
Abstract
Background:
This is the third in a series of publications presenting formal method validation for biospecimen processing in the context of accreditation in laboratories and biobanks. We report here optimization of a stool processing protocol validated for fitness-for-purpose in terms of downstream DNA-based analyses.
Methods:
Stool collection was initially optimized in terms of sample input quantity and supernatant volume using canine stool. Three DNA extraction methods (PerkinElmer MSM I®, Norgen Biotek All-In-One®, MoBio PowerMag®) and six collection container types were evaluated with human stool in terms of DNA quantity and quality, DNA yield, and its reproducibility by spectrophotometry, spectrofluorometry, and quantitative PCR, DNA purity, SPUD assay, and 16S rRNA gene sequence-based taxonomic signatures.
Results:
The optimal MSM I protocol involves a 0.2 g stool sample and 1000 μL supernatant. The MSM I extraction was superior in terms of DNA quantity and quality when compared to the other two methods tested. Optimal results were obtained with plain Sarstedt tubes (without stabilizer, requiring immediate freezing and storage at −20°C or −80°C) and Genotek tubes (with stabilizer and RT storage) in terms of DNA yields (total, human, bacterial, and double-stranded) according to spectrophotometry and spectrofluorometry, with low yield variability and good DNA purity. No inhibitors were identified at 25 ng/μL. The protocol was reproducible in terms of DNA yield among different stool aliquots.
Conclusions:
We validated a stool collection method suitable for downstream DNA metagenomic analysis. DNA extraction with the MSM I method using Genotek tubes was considered optimal, with simple logistics in terms of collection and shipment and offers the possibility of automation. Laboratories and biobanks should ensure protocol conditions are systematically recorded in the scope of accreditation.
Introduction
T
The human gastrointestinal microbiome and its interactions with the host is of major importance, not only in the context of gastrointestinal disorders, but also multifactorial chronic diseases. 5 Microbiome profiles have been associated with various pathologies, including obesity. 6 Molecular analytes that can be extracted from stool include human and microbial DNA, microbial RNA, proteins, and metabolites, all of which can be used in downstream metagenomic, metatranscriptomic, metaproteomic, and metametabolomic analyses.7–9 The main downstream application of stool biobanking is the study of microbiome profiles by either random shotgun or 16S rRNA amplicon sequencing, with current research focusing on 16S rRNA profile-derived microbiome biomarkers.10–12 Best Practices 13 and Standard Operating Procedures13–15 for processing of biobank samples have been published, but data on stool samples are minimal, although comprehensive SOPs have been published by the Human Microbiome Project. 16
Manual DNA extraction from stool is laborious and represents a bottleneck for the integration of stool specimens into large scale cohort collections, although semi-automated DNA extraction has been shown to be effective. 17 Another bottleneck is the practical difficulty of stabilizing stool by freezing and maintenance of the cold chain during shipment. Data on the effectiveness of different stabilizers for ambient shipment, such as RNAlater, Whatman FTA cards, 70% alcohol, or 2.5% potassium dichromate, are rare.18,19 However, 24 h pre-processing stability of stool at room temperature (RT) has recently been demonstrated in the scope of microbiome and organic volatile compound metabolome. 20
The current study was designed to identify the optimal combination of stool collection container type and DNA extraction method. Extraction of proteins, metabolites, and RNA was not evaluated. Fitness-for-purpose for human and microbiome DNA analysis was assessed by quantitative real-time PCR (qPCR) and bacterial 16S rRNA gene profiling. Several measures of DNA integrity in terms of downstream applications were assessed. DNA concentrations were determined with spectrophotometry (assessing both single and double-stranded) and spectrofluorometry (double-strand specific). Bacterial 16S rRNA genes were quantified with qPCR, with a primer combination which allows resolution of more than 90% of all bacterial strains.21,22 DNA template quality, which affects the accuracy of qPCR, 23 was assessed with the SPUD assay that detects inhibitors of nucleic acid amplification. 24
Materials and Methods
Study design
Three stool DNA extraction methods were compared: 1) a Chemagic Magnetic Separation Module with a 24-rod head (MSM I; PerkinElmer) with a modified blood extraction automated kit; 2) column-based manual extraction, the All-In-One purification kit (totalRNA, microRNA, total proteins, genomic DNA; Norgen Biotek, Ref. 24200) with adaptations, as described below; and 3) manual magnetic bead isolation with the PowerMag® Soil DNA Isolation Kit (MoBio, Ref. 27000-4-KF). In an initial step, the MSM I extraction method was optimized in terms of DNA yield for 1) stool collection quantity (200 mg, 10 or 20 g); and 2) supernatant volume (300 or 1000 μL). In a second optimization step, the three extraction methods were compared in terms of 1) DNA quantification and purity by spectrophotometry; 2) DNA quantification by spectrofluorometry; 3) DNA quantification by qPCR for human and bacterial DNA; 4) presence of DNA inhibitors by the qPCR SPUD assay; 24 and 5) microbiome signature by 16S rRNA characterization. The effect of six collection containers was also analyzed for these parameters for each extraction method (Table 1). Finally, reproducibility of the optimal protocol was determined in terms of DNA yield (total, bacterial and human).
The study was performed in accordance with the Declaration of Helsinki (1975, revised in 2008). Human stool samples were collected from healthy infants with written informed consent provided by their legal guardian (CNER approval #201107/02).
