
Abstract
We have previously demonstrated storage of ice-free cryopreserved heart valves at −80°C without the need for liquid nitrogen, with the aims of decreasing manufacturing costs and reducing employee safety hazards. The objectives of the present study were a further simplification of the ice-free cryopreservation method and characterization of tissue viability. Porcine pulmonary heart valves were permeated with an 83% cryoprotectant solution (VS83) followed by rapid cooling and storage at −80°C. The cryoprotectants were added and removed in either single or multiple steps. Fresh untreated frozen controls employing 10% dimethylsulfoxide and controlled rate freezing to −80°C, and storage in vapor phase nitrogen were also performed. After rewarming and washing, cryopreserved leaflets were compared with fresh controls using the resazurin reduction metabolism assay. Comparison of valve tissues in which the cryoprotectants were added and removed in either single or multiple steps demonstrated similar viability results for the muscle, conduit, and leaflet components. The ice-free cryopreserved conduit and leaflet components were significantly less viable than either fresh or frozen tissues. The muscle component, although less viable, was not significantly different. The changes in tissue viability were a function of cryoprotectant exposure, and resulting cytotoxicity, not temperature reduction during storage. TUNEL staining showed that ice-free cryopreservation did not induce significant amounts of apoptosis, suggesting that necrosis is the predominant cell death pathway in ice-free cryopreserved heart valves. There was very little difference in cell viability when the cryoprotectants were added and removed in a single step versus multiple steps. Ice-free cryopreserved valve tissues demonstrated very low viability compared with controls. These results support further simplification of the ice-free cryopreservation method.
Introduction
The potential advantages of IFC heart valve storage at −80°C include reduced infrastructural needs for preservation, storage, and shipping, in comparison with traditional freezing methods, while maintaining extracellular matrix integrity and material properties. 3 The typical methods employed for cryopreserved heart valves require a controlled-rate freezer, storage in nitrogen-cooled tanks, and continuous supply of liquid nitrogen. Furthermore, traditionally cryopreserved valves also require shipment in nitrogen dry shippers to the implantation site where the valves need to be kept in nitrogen-cooled freezers until implantation. Liquid nitrogen is also a safety hazard for employees and precautions must be taken during operation of nitrogen-cooled equipment. In contrast, expensive equipment and liquid nitrogen are not required for the IFC storage method.
The aims of this study were further reduction of complexity of the IFC method by adding and removing the cryoprotectants in single rather than multiple steps,3,4 and characterization of cell viability.
Materials and Methods
Tissue preparation
No animals were sacrificed for these studies. Bona fide excess tissue was employed. (Bona fide excess tissue is a term used to describe animal-derived materials obtained after the animals have been sacrificed for other uses.) In this case, pig hearts were procured from local slaughter houses from pigs aged 4–6 months, weighing approximately 90–120 Kg. All tissues were collected and processed within the Standards of the American Association of Tissue Banks. 5 Cooled tissue was transported to the laboratory. Pulmonary valves were immediately dissected from the hearts and rinsed in cold phosphate-buffered saline (PBS, Lonza, Germany and Gibco/Invitrogen, USA). The valves were then treated with an antibiotic cocktail in Dulbecco's Modified Eagle Medium (DMEM, 4.5 g/L glucose, pyruvate-free, Lonza, Germany, and Mediatech, USA) with 10% fetal bovine serum (FBS, Biochrome, Germany and JR Scientific, USA) for 24 h at 4°C. Antibiotic-treated tissues served as fresh control samples, and valves further subjected to preservation were randomly distributed to IFC or CFC groups.
Ice-free cryopreservation
Heart valves subjected to IFC were permeated in a single step with 80 mL of VS83, a EuroCollins-based 12.6 molar solution containing 4.65 mol/L formamide (Sigma-Aldrich), 4.65 mol/L DMSO (Sigma-Aldrich), and 3.31 mol/L 1,2-propanediol (Sigma-Aldrich). The valves were placed in sterile polyethylene bags (Ampac Flexible SealPack, Fisher Scientific, USA) containing VS83 and incubated for at least 1 h at room temperature prior to cooling. The cooling process was achieved by placing the bags for 10–15 min in a pre-cooled methylbutane bath (−130±10°C). A sample cooling curve is shown in Figure 1. The bags containing the valves were then stored at −80°C in a storage freezer for at least 24 h. Warming of the tissue was realized by submersion of each bag with its valve in a 37°C water bath for 1–2 min, followed by five washes in 4°C cold EuroCollins solution for 5 min each.

