
Abstract
The comet assay measures DNA damage in individual cells (usually lymphocytes) and is widely used in biomonitoring studies. Lymphocytes are harvested and are usually cryopreserved for batch testing. We investigated cell loss during harvesting, cryopreservation, thawing, and washing of human peripheral lymphocytes and compared DNA damage, using the Fpg-assisted comet assay for oxidation-induced DNA lesions, in freshly harvested cells and cells that were thawed and tested after cryopreservation of 2–3 days and 4 weeks. Lymphocyte numbers were measured in fresh venous blood and after the steps of harvesting, cryopreservation, and washing. Results showed that >50% of lymphocytes in whole blood were harvested, but ∼60% were lost during washing. Loss during washing was not different (P>0.05) between fresh cells and cells thawed and washed after 2–3 days or 4 weeks cryopreservation. No change in DNA damage was seen after cryopreservation and thawing: mean (SD) % DNA in comet tail was 11.2 (1.53) in freshly harvested cells, 12.9 (1.39) in 2–3 days cryopreserved cells, and 12.9 (2.0) in cells tested after 4 weeks cryopreservation (P>0.05). Results indicate that there is no predominant loss of more highly damaged cells during cryopreservation and thawing and there is no induction of oxidation-induced DNA lesions in cryopreserved cells stored for up to 4 weeks.
Introduction
DNA damage can be measured by several techniques, but a widely used, validated method is the comet assay.4–6 This assay, which exists in several versions or modified forms, measures damage as single-strand breaks in DNA. The formamidopyrimidine–DNA glycosylase (Fpg)-assisted version of the comet assay measures oxidation-induced DNA damage more sensitively and specifically than the standard alkaline version, because the enzyme predominantly recognizes lesions that contain oxidized purines, mainly 8oxoGua, within nuclear DNA and creates strand breaks at these lesions.6,7
The most widely used cell type in human studies using the comet assay is the peripheral lymphocyte, which can be harvested easily from venous blood.4–6,8,9 The amount of damage to nuclear DNA in peripheral lymphocytes is thought to provide a general overview of global oxidative stress within the body. 6 For logistical and technical reasons, most investigators test lymphocytes that were harvested from fresh venous blood and then cryopreserved and stored at low temperature for days or weeks. However, there are 2 areas of possible concern in assessing DNA damage in such cells. One is that during the steps of cryopreservation, storage, and subsequent thawing and washing of cells, cell loss is likely to be very high, and it is possible that a certain subset of susceptible cells may die. For example, cells that are highly damaged may be more easily lost during freezing, storage, and thawing, and therefore, the DNA damage in the surviving stored cells may not be representative of that in freshly harvested cells before cryopreservation. The other concern relates to the duration of cryopreservation: damage may accumulate during storage, or cells may be more susceptible to oxidation-induced damage during thawing and washing when they have been stored for a longer period. Therefore, the aims of this study were to investigate the magnitude of cell loss during the harvesting, cryopreservation, thawing, and washing steps of lymphocyte preparation for comet assay testing and to compare the amount of DNA damage, measured using the Fpg-assisted comet assay, in cells that were tested soon after harvesting and without freezing, with that in cells that were thawed and tested after 2–3 days and 4 weeks of cryopreservation.
Materials and Methods
Lymphocytes from 3 healthy adult volunteers were used in independent experiments, and samples were processed independently but in parallel by 2 investigators. For each volunteer, 8 mL of venous blood (2 mL in EDTA and 3 mL in each of 2 heparinized blood collection tubes) was collected. The total lymphocyte number was counted in an aliquot of fresh EDTA blood using the Cell-Dyn3200 (Abbott Diagnostics). One tube of heparinized blood from each volunteer was used by each investigator, who harvested, cryopreserved, stored, thawed, and washed the lymphocytes following the same, standardized protocol.10,11 Each investigator counted the cells using a hemocytometer with trypan blue to assess cellular integrity (see below) and calculated the percentage of cell loss at each stage of processing. This was to see whether cell loss or effects of processing on DNA damage were operator dependent as well as to look at loss at each stage. One operator was highly experienced and 1 was recently trained in harvesting and processing lymphocytes. The Fpg-assisted version of the comet assay was performed on freshly harvested, nonfrozen cells and on cryopreserved cells after 2–3 days' and after 4 weeks' storage. Comet assay testing and scoring were performed by 1 of the investigators only. The experimental design is summarized in Fig. 1.

