
Abstract
Preanalytical conditions applied during sample collection and processing can affect the detection or quantification of unstable phosphoprotein biomarkers. We evaluated the consequences of tissue stabilization and protein extraction methods on phosphoprotein analysis. The effects of stabilization techniques (heat stabilization, snap-freezing) and time on the levels of phosphoproteins, including phospho-Akt, p-ERK 1/2, p-IkBα, p-JNK, and p38 MAPK, were evaluated using a BioPlex phosphoprotein assay. Additionally, two different protein extraction protocols, using different extraction buffers (8 M urea buffer, or Bio-Rad buffer without urea) were tested. For snap-frozen samples, protein extraction yields were comparable with the two buffer systems. For heat-stabilized samples, total protein yields were significantly lower following extraction in non-urea buffer. However, the concentrations of specific phosphoproteins were significantly higher in heat-stabilized samples than in the corresponding snap-frozen samples, indicating that this tissue processing method better preserved phosphoproteins. Significant differences were found between the measured phosphoprotein levels in heat-stabilized and snap-frozen tissue, suggesting that alterations occur very rapidly after tissue excision. Our results suggest that heat stabilization can be used as a tissue processing method for subsequent phosphoprotein analyses, but also suggest that the BioPlex phosphoprotein assay could be used as a possible quality control method to assess tissue sample integrity.
Introduction
A general rule in tissue processing is the faster the tissue fixation, the closer the quality of the tissue sample to its in vivo state. Formaldehyde is the gold standard of fixatives for routine histology and immunohistochemistry (IHC). Formaldehyde mainly preserves the general structure of cellular organelles. The final result of formaldehyde fixation is a profound change in the conformation of macromolecules that can make the recognition of proteins (antigens) by antibodies impossible or difficult. 8 Formalin fixation is a progressive, time- and temperature-dependent process. Overfixation in IHC can produce false negative results from excessive cross-links; 9 however, underfixation can also produce intra-tissue molecular cross-link inhomogeneity.9,10 Recently, a novel fixation method has been proposed for both histomorphology and phosphoprotein analysis. 11 If the tissue is not fixed chemically, it can be cryo-stabilized. Snap-freezing works quickly, but the cryogenic equipment needed to process and store the samples is expensive and not available in many clinical environments. A newly-developed, novel tissue stabilization method that is based on a combination of heat and pressure under vacuum rapidly inactivates protein-modifying enzymes.12,13
In this study, mouse brain tissue was used to compare two different tissue stabilization methods (snap-freezing and heat stabilization), and two different protein extraction protocols using different extraction buffers (8 M urea buffer and Bio-Rad buffer without urea). In particular, we assessed phosphoprotein preservation in samples subjected to these processing methods (Fig. 1). We used a multiplex, bead-based assay (xMAP technology) for quantification of phosphorylated and total protein in tissue lysates. 14 We compared the heat-stabilized and paired snap-frozen samples by assaying phosphoproteins, including p-Akt, p-ERK 1/2, p-IkB-α, p-JNK, and p38 MAPK.

Experimental schema of tissue treatment groups, stabilization and extraction methods. DEN, heat-stabilized with Stabilizor T1 instrument (Denator AB); SNAP, snap-frozen samples.
Materials and Methods
Tissue preparation and stabilization
Adult (250–300 g) male NOD/Scid mice were obtained from the Department of Oncology, Norlux Neuro-Oncology Laboratory, Centre de Recherche Public Santé (CRP-Santé), Luxembourg. The handling of the animals and collection of tissues were performed in accordance with the regulations of the national authorities responsible for animal experiments in Luxembourg. Five mice were sacrificed by cervical dislocation, and each brain was immediately excised and cut in two hemispheres with a scalpel. Separated hemispheres were stabilized by one of three protocols: snap-freezing immediately after specimen excision; heat stabilization immediately after specimen excision; or snap-freezing after 20 min ischemia delay at room temperature (RT around +20°C) (Fig. 1). For heat-stabilization, the dissected tissue was placed on the flexible Teflon window in the center of the polycarbonate thermoplastic cassette. The cassette was placed in the Stabilizor T1 instrument (Denator AB, Gothenburg, Sweden), and the air removed by automated vacuum to minimize protein oxidation. The samples were then subjected to 5 mbar of pressure and heated to 95°C for 20 sec to eliminate residual enzymatic activity. For snap-freezing, the dissected tissue was placed in a cryovial and immersed in liquid nitrogen in a dewar. All samples were transferred to −80°C for long-term storage.