Stool collection
For MSM I optimization, fresh stools were obtained from three dogs on the same day. Three samples of different quantities (200 mg in Brw tubes, ∼10 g and ∼20 g in Yel tubes) were prepared from each stool, each quantity in triplicate (i.e., nine aliquots per dog). The collection container coding scheme is explained in Table 1. After a maximum of 20 min at room temperature (RT), aliquots were stored at −20°C, shipped the following day in dry ice, and were then stored at −80°C.
For comparison of the extraction method and container types (Table 1), stool samples were collected on the same day at home from three children aged 1 to 4 years (two males, one female). Six aliquots were prepared per stool donor for each container type (i.e., three aliquots per extraction method per donor). Samples with liquid stabilizer (Str, Blue, Red, RNL) were shaken vigorously immediately after collection, then stored at RT, and those without stabilizer were stored immediately after collection on dry ice. The following day, samples with stabilizer were shipped at RT, and those without stabilizer were shipped on dry ice, then all samples were stored at −80°C. A total of 54 samples were used for DNA extraction by MSM I, 63 samples for All-In-One, and 34 samples for PowerMag.
DNA extraction (MSM I, PerkinElmer)
A Chemagic DNA blood protocol was used with the MSM I instrument using the Chemagic Blood kit special 4 ml (Ref. CMG-1074) with a lysis buffer for fecal samples, and MSM I software. Five glass beads were added to frozen stool samples, thawed at 50°C, then vortexed for 1 min. Samples were lysed (lysis method, according to each tube manufacturer's instructions) and vortexed to obtain a homogenous suspension that was incubated for 10 min at 70°C, then 5 min at 95°C. Lysate (1.5 mL) was centrifuged for 5 min at 10,000 g at RT. Supernatants were transferred to a 24XL deep-well plate and the well volume was made up to 2 mL with MilliQ water. Plates were processed using the MSM I automated protocol.
DNA extraction (All-In-One-based method, Norgen Biotek)
Three cold 3 mm stainless-steel beads (Retsch) were added to each tube, milled at 50 Hz for 2 min (Tissue Lyser LT, Qiagen, Ref. 23.1001/05844), then centrifuged at 700 g for 2 min at 4°C with brake (Eppendorf 5417R). Supernatant was transferred to a 2 mL Eppendorf tube and centrifuged at 14,000 g for 5 min at 4°C with brake. Larger pellets (>30 mg) were re-suspended in RNAlater ICE (Life Technologies), split into two portions and the centrifugation repeated. The supernatant was discarded and the pellet maintained at 2°–8°C. Two 5 mm and five 3 mm cold stainless steel milling beads were added to the sample along with 400 μL cold modified lysis solution (10 μL 2-β-mercaptoethanol/ml lysis solution) and 100 μL cold 1X Tris-EDTA buffer, and the pellet was briefly vortexed. The sample was homogenized by shaking at 2°–8°C for 30 sec at ∼25 Hz, then stored at −80°C if necessary.
All subsequent procedures were performed at RT. 500 μL gDNA wash solution was applied to an All-in-One spin column with an elution tube and centrifuged for 2 min at 12,000 g, RT with brake. The flow-through was discarded and the spin column reassembled with the same collection tube and spun again to completely dry the resin. The column was placed in a fresh 1.7 mL elution tube, 100 μL gDNA elution buffer was added and then centrifuged for 2 min at 400 g, then 2 min at 12,000 g with brake. A second 100 μL gDNA elution buffer was added to the column, which was then centrifuged for 2 min at 400 g, followed by 2 min at 12,000 g with brake and collected in the same tube as the first centrifugation. The total purified gDNA elution was stored at −80°C.
DNA extraction (PowerMag, MoBio)
A 0.25 g stool sample was added to each well of the PowerMag bead plate with the aid of an anti-static polypropylene weighing funnel (Ref. 23302-50) to minimize cross-contamination. 750 μL PowerMag bead/RNase A solution (400 μL RNase A to 75 mL beads) was added per well, then 60 μL lysis solution, and a Square Well Mat was tightly secured. Plates were placed between two adapter plates (Ref. 11990) on the 96-well plate shaker (Ref. 11996) at speed “20” for 10 min twice, turning in between. Plates were centrifuged at RT for 6 min at 4500 g and supernatant transferred to a 1 mL collection plate. IRT solution (450 μL) was added to each well, and plates were sealed with Sealing Tape, then vortexed horizontally for 5 sec and incubated at 4°C for 10 min.
Plates were centrifuged at RT for 6 min at 4500 g and the supernatant transferred to a fresh plate, then centrifuged at 4500 g for 6 min. A maximum of 850 μL supernatant per well was transferred to a 2 mL deep 96-well plate, then 870 μL ClearMag beads/binding solution (20 μL resuspended beads plus 850 μL binding solution) was added and mixed at 1000 rpm at 55°C for 10 min (Eppendorf Thermomixer), then the mixing step was repeated at RT. Plates were moved to the magnet location for 15 min and the maximum lysate volume was discarded. Plates were returned to the magnet with 500 μL wash solution for 10 min, and the wash solution was discarded; this wash process was repeated twice. 100 μL elution buffer was added, mixed at 1000 rpm for 15 min at RT, then moved to the magnet location for 10 min. 100 μL eluted DNA was collected per well and DNA samples were stored at −80°C until analysis.