Schematic diagram of the experimental design.
Extraction and cryopreservation of lymphocytes from whole blood were performed using established protocols.10,11 For each subject, 3 mL heparinized venous blood was transferred to a 15-mL centrifuge tube and mixed with an equal volume of phosphate-buffered saline (PBS), after which 3 mL of Histopaque® (Sigma Aldrich) was added carefully to the bottom of the centrifuge tube. The tube was centrifuged at 1200 rpm for 20 min at 20°C to isolate the mononuclear cells (lymphocytes and monocytes) from whole blood. As the majority (>70%) of mononuclear cells in human blood consist of lymphocytes, the isolated cells are referred to as lymphocytes in this report. The isolated lymphocytes (which appeared as a blurry milky layer in between the Histopaque® and plasma/PBS layers) were transferred to a clean centrifuge tube, washed with ∼10 mL cold PBS, and centrifuged at 1200 rpm for 5 min at 4°C. The supernatant was discarded. After a second wash, the cell pellet was resuspended in 3 mL freezing medium (FBS and DMSO in a ratio of 9:1), and the cell suspension was aliquoted into 1.5-mL Eppendorf tubes. A total of six 0.5 mL aliquot samples were prepared by each investigator from each of the 3 volunteers, and an assumption was made that all 6 aliquots of cells from each individual blood sample contained very similar numbers of lymphocytes. For each blood sample, 1 of the aliquots (referred to as “fresh cells”) was used immediately in the comet assay and the other aliquots were frozen slowly in a thick-walled polystyrene box in a −80°C freezer. Using this procedure, the cells are exposed to a decrease in temperature of ∼1°C per minute. Aliquots of cryopreserved cells were removed and quickly thawed after 2–3 days and after 4 weeks of storage. Fresh cells and thawed cryopreserved cells were treated identically in the subsequent procedures of washing, cell counting, embedding, and Fpg comet assay testing.
To count the cells, 10 μL of cell suspension was removed and mixed with the same volume of trypan blue dye. Cells with a damaged membrane take up this stain. Using a hemocytometer and a light microscope, stained cells can be distinguished from unstained cells during visual cell counting. Only the unstained cells were counted. Some stained cells may in fact be viable but, generally, the trypan blue-stained cells are regarded as “nonviable.” Eppendorf tubes were centrifuged at 1200 rpm for 5 min at 4°C and the freezing medium was discarded. The cell pellet was then resuspended and washed with cold PBS twice. Cells were finally resuspended in 0.5 mL cold PBS, 10 μL was removed and used for cell counting and viability, as earlier, and the remaining cells were used for the Fpg-assisted comet assay after centrifugation and removal of the PBS.
The materials, equipment, and procedures for the Fpg-assisted comet assay were as described in detail in previously published reports.5–8,10,11 In brief, lymphocytes were embedded in low-melting-point agarose on a microscope slide that had been previously coated with standard agarose. Slides were placed in a solution of high salt and detergent (lysis solution) to lyse the cells. The resulting nucleoids from lysed cells were treated with Fpg, which creates single-strand breaks at sites of oxidation-induced lesions. During electrophoresis at high pH, loops of relaxed DNA around the sites of single-strand breaks are pulled from the nucleoid in the direction of the anode, leaving less DNA in the comet “head” and forming a comet “tail.” The DNA was stained by a DNA-intercalating fluorescent dye (in this case, ethidium bromide). The relative intensity of the fluorescence in the comet tail was measured (as %DNA in comet tail) using a fluorescence microscope (Nikon Eclipse E600; Nikon) linked to a computerized comet assay image capture and analysis system (Komet 5.5; Kinetic Imaging Ltd.). A total of 50 nucleoids per gel and 200 nucleoids per treatment were scored. Results are presented as mean and standard deviation.
Statistical analysis
GraphPad software PRISM (ver5.0) was used. For cell numbers, one-way ANOVA was performed for results at each stage. The Friedman test with Dunn's post hoc test was used to compare cell loss and DNA damage in samples processed by each investigator. A P value of <0.05 was considered to be statistically significant.