Protein extraction
Brain tissue samples were thawed in 1.5 mL of cell lysis buffer (Bio-Rad Cell Lysis Kit #171-304012) containing protease inhibitor cocktail (#171-304012; Bio-Rad) and 3 μL of a stock solution of 500 mM phenylmethylsulfonyl fluoride (Sigma #P-7626) in dimethyl sulfoxide (Sigma #D2650). Samples were then processed in either 8 M urea buffer (8 M urea, 30 mM Tris-HCl, pH 8.0) or cell lysis buffer from Bio-Rad as described above. Tissue specimens weighing 70±5 mg were disrupted for 15 min with a steel-bead Tissue Lyser (Qiagen) and centrifuged at 10,000 g for 15 min at 4°C; the supernatants were collected and stored at −80°C.
Total protein concentrations were determined using a DC Protein Assay Kit (#500-0113 Bio-Rad) and spectrophotometer (Synergy, BioTek). All protein samples were diluted in Bio-Rad phosphoprotein assay buffer to a final total protein concentration of 500 μg/mL, according to the manufacturer's protocol.
Quantitative Bio-Plex phosphoprotein assay
Phosphoprotein concentrations were evaluated with a BioPlex phosphoprotein assay according to the manufacturer's protocol. In brief, the antibody-conjugated beads were added to each well and incubated with the samples overnight. The plates were washed and incubated with another set of target phosphoprotein-specific biotinylated antibodies, and a streptavidin–phycoerythrin solution was added. The analysis consisted of double-laser fluorescence detection, which allows simultaneous identification of the target protein through the red fluorescence emission signal of the beads and quantification of the target phosphoprotein through the fluorescence intensity of phycoerythrin. A standard protein extract from a control cell line (included in the kit) was included in each run as a positive control. Results were recorded as mean fluorescence intensity and normalized against the total protein fluorescence intensity detected by the Bio-Plex total target assay, according to the manufacturer's instructions. BioPlex Manager software version 6.0 was used for data acquisition and analysis.
Statistical analysis
SigmaPlot 11.0 was used for descriptive statistics and t-tests (paired testing). Data are presented as means±standard error of the mean. The statistical significance level was set at p<0.05.
Results and Discussion
To evaluate preservation of phosphoproteins following stabilization and extraction under different conditions, we measured the relative amounts of phosphoproteins using the multiplex BioPlex assay. Differences in protein extraction efficiency may result in different end-point protein profiles. Therefore, we first compared total protein yields from heat-stabilized tissue samples with those from liquid nitrogen snap-frozen tissue samples, following extraction with two different extraction buffers (Fig. 2). Total protein extraction yields from snap-frozen tissue samples were comparable with the two extraction buffers: there was no significant difference in protein yield between urea- and non-urea buffers for samples that had been snap-frozen immediately or snap-frozen with a 20-min delay. However, total protein yields from heat-stabilized samples were significantly lower (p<0.001) when non-urea buffer was used for extraction compared to use of the urea-based buffer. Total protein extraction efficiency was found to be dependent on the extraction buffer used. Although lower with BioRad (non-urea) buffer than with the urea-based buffer, the protein yields in all cases were sufficient for phosphoprotein detection by BioPlex analysis.

Protein concentrations in different stabilized mouse brain tissue extracts: DEN, heat-stabilized with Stabilizor T1 instrument (Denator AB); SNAP, snap-frozen samples. (See Fig.1 for the exact conditions).
The stability of each phosphoprotein was differently affected by each combination of stabilization method/protein extraction method used (Fig. 3). In non-urea-based buffer (Bio-Rad Buffer), despite the lower total protein yield from heat-stabilized tissue samples, the ratio of p-Akt, p-ERK1/2, p-IkBα, and p38 MAPK to the corresponding total protein was significantly higher in the heat-stabilized samples than in the snap-frozen samples. These observations confirm the previously reported phosphoprotein stabilization effect of the heat-stabilization method for tissue samples. 12 In urea-based extracts, we found significant differences between the p-ERK1/2 and p38 MAPK quantities observed in heat-stabilized and snap-frozen tissue samples, suggesting that phosphoprotein alterations can occur very rapidly, within the 3 minutes between tissue excision and snap-freezing (Fig. 3).