Spectrophotometry and spectrofluorometry
DNA quantification by spectrophotometry was performed using a Synergy Mx Monochromator-Based Multi-Mode microplate reader (Biotek) with Gen 5 version 2.0 software and Take 3 plate. DNA quantification and purity (OD260/OD280 ratio) measurements were performed in the same assay after DNA extraction. Spectrofluorometry was performed with Quant-iT™ PicoGreen® dsDNA Assay Kit (Invitrogen) using 96-well plates.
qPCR of human and prokaryotic DNA
DNA was quantified by real-time PCR in duplicate. For human DNA, fluorescent SYBR Green with 7500 Fast Real-Time PCR System (Applied Biosystems) was used to analyze the phenylalanine-tRNA-synthetase gene. 16S rRNA qPCR was used to quantify bacterial DNA. The following primer pairs were used, human: PHA_Syn5-FWD: AAGCCCAACGATGATCAAAT/ PHA_Syn5-REV: CAGGGGACTGTCATACACCA and bacterial: F_Bact1369 CGGTGAATACGTTCCCGG/ R_Prok1492 TACGGCTACCTTGTTACGACTT (amplifying approximately 90% of all bacterial strains). 22 The qPCR SPUD assay was used to identify DNA inhibitors (samples tested in triplicate). 24
16S rRNA gene amplification and sequencing
Metagenomic 16S rRNA gene sequencing was performed at Metanome (Baylor College of Medicine; Houston, TX, USA). Identification and classification of bacteria in a sample was achieved by profiling the 16S rRNA gene, a standard target gene in microbial ecology, employing methods consistent with those developed for the NIH-Human Microbiome Project. 25 The 16S rDNA V4 region amplicons (single index) were produced by PCR and sequenced on the MiSeq platform (Illumina) using the 2x250bp protocol yielding pair-end reads that overlap by ∼247 bps. 26 Raw BCL files were retrieved from the MiSeq platform and called into fastqs by Casava v1.8.3 (Illumina). The read pairs were demultiplexed based on unique molecular barcodes, filtered for PhiX using Bowtie2 v2.2.1, 27 and reconstituted into two fastq files for each read using standard BASH. A barcodes file was generated from a raw fastq base called previously to preserve the original barcode qualities associated per read cluster. Reads were merged using USEARCH v7.0.1001 28 (four mismatches per ≥50 bases) and a barcodes file is generated for the merged set in the same manner they were generated for the raw reads.
Processing of sequencing reads include quality-filtering using USEARCH v7.0.1001 (maximum expected error method). Sequences were demultiplexed using QIIME v1.8.0 29 and then clustered using the UPARSE pipeline. Operational taxonomic unit (OTU) classification was achieved by mapping the UPARSE OTU table to the SILVA database. 30 Abundances were recovered by mapping the demultiplexed reads to the UPARSE OTUs. A custom script was used to construct an OTU table from the output files generated in the previous two steps. This OTU table was used to provide taxonomic summaries, and calculate alpha-diversity, beta-diversity.31–33
Bacterial diversity in each sample was assessed by determining richness based on the calculation of alpha-diversity.31–33 Alpha diversity was calculated based on observed OTUs by the amount of sequences per sample. Beta diversity analysis, or comparisons across samples and sample groups, was achieved by the UniFrac method,34,35 which calculates a distance measure between communities using phylogenetic information. UniFrac distances are calculated on an unweighted fashion based on presence or absence of OTUs attributing the same weight to low and high abundance. Taxa summary tables were generated for each sample and bacterial relative abundance was calculated at taxonomic levels of genus and above. The mean abundance at each taxonomic level between different conditions was compared by a nonparametric t-test to identify differences in each taxon between sample groups.
Statistical analyses
Mean, SD, and CV% were calculated using Excel. Significance was calculated using Sigma Plot v.12.0 (Systat Software) and R.3.0.0, with a 5% significance threshold using Tukey and Dunn tests.
Results
Optimization of MSM I automated DNA extraction
The initial quantity of stool sample and volume of supernatant used for extraction were optimized using canine stool in Brw and Yel tubes, to obtain the highest absolute DNA yield. Canine stool has been shown to be commutable with human stool, in the context of metagenomic analyses.36,37 Absolute DNA yields were higher with smaller stool quantities (Table 2) and yields were over 1 μg DNA when the supernatant volume was at least 100 μL. DNA OD 260/OD 280 values were approximately 2.0, with the exception of four samples, all of which had a supernatant volume below 100 μL. Comparison of 300 and 1000 μL supernatant volumes showed the DNA yield was directly proportional to volume. The 200 mg starting quantity and 1000 μL volume were selected as optimal for subsequent experiments.