Results
Table 1 shows the average cell counts and % losses at each stage for the 3 blood samples, with results for investigators 1 and 2 shown separately and averaged for both. Some explanation of the numbers in the table is warranted. The average total lymphocytes count in 3 fresh, whole blood samples was 2200 cells/μL. The number of cells harvested was 1230 cells/μL for investigator 1 and 1197 cells/μL for investigator 2, giving an average of 1214 cells/μL. This represents 55% of the 2200 cells/μL in fresh whole blood, that is, an average of 45% of the cells in whole fresh venous blood samples had been lost during harvesting after gradient separation. The rest of the table gives the averaged absolute numbers of cells from the 3 blood samples for each investigator and the average for both investigators, comparing numbers after washing in freshly harvested and cryopreserved cells. The % cell loss after different steps or conditions is expressed in different ways. For example, the overall % loss in freshly thawed aliquots after 2–3 days cryopreservation in relation to the cell numbers in fresh whole blood was calculated as 1051/2200 cells/μL, which equals a loss of 52% (ie, the number of viable cells remaining represent 48% of those in fresh whole blood). The average cell loss due to “freeze and thaw” of these cells was calculated by taking the number in the freshly thawed aliquots of 2–3-day cryopreserved cells (1051 cells/μL) in relation to the number of freshly harvested cells (1214 cells/μL). This revealed that 86.5% of harvested cells remained, indicating a loss of 13.5% due to the freeze/thaw cycle. Cell loss due to washing was calculated for freshly harvested and cryopreserved cells by comparing the number of cells after removal of medium and following 2 washes in PBS (eg, 540 cells/μL in aliquots of washed, freshly harvested cells, or 440 cells/μL in the washed cells that had been cryopreserved and stored for 4 weeks) with the number in the same aliquot before removal of medium and washing. Thus, in freshly harvested cells after washing, 540/1214, 44.5% of the harvested cells remained, representing a loss due to the washing step of 56.5%, whereas in the 4-week cryopreserved cells, the figures were 440/1143 remaining (38.5%), giving a wash-related loss of 61.5%. The pattern of change in cell numbers in freshly harvested, 2–3-day cryopreserved, and 4-week cryopreserved samples before and after washing is given more clearly in Fig. 2.
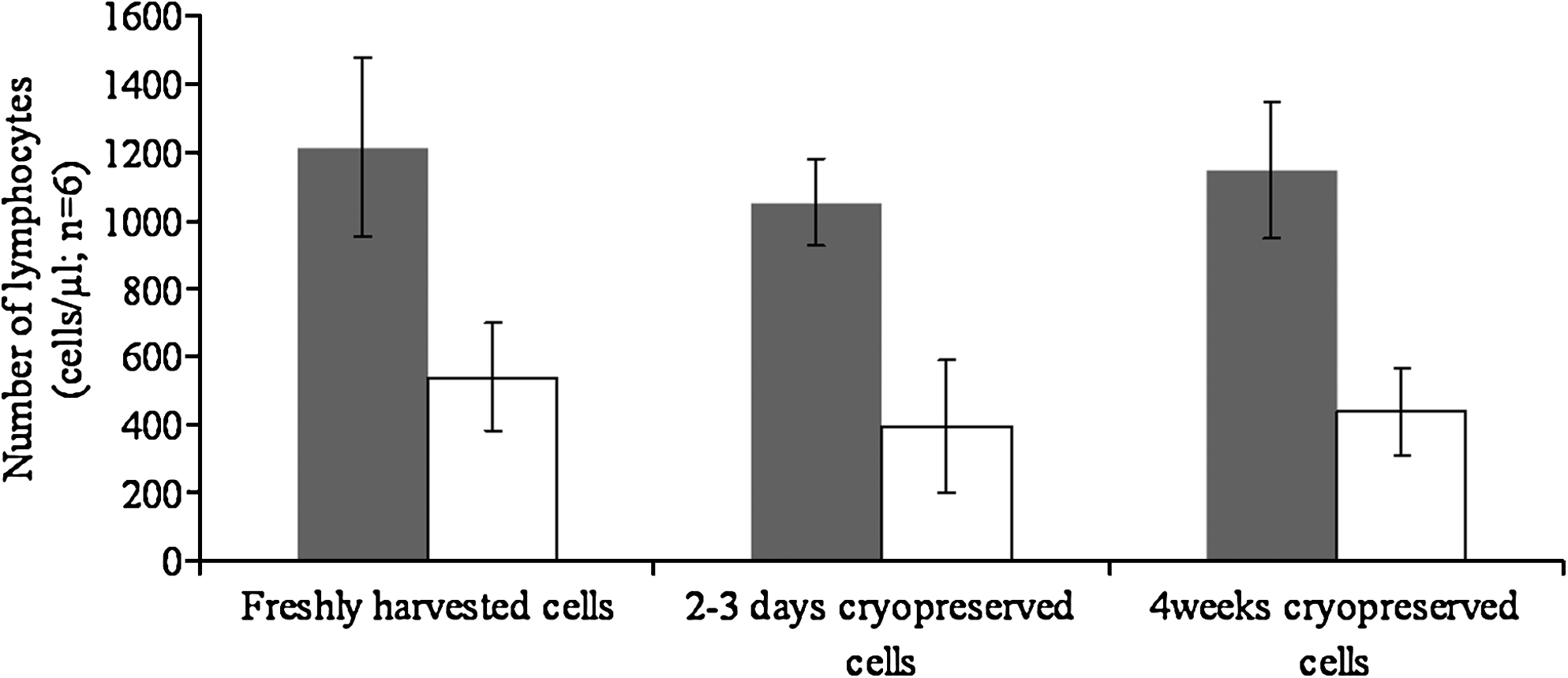