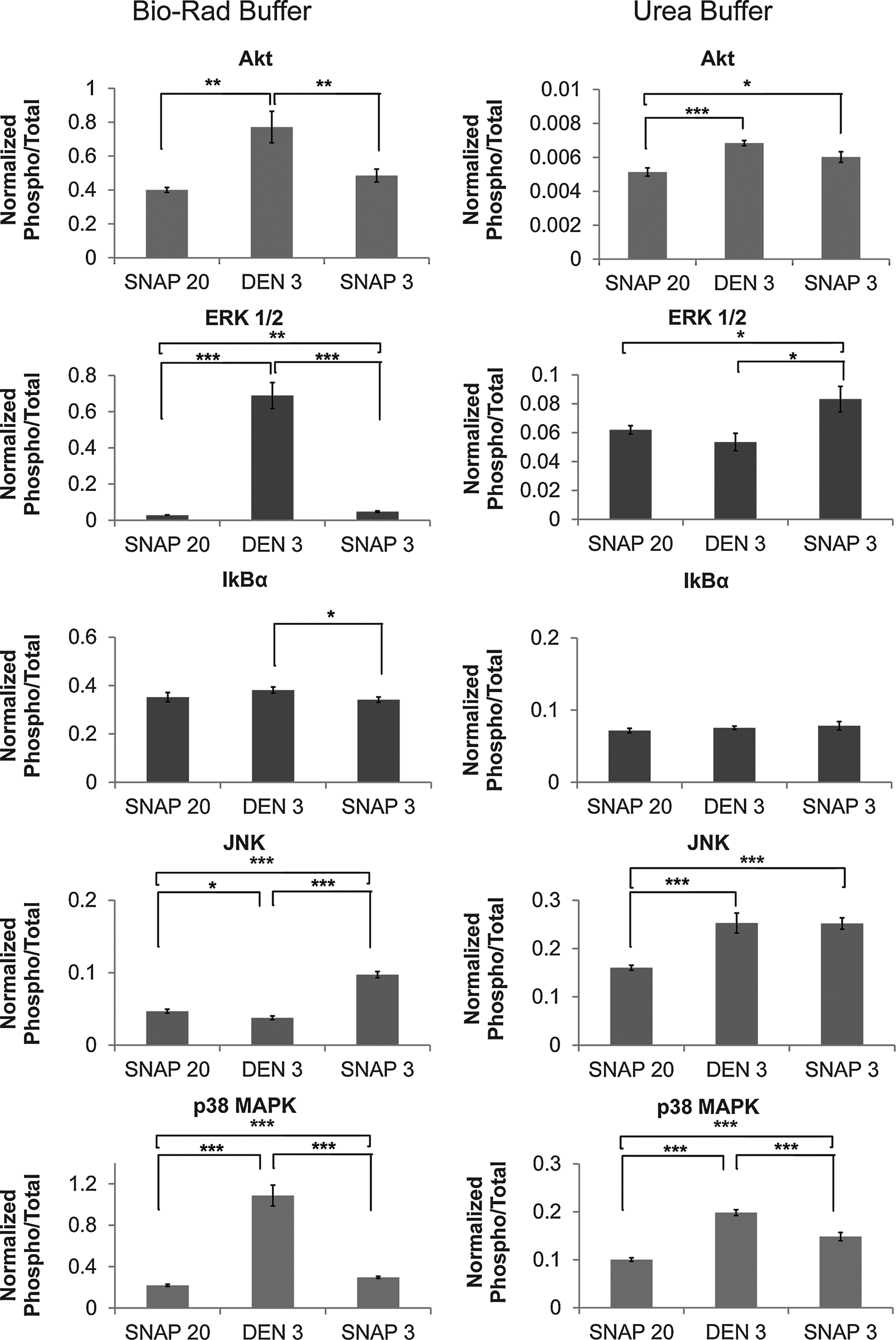
BioPlex analysis of phosphoroteins extracted in different buffers. Data are presented as means±S.E.M. (mean fluorescence intensity normalized against the total protein fluorescence intensity), n=12, *t-test (*p<= 0.05, **p<= 0.01, ***p<= 0.001).
We examined the effects of immediate stabilization on the results for individual phosphoproteins. The values for p-Akt, p-ERK1/2, p-IkBα, and p38 MAPK were significantly higher after immediate heat-stabilization than those for immediate snap-frozen samples when extraction was performed in non-urea buffer. The values for p-Akt, p-ERK1/2, p-JNK, and p38 MAPK were significantly higher for immediate snap-frozen samples than those for snap-frozen with delay when extraction was performed in urea buffer.
The results for urea-based extraction of p-IkBα did not differ between heat-stabilized and snap-frozen samples. The findings for p-IkBα extracted in urea buffer did not differ according to the stabilization method, whereas using extraction with non-urea buffer, lower levels were observed after immediate snap-freezing than after heat stabilization.
Higher levels of p-JNK were observed after quick snap-freezing than heat stabilization when proteins were extracted in non-urea buffer. Similarly, higher p-JNK levels were observed after either quick snap-freezing or heat-stabilization relative to late snap-freezing when proteins were extracted in urea buffer.