Comparison of DNA extraction methods and collection containers
Quantification of total DNA by spectrophotometry
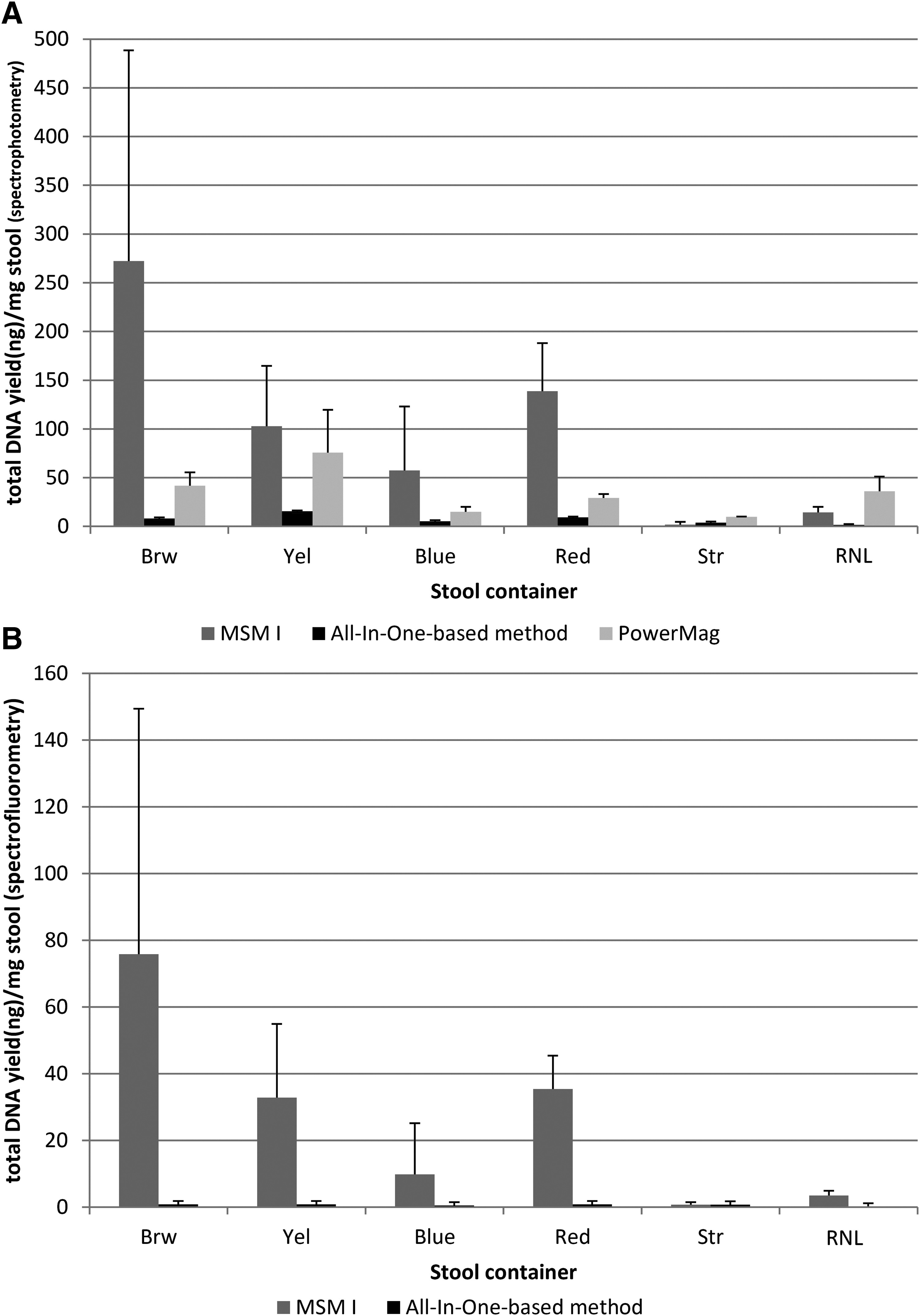
DNA quantification of the 117 samples obtained with the MSM I and All-In-One DNA extraction methods and the 34 PowerMag samples is presented in Figure 1A by collection container type. A wide range in yields was seen with different methods and container types. The MSM I extraction gave higher DNA quantities than the other two methods for four of the six collection tubes. Yields were low with All-In-One-based method and the PowerMag extractions, irrespective of container type. The highest yield with the MSM I method was obtained with Brw tubes, and was significantly higher (p<0.05) than with Str or RNL. High inter-donor variability likely influences the absence of significant difference between Brw yields and Red, Yel or Blue yields. Yields with Red tubes, containing stabilizer, were significantly higher than with Str and RNL tubes.
DNA extraction yields assessed with
Quantification of total DNA by spectrofluorometry
DNA quantification of 117 samples obtained with MSM I and All-In-One DNA extraction is presented in Figure 1B by collection container type. Fourteen samples (all but one extracted by All-In-One; six with RNL tubes) did not have quantifiable DNA (below the detection limit of 0.2 pg DNA/μL). A similar profile was seen to that identified with spectrophotometry (i.e., higher yields with MSM I extraction and lower yields with all methods in Str and RNL tubes). Statistical analyses also showed high inter-donor variation; statistical significance was seen comparing Brw to RNL, Str or Blue (p<0.05) with the MSM I method.
In samples obtained using the MSM I extraction, DNA yields determined by spectrofluorometry were on average four times lower than those determined by spectrophotometry, indicating that one-quarter of the extracted DNA was double-stranded. For the All-In-One-based method, they were on average ten times lower, indicating that only one-tenth of extracted DNA was double-stranded (Table 3).
DNA purity by spectrophotometry
DNA OD260/OD280 ratios of total DNA were higher with MSM I extraction than the two other methods (Fig. 1C). With the exception of DNA obtained by the MSM I method using the Brw and Yel tubes, mean ratios were well below the expected ratio of 1.8 (representing a pure DNA solution). For DNA samples extracted by MSM I, mean ratios with Brw or Yel tubes were significantly higher (p<0.05) than with Blue, Red, and Str. Likewise with the All-In-One-based method, DNA from Yel tubes was significantly more pure than with Str or RNL containers (p<0.001). Significance was not evaluated with the PowerMag method.