Number of lymphocytes in samples at different stages of processing. Data are shown before (shaded bars) and after (open bars) 2 washes in PBS. Results are presented as mean and SD of cell numbers in freshly harvested and cryopreserved aliquots of cells from 3 venous samples processed and counted at each stage separately and by 2 investigators independently but in parallel.
Data from samples of freshly harvested lymphocytes and freshly thawed cryopreserved aliquots from the same blood samples (n=3) before and after washing are shown, as measured independently using trypan blue exclusion assay by 2 investigators and overall.
PBS, phosphate-buffered saline.
In summary, ∼55% of lymphocytes were harvested from venous blood. The amount of cells harvested and lost at each step was similar for each investigator. There was some loss associated with a freeze/thaw cycle, but this was relatively small. Most cell loss (∼60% of cells present in aliquots prewash) occurred during washing. The wash-related losses were not significantly different (P>0.05) between fresh cells and cells thawed and washed after 2–3 days or after 4 weeks of cryopreservation. Comparing the cell count of fresh whole blood with those of washed aliquots just prior to embedding the cells in agarose ready for comet assay testing revealed a total cell loss of ∼80% regardless of whether cells were freshly harvested and washed, or cryopreserved for 2–3 days or 4 weeks before being thawed and washed. Further, the loss was similar whether the procedures were performed by a highly experienced operator or by someone fairly new to the technique.
With reference to DNA damage in freshly harvested compared with cryopreserved cells, the Fpg-assisted comet assay results are presented in Fig. 3. Results indicated no significant change in DNA damage due to cryopreservation and thawing prior to testing. There were no significant differences in DNA damage scores between cells harvested, cryopreserved, and washed by each of the 2 investigators (Table 2).

DNA damage (mean and SD of %DNA in comet tail; n=6). Data are shown from samples in freshly harvested cells and cells tested after 2–3 days and 4 weeks of cryopreservation. Black bars show the comet assay results without the use of Fpg (ie, pre-existing strand breaks in DNA); shaded bars show the results with Fpg treatment (ie, pre-existing strand breaks plus those created at Fpg-sensitive lesions).
Values are mean (SD) %DNA in comet tail; n=3.
Data from freshly harvested lymphocytes and matched lymphocytes thawed after 2–3 days and 4 weeks of cryopreservation. Lymphocytes were harvested from the blood of 3 healthy volunteers and cryopreserved, thawed, and washed separately by 2 investigators independently but in parallel. The comet assay was performed on all samples by 1 investigator.