Cooling curve of IFC tissue. Representative sample cooling curve for a bag containing 80 mL of VS83 after placement in a −140°C methylbutane bath. The thermocouple was placed in the center of the bag.
Conventional freezing cryopreservation
CFC was achieved in DMEM with 10% FBS and 10% DMSO. The valves were held in the freezing media for 30 min at 4°C prior to initiating controlled rate freezing at 1°C per min to −80°C. The bags were then stored in the vapor phase above liquid nitrogen for at least 24 h. After a maximum storage time of 2 months, the tissues were thawed in a 37°C water bath until all visible ice was gone, and subsequently washed for 5 min each in 4°C cold DMEM with 0.5 M mannitol (Sigma-Aldrich), DMEM with 0.25 M mannitol, and finally in DMEM without mannitol.
Cell metabolic activity (resazurin assay)
Cell metabolic activity was assessed with a resazurin reduction assay. 6 Leaflets were divided into three parts; small pieces of muscle tissue as well as round transmural pulmonary conduit biopsies were incubated in DMEM with 10% FBS plus 1% PenStrep (Gibco/Invitrogen) and 10% resazurin reagent (AbD Serotec, Germany and Biotium, USA). After 4 h, resazurin reduction to resorufin was measured using a fluorescence reader (excitation wavelength 544 nm and an emission of 590 nm, Molecular Devices, Sunnyvale, CA, shown in Fig. 2, or excitation wavelengths of 530 nm and emission of 600 nm, Mithras, Berthold Tech GmbH, Bad Wildbad, Germany, in Figs. 3 and 4). The signal intensity was normalized to tissue dry weight.

Comparison of single and multiple step cryoprotectant addition and removal. The impact of single step and stepwise addition and removal of VS83 upon cell viability was investigated: The cryoprotectant was added and removed from the valves in either a single step or in 7 steps of increasing cryoprotectant concentration. Metabolic activity of tissue samples was assessed either directly after warming

Viability after incubation in VS83 without cooling. Tissue metabolic activity was tested immediately (fresh) or after incubation in VS83 at room temperature for 1.5 h and 2.5 h, respectively, and washing. The results are expressed as fluorescent units per mg tissue dry weight, *p<0.05 vs. fresh controls, n=4–5.

Apoptosis detection
Paraffin-embedded tissue sections were stained with a terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) apoptosis detection kit (GeneScript, Piscataway, NJ). The tissue sections for 24 h samples were incubated after warming/thawing in DMEM with FBS (10%) and PenStrep (1%) at 37°C and 5% CO2 and then para-formaldehyde-fixed and embedded. Apoptosis detection was performed as described by the manufacturer. Digital images were captured using an Axio Observer Z1 (Carl Zeiss, Jena, Germany).
Statistical methods
All quantitative results were expressed as the mean±1 standard deviation. An independent sample t-test was performed between paired groups to determine whether significant statistical differences existed. P values <0.05 were considered statistically significant.
Results
Preliminary metabolic activity experiments were performed to evaluate the impact of stepwise cold addition and removal of the cryoprotectant solutions in combination with cryopreservation (Fig. 2A and 2B). These studies demonstrated that most leaflet cell viability was lost, regardless of how many steps were used to add or remove the cryoprotectants. The only significant difference observed was that the conduit samples were consistently more viable after the one-step addition and removal protocol (p<0.05) both immediately after rewarming (Fig. 2A) and after 24 h of incubation under physiological conditions (Fig. 2B). In further experiments, heart valve segments incubated in VS83 at room temperature for 1.5 h without subsequent cooling and storage also demonstrated significant declines in leaflet and conduit viability (Fig. 3). Therefore, for further experiments the simplified protocol described in the Materials and Methods with one-step room temperature addition and cold removal was used.
Employing the simplified one-step addition and removal protocol, cell metabolic activity assessed with a resazurin reduction assay showed a significant decrease in IFC leaflet biopsies. A decline was also observed for CFC tissue but it was not statistically significant (Fig. 4A). The absolute fluorescence intensities per mg tissue dry weight were lower in the muscle for both preservation groups, but the differences in viability decrease did not achieve statistical significance (Fig. 4B). Statistically significant viability decreases were measured for both CFC and IFC pulmonary conduit compared to fresh controls. IFC conduit viability was also significantly lower than the CFC viability (Fig. 4C). The degree of metabolic activity decline was directly proportional to the thickness of the tissue type. Leaflets, the thinnest tissue, were the most impacted, while the pulmonary artery, intermediate, and cardiac muscle (the thickest area, associated with the base of the valve) demonstrated more viability, respectively.