DNA quantification by qPCR
Estimated bacterial DNA yields varied considerably (Fig. 2A) between samples and container types. In 18 cases, bacterial DNA could not be amplified, despite spectrophotometric confirmation of its presence. For another three samples, DNA concentrations measured by qPCR were >50 times lower than by spectrophotometry. Estimated bacterial DNA yields were higher for DNA samples extracted with MSM I compared to the All-In-One-based method, except for Blue and Str tubes (Fig. 2A). DNA from samples collected in Brw tubes gave the highest bacterial DNA yield, with similar results to spectrofluorometry quantification (Fig. 1B).
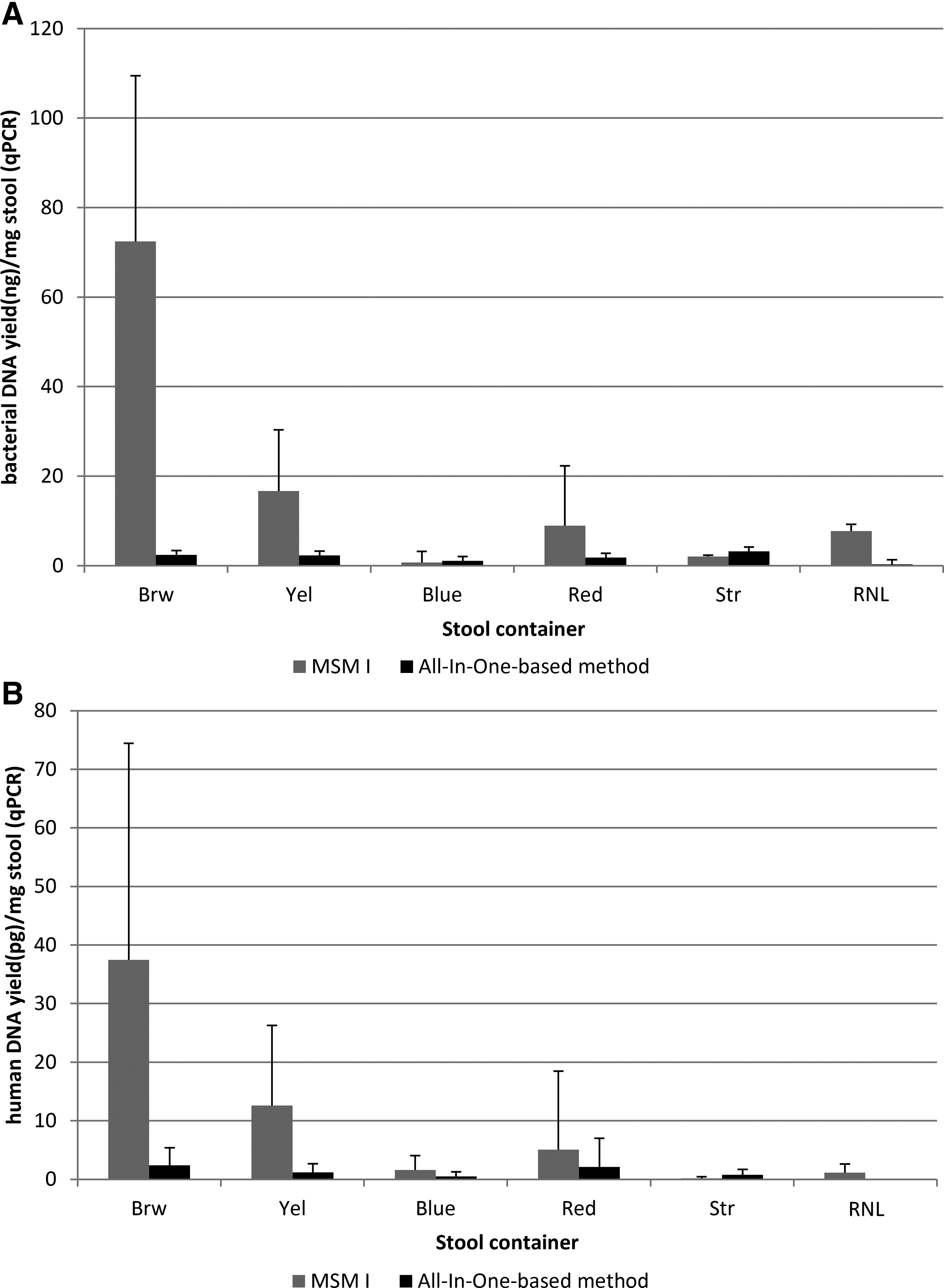
Human DNA content was close to the detection limit of 6.4 pg/μL. With the exception of Str tubes, the highest yields were obtained with MSM I extraction using Brw tubes (Fig. 2B). Table 4 shows human DNA yields expressed in terms of total DNA (determined by spectrophotometry). In all fecal DNA extracts, the percent human DNA was lower than 0.12%, the remainder being microbial, mainly bacterial.
ND, not determined.
