. Importantly, this is a dynamic process that should be evaluated and adjusted constantly.
Key to the above processes and strategies is the involvement and feedback from experts from many diverse disciplines. In view of this, the EORTC headquarters is actively involved in engaging pathologists, biobankers, biostatisticians, basic scientists, physicians, and others from in and outside the EORTC network. All this expertise should be involved in the early stages of concept development, rationale, and design of TR studies. Only through such intense collaboration, HBM collection for TR can be further improved thereby greatly contributing to further progress in the management of cancer patients.
Footnotes
Acknowledgments
We would like to acknowledge all the members of the EORTC TRAC, Pharmacology and Molecular Mechanisms Group, PathoBiology Group, TRU, and EORTC headquarters staff for their expertise and contribution to this work in addition to the long-standing expertise of the EORTC groups. We would like to acknowledge all the physicians, pathologists, and biobankers and the staff at the clinical centers involved in HBM collection in EORTC studies. In particular, we are indebted to all the patients and their families for participating in EORTC clinical trials and for donating their biospecimens for TR. This publication was supported by Fonds Cancer (FOCA) from Belgium.
Author Disclosure Statement
No competing financial interests exist.