No apoptotic cells were detected in fresh leaflets using the TUNEL assay (Fig 5a). Both CFC and IFC cryopreserved samples stained for DNA fragmentation, either immediately after warming or after 24 h incubation under cell culture conditions, revealed few apoptotic cells (Fig. 5b–5e). Although in some areas of IFC leaflets an accumulation of cells undergoing apoptosis was observed, the total number of apoptotic cells was low. CFC leaflets showed only singular apoptotic cells. These results suggest that the majority of cells in IFC leaflets die by necrosis, not apoptosis.
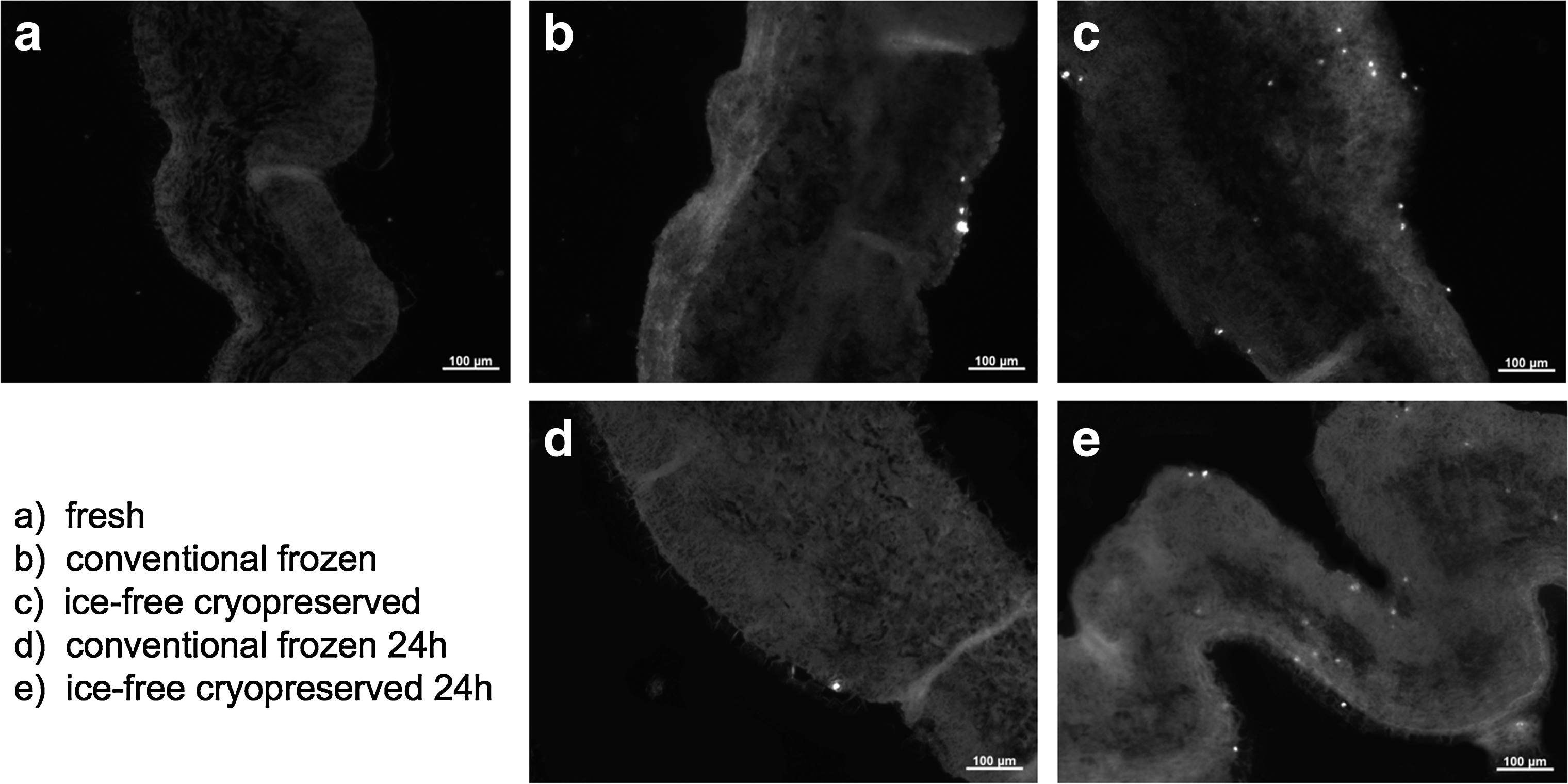
Apoptosis detection. Representative leaflet sections stained using the TUNEL assay to identify apoptotic cells.