Detection of inhibitors by qPCR
Samples were tested with the SPUD assay at their initial concentration and after dilution to 10 ng/mL. The DNA concentration required in Miseq metagenomic applications is approximately 10 ng/μL. Low to moderate inhibition was found in some undiluted samples which had DNA concentrations >40 ng/μL (Table 5), however, there was no inhibition in any sample at 10 ng/mL.
ND, not determined; NI, uninterpretable.
16S rRNA gene amplicon-based community composition
Microbial profiling by 16S rRNA gene amplicon sequencing was performed on 17 DNA samples to assess the relative abundances of specific OTUs, and whether any OTUs had been lost in any tube/extraction method combinations. Only the two tube types with optimal results in terms of DNA yield and quality were evaluated (Red with stabilizer and Brw without), with the MSM I and the All-in-One-based extraction methods. In total, 550,987 raw reads were obtained, of which 90.7% could be mapped (Fig. 3). A total of 1106 OTUs were assigned. In terms of α-diversity, more OTUs were reported with All-In-One-based extraction compared to the two other methods (Fig. 4A), but the difference was not significant. More species were identified with Red tubes than Brw tubes, however again without significance (Fig. 4B). For β-diversity plots, different samples could be clustered by donor, but not by tube type or extraction method (data not shown).
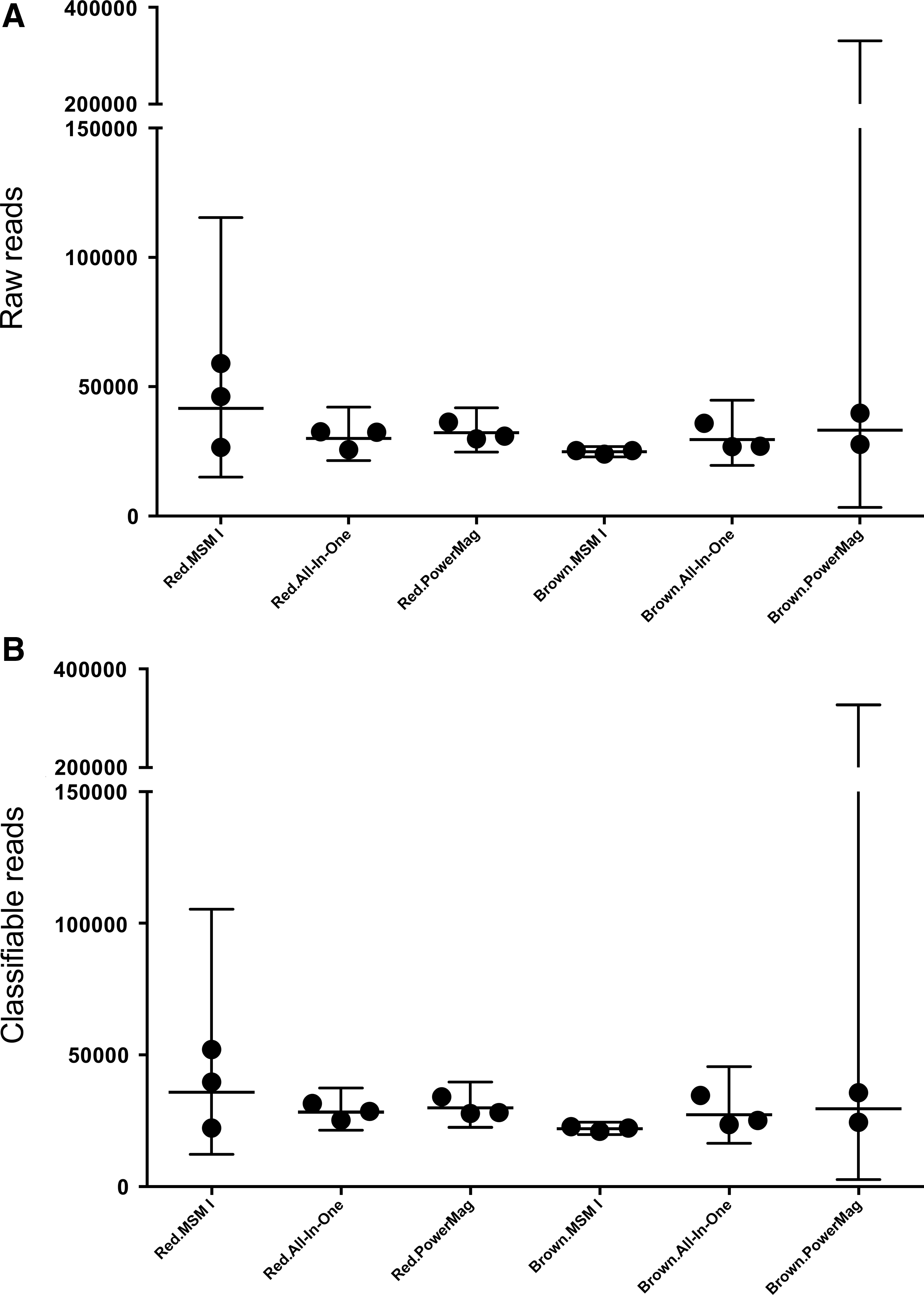
Number of total reads

Alpha-diversity plots. Operational Taxonomic Units (OTUs) observed by extraction method
Different microbiome profiles were observed for the three donors (Fig. 5A). Differential abundances of taxa were observed for Eggerthella, Anaerostipes, Blautia, and Incertae_Sedis, which were less abundant with MSM I extraction than with PowerMag or All-In-One-based method (Fig. 5B). Of note, several species could not be identified with Brw tubes, but were with Red, including Collinsella, Peptoniphilus, Barnesiella, Roseburia, Lachnospiraceae_g, Methylobacterium, Burkholderia, Escherichia, Shigella, Serratia, and Enterobacteriaceae_g. (Fig. 5C). Thus, in terms of resolution of 16S rRNA gene-based community composition, the combination of a Red tube with MSM I extraction was comparable to the combinations Red tube/All-In-One-based method, Brw tube/MSM I, Brw tube/All-In-One-based method.