The yield of p-ERK1/2 was significantly higher from heat-stabilized than snap-frozen samples in the non-urea buffer, but not in the urea buffer. The yield of p38 MAPK was significantly higher from heat-stabilized samples with both extraction buffers; however, the difference between heat-stabilized and immediately-snap-frozen tissue was less pronounced in urea buffer extracts. The findings for p-Akt were similar to those of p38 MAPK; in the case of p-Akt, there was no difference between heat-stabilized and immediately-snap-frozen urea extracts. These results are in agreement with previous findings for brain, lung, and liver samples, assessed by SDS-PAGE and SyproRuby and ProQ Diamond gel staining. 15 Analysis of stabilization techniques revealed that the level of phosphorylated forms of particular proteins can be very unstable and sensitive to cold ischemia time.
We assume that profiles of phosphoproteins in immediately snap-frozen tissue are the most similar to those in the in vivo state. The use of urea extraction buffer allowed extraction of proteins from heat-stabilized tissue with similar yields of phosphoproteins as from corresponding immediately snap-frozen specimens, although there were exceptions: a difference was found between heat-stabilized and immediately-snap-frozen tissue for p-ERK1/2 and p38 MAPK, and this difference was much larger when non-urea buffer was used for extractions. This suggests that heat stabilization effectively preserves the phosphoproteins in their in vivo state, and non-urea buffer probably does not allow efficient extraction of all proteins, either from heat-stabilized or frozen samples. The use of urea buffer is necessary to exploit the phosphoprotein preservation capacity of the heat stabilization method appropriately.
Collection of stable biospecimens is very important to basic and clinical research, for example, proteomics. There is a critical need for stabilization technologies and in this respect, the technology based on heat-stabilization is promising. Optimized and validated protocols save time and costs, and help ensure consistent analytical results. The Denator Stabilizor system uses instant +95°C heating to preserve tissue samples as close as possible to their in vivo state (e.g., permanently prevents biological changes from the moment of heat application, inactivates phosphatases, stops degradation).12,16 The ability to stabilize biospecimens and preserve protein molecules represents a cost-efficient alternative method for tissue processing and storage. In our study, we have shown that depending on the downstream application, the heat-stabilization method could advantageously be integrated in the biobank workflow. A future comprehensive study using more tissue types could provide full validation, since in our study the results were obtained from brain tissue, which has a higher lipid content than that of other tissues.
In this study, we show that heat stabilization is a tissue processing method that facilitates downstream phosphoprotein analyses, most likely due to rapid inactivation of phosphatase and kinase activities. The decrease in specific phosphoprotein levels can theoretically be attributed to either proteolytic degradation of the epitope corresponding to the antibodies used in the assay, or to enzymatic dephosphorylation. The latter is more probable, given the time necessary for enzymatic phosphor-/dephosphorylation (minutes) and for proteolysis (hours). The increase observed for some phosphoproteins further suggests phosphorylation/dephosphorylation mechanisms rather than proteolysis, which would have led to a global decrease of all phosphoproteins. Our results also suggest that a BioPlex phosphoprotein assay may be of value as a quality control method to assess tissue sample integrity. For example, high levels of p-Akt, p-ERK1/2, or p38 MAPK could serve as indicators of phosphoprotein integrity in tissue samples processed in non-urea buffer. High levels of p-JNK may be an indicator of phosphoprotein integrity in tissue samples processed in urea buffer. Exploiting phosphoproteins as quality control tools for tissue specimens has already been suggested. 7 However, further studies are needed, particularly in tissue types other than brain, to assess the more general utility of such quality control tools. Further studies on development of novel phosphoprotein extraction methods from tissues could also improve the reliability of phosphoprotein analysis for new clinically relevant biomarkers.
Our data also indicate that protein stabilization during processing and the protocols for tissue handling and protein lysate preparation are important factors that can lead to significantly different protein profiles. In conclusion, specific phosphoprotein detection is affected by both the stabilization method (heat-stabilized vs. snap-freezing) applied during sample processing and the protein extraction method used (buffer with or without urea). Urea buffer is recommended for extractions from either heat-stabilized or snap-frozen tissues as it gives better agreement between phosphoprotein measures with heat-stabilized and immediately-snap-frozen tissues. These issues may need to be considered in the development of phosphoprotein-based quality control tools for specific tissue types. The BioPlex platform offers a convenient and standardized assay format for these purposes.
Footnotes
Acknowledgments
We thank Denator AB for providing the Stabilizor T1 instrument, and Katarina Alenäs for her constant support.
Author Disclosure Statement
No competing financial interests exist.