Discussion
At this time there is no convincing evidence in the literature suggesting that maintenance of graft cell viability after cryopreservation is beneficial in allogeneic heart valve recipients. Analysis of explanted heart valves supports the assumption that living cells are not necessary in a functioning transplant. Explant analyses indicate that most of the original cells either vanished, especially in the leaflet cusps, or were replaced by donor fibroblasts.7,8 Only very few living cells from the original donor could be detected after long-term transplantation, and normal graft leaflet cellularity was not observed in a single explants. 9 Furthermore, many groups are decellularizing heart valves in order to reduce their concerns regarding immunogenicity and its consequences in patients.10,11
Therefore, we did not attempt to preserve cell viability during the development of the IFC method. The metabolic assay results clearly demonstrated that the VS83 cryoprotectant formulation is cytotoxic for the cells in porcine heart valve tissues. This is in agreement with previous studies demonstrating that the VS83 cryoprotectant components are cytotoxic for cultured porcine myofibroblasts. 12 Varying the number of addition or removal steps (Fig. 2A/B) in combination with cooling made little if any difference for cell survival. Room temperature incubation in VS83 without subsequent cooling and storage had similar effects on cell viability. Therefore, the simple one-step addition at room temperature was used in further experiments in this study. Multiple changes of washing solution were employed to minimize the residual cryoprotectant concentrations.
It should be noted that the reason the steps in IFC tissue processing may appear illogical is because the process was initially a vitrification process based upon the VS55 cryoprotectant formulation originally developed for kidney preservation 13 in which we could keep vascular tissue samples viable below Tg (−119°C). 14 We increased the formulation from 55% to 83% in order to avoid ice formation, above the VS83 formulation Tg at −80°C. 3 The rapid cooling to below −100°C and the warming and washing conditions are holdovers from the original VS55 method where retention of viability was an issue. Rapid cooling and washing with 4°C cold EuroCollins solution are probably not necessary but we have not yet had the opportunity to remove these steps.
We believe the higher relative viability of conduit tissue and muscle compared with leaflets can be explained by tissue thickness. It is likely that the cryoprotectants permeation was incomplete in the thicker tissues compared with the relatively thin leaflets where essentially all the cells were dead due to the cytotoxic VS83 formulation. Comparison of studies using different thicknesses of articular cartilage from various mammals permits the conclusion that thin pieces of tissue are permeated by DMSO faster than thick pieces of tissue.15–17 Longer incubation periods in VS83 before cryopreservation are suggested in order to achieve lower viability in the thicker valve components.
Apoptosis detection showed only a few cells undergoing programmed cell death within either cryopreservation group, indicating that the loss of viability is primarily due to necrosis. It has previously been reported that necrosis is the predominant cell death pathway after CFC. 18 Our findings for CFC heart valve leaflets are in agreement and further indicate that a similar process is occurring within IFC leaflets.
It is possible that the loss of cell viability may have in vivo benefits since any viable “foreign” cell is susceptible to attack from the recipients' immune systems. 19 However, since a nonviable cell still possesses the same cell surface antigenic determinants as a viable cell, further research is needed to support this potential interpretation. Most heart valve processors and research groups now remove the cell populations (decellularization) of allogeneic heart valves, employing various techniques including water, detergent, surfactant and enzymatic treatments to reduce potential immunogenicity.10,20,21
In conclusion, we have demonstrated that the IFC process can be performed with either single or multiple cryoprotectant addition and removal steps, resulting in leaflet and conduit tissues with very low cell viability. The muscle component viability was also reduced but did not achieve statistical significance. Preclinical large animal trials will also be needed to verify that simplification of the procedure has not resulted in loss of the previously observed in vivo benefits.
In the early years of allograft heart valve transplantation, cell viability was considered to be very important. Nowadays an opposite trend has been observed. The success of decellularized heart valve transplants10,20,21 indicates that living cells in transplants are not necessary. IFC valve tissues are minimally viable and sheep implant studies suggest that they are also less immunogenic. 4 Further research on IFC valve tissue hemocompatibility and immunogenicity are mandatory and in progress.
Footnotes
Acknowledgments
The authors would like to thank Ilka Degenkolbe for her excellent technical assistance, the Slaughterhouse Gärtringen, BW, and Mrs. Quindt, and the staff and owners of Burbage Meats, Ravenel, SC, for providing tissues.
Author Disclosure Statement
This work was supported by the Deutsche Forschungsgemeinschaft (STO359/7-1). Dr. Brockbank is an owner and employee of Cell and Tissue Systems, Inc., and Mrs. Greene and Mrs. Chen are both employees of the company. No competing financial interests exist for any other authors.