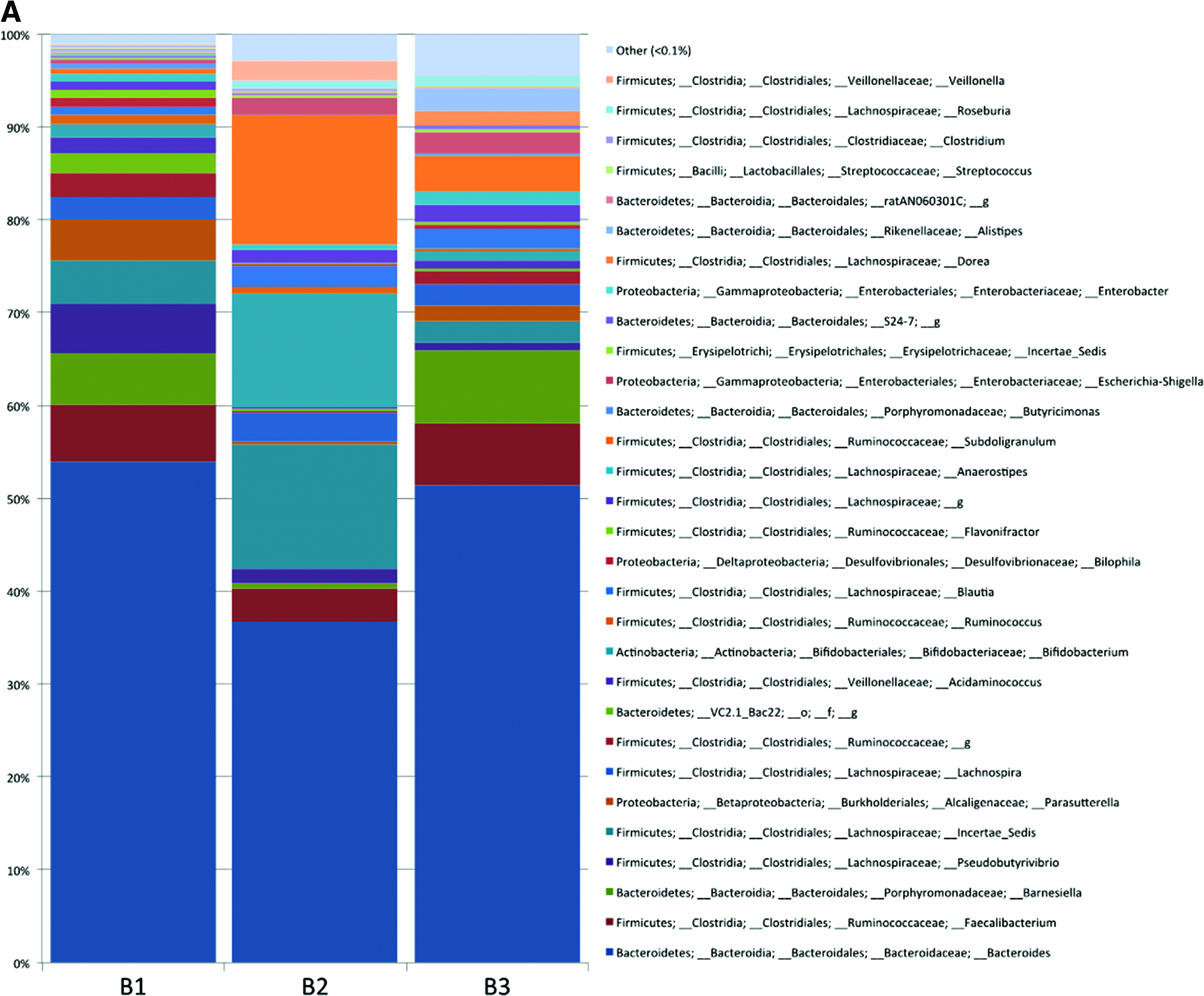
Relative abundance of OTUs in 16S rRNA gene amplicon sequencing profiles in
Reproducibility of the stool collection protocol
Reproducibility of stool collection (Red and Brw tubes) and DNA extraction (MSM I) workflow was evaluated in terms of intra-donor CV% for total, prokaryotic, and human DNA yields (Table 6). CV% for total DNA concentrations were between 18% and 35% for Red tubes and 10% and 91% for Brw tubes, and for bacterial DNA concentrations were 16% for Red tubes and between 16% and 30% for Brw.
B1/2/3, donor 1/2/3.
Discussion
The initial optimization step of this study demonstrated that DNA extraction was feasible with the MSM I method, giving yields over 1 μg, which is sufficient for all standard downstream analyses. Absolute DNA yield increased with supernatant volume, and, of the volumes tested, 1000 μL gave the highest yield. Surprisingly, this optimization step demonstrated clearly that a low stool quantity (0.2 g) gave the highest absolute DNA yields. The most likely explanation for this is that an insufficient lysis buffer volume for the 10–20 g sample quantity in the Yel tubes resulted in incomplete sample solubilization and small supernatant volumes.