Discussion
For convenience, and to test paired samples in controlled biomonitoring studies, cryopreserved cells (generally lymphocytes harvested from venous blood) are commonly used in the comet assay.5,8 This raises several issues of potential concern. The first relates to cell numbers for testing: it is important that there are enough cells in each gel being scored and that there is little chance of the same cell being counted more than once. The second, more important, issue relates to the effect of cryopreservation and subsequent thawing on single-strand breaks or oxidation-induced lesions in DNA. This effect could be a process-related (cryopreservation/storage/duration/thaw) increase in damage, or it could be an apparent decrease in damage due to the predominant loss during storage and thawing of more “labile” cells containing a higher basal level of DNA damage at collection. Either of these effects would lead to results on stored cells that were not representative of fresh cells. There is also the issue of operator dependence in loss and induction of DNA damage during processing.
With reference to the first issue, it is clear that cell loss is substantial as a result of harvesting and, in particular, washing steps, with only ∼40% of harvested lymphocytes (<20% of lymphocytes in venous blood) remaining to be embedded in agarose and tested using the comet assay. Loss is likely to be due mainly to cells attaching to the sides of the tube during the centrifugation steps needed in harvesting and washing of cells. Previous studies with stem cells have shown a cell loss of up to 30% caused by post-thaw washing alone. 12 Additional cell loss due to freeze/thaw is relatively low, and the level of experience did not significantly influence cell loss at any stage or overall. Based on the results presented here, it can be calculated that the 85 μL of agarose-based cell suspension used to form each gel contained ∼1200 lymphocytes. This indicates that even with the unavoidably high loss during processing, enough cells remain in each gel to score 50 different cells with confidence that they are “independent” (ie, the risk of counting the same cell more than once is low). With reference to effects of freeze/thaw and duration of cryopreservation on DNA damage, no significant differences were seen between freshly harvested lymphocytes and lymphocytes tested after 2–3 days or 4 weeks of cryopreservation. This indicates there is no predominant loss of susceptible, more highly damaged cells during cryopreservation and thawing. Further, storage for 4 weeks did not result in a significant increase in single-strand breaks or oxidation-induced DNA lesions.
These findings are in accord with those recently published by Muniz et al. on fresh and cryopreserved leukocytes from the buccal cavity. 13 No significant differences were found in DNA tail length or tail moment, although a significantly lower (P=0.03) comet head intensity was seen in cryopreserved cells (median: 54.5) compared with fresh cells (median: 60.9). 13 The head intensity is a measure of the amount of DNA remaining in the comet head and so is the opposite to the %DNA in comet tail. These results on buccal lymphocytes were obtained using the standard alkaline comet assay for pre-existing single-strand breaks, rather than the Fpg-assisted comet assay used here. As can be seen from Fig. 3, the Fpg-assisted comet assay has a 3–4-fold higher sensitivity than the standard alkaline version as well as enhanced specificity for oxidation-induced DNA damage. Further, the buccal lymphocytes were cryopreserved for only 96 h, compared with 2–3 days and 4 weeks in the present study. In an earlier study, Duthie et al. used several versions of the comet assay, including the Fpg-assisted comet assay, on fresh and cryopreserved lymphocytes. 14 Measurement of DNA damage (oxidized purines) was performed manually using a 0–400 visual scoring system, so the results cannot be compared directly with % tail DNA values. However, there was a slight, nonsignificant increase in visual damage scores in frozen (mean [SD]: 62 [3.6]) compared with fresh (57.4 [5.8]) lymphocytes. 14
In summary, this study confirms that oxidation-induced DNA damage in human lymphocytes is not significantly affected by cryopreservation followed by storage for up to 4 weeks. The results highlight also the sensitivity of the Fpg-assisted version of the comet assay and show that the effect of harvesting and washing on cell numbers (which is marked) and on DNA damage is not operator dependent, provided basic training and established protocols are followed.
Footnotes
Acknowledgments
The authors thank The Hong Kong Polytechnic University for financially supporting this work and are grateful to Professor A.R. Collins for kindly providing the Fpg.
Author Disclosure Statement
No competing financial interests exist.