Mechanical disruption of stool has been shown to increase the efficiency of DNA extraction using silica-based methods. 38 One study has shown that optimal fecal specimen input using various silica-based extraction methods is 10–50 mg. 39 A FastPrep gave DNA yields up to 300 μg/mg dry stool after manual disruption with ceramic beads, while without ceramic disruption, 25–200 mg stool gave DNA yields of only 50 μg/mg dry stool. 39 The MSM I magnetic bead-based extraction method used here gave up to 300 ng DNA/mg wet stool. Published data on relative DNA yields from silica and magnetic bead-based methods are conflicting.40,41
Spectrophotometry and spectrofluorometry results correlated in terms of total DNA quantification. Also qPCR results correlated in terms of human and bacterial DNA quantification. Overall, samples extracted using Red tubes with MSM I generated the best outcome, with higher DNA yields and better DNA purity than with All-In-One-based method or PowerMag extraction (Table 7). Furthermore, starting material for Next Generation Sequencing library construction is usually double-stranded DNA. DNA samples extracted by MSM I contained a higher percent of double-stranded DNA (25% versus 10% with All-In-One-based method). High milling frequency of 50 Hz might have negatively affected the percentage of double-stranded DNA, and milling at 10Hz with the All-In-One-based method might have resulted in higher percentage of double-stranded DNA. DNA extracted by all three methods showed electrophoretic fragments >17 kb (data not shown). DNA extraction yields with All-In-One-based method and PowerMag were generally low.
ND, not done; +++, >250 ng/mg stool; ++, >1.8.
When comparing the six collection containers, only results obtained with MSM I extraction were truly meaningful, the low extraction yields with the other two methods making differences between collection tubes difficult to detect. The Brw tube (0.2 g stool sample) is the best match for the Chemagic kit lysis buffer volume (used with the MSM I method), and gives the highest DNA yield (with all evaluation parameters). The Yel tube also performed well; however, the large stool input quantities may have diminished DNA extraction efficiency with MSM I, with yields approximately three times lower than the Brw tube, as seen initially in optimization of the MSM I method. Red tubes (containing stabilizer for both DNA and RNA) gave DNA yields higher than Yel tubes with spectrophotometry or spectrofluorometry. OD260/OD280 ratios below 1.8 suggest the presence of contaminants such as carbohydrates or proteins. Brw and Yel tubes gave the best DNA purity results (1.8 or above). Of the tubes with stabilizer, all OD260/OD280 ratios were below 1.8, RNL gave the best result (1.5). MSM I extraction gave the highest purity DNA, probably because only this DNA extraction method uses proteinase K so that no protein contaminants decreased the ratio.
The percent of human DNA in stool-extracted total DNA was extremely low (<0.13%). For endpoint applications focused on human DNA, Brw tubes provided the highest yield. Given the low DNA quantities reported in successful human qPCR reactions, unsuccessful reactions were likely due to concentrations being below the detection limit. The extremely low yield of human DNA was probably linked to the low epithelial cell exfoliation in infants. If the foreseen end-application is analysis of human DNA, a specific extraction method, based on sequence-specific capture with acrylamide gel immobilized capture probes, which increases the yield of human DNA by 5-fold, has been described. 42
Samples in Yel and Brw tubes were frozen immediately after collection, while the other four collection containers, all containing stabilizers, were kept overnight at RT. One study suggests that stool DNA and RNA is stable for up to 24 h at RT, and highlights the detrimental effect on their metagenomic profile of thawing unstabilized stool specimens. 43 Thawing unstabilized stool samples also caused an increase in the 16S rRNA gene ratio of the bacterial phyla Firmicutes to Bacteroidetes. 44 Recently, it was suggested that up to 3 days at RT does not substantially influence the assessment of gut microbiome. 45 However, it is logical to assume that differential bacterial growth induction and growth inhibition take place at aerobic conditions at RT, and for this reason, stabilization is considered as a better option.
DNA amplification can be inhibited by ions, bilirubin, or bile acids, present in stool. In the scope of metagenomic analyses, the critical PCR application is 16S rRNA amplification using an amplification method, such as the BactQuant method. 21 When samples were diluted to the concentration required by this method (10 ng/mL), no inhibition was observed. The presence of inhibitors in higher concentration DNA extracts was not associated with donors, tubes, or extraction method.
Our study evaluated only DNA extraction, however sequential isolation of metabolites, RNA, DNA, and proteins from the same sample has been successfully applied to environmental specimens.7,8 This multianalyte extraction method, based on the All-In-One kit, is fit-for-purpose when end-users wish to extract not only DNA, but also a range of analytes (DNA, RNA, proteins, and metabolites) from a single stool sample.7–9
Conclusion
In conclusion, the best results in terms of total DNA yield, purity, percent double-stranded, and amplification were obtained with Brw tubes (without stabilizer, requiring immediate freezing and storage at −20°C or −80°C) and Red tubes (with stabilizer and RT storage). Taking into account all analytical results and logistics, the Red tube prototype from DNA Genotek with the PerkinElmer MSM I extraction is the preferred workflow, providing high and reproducible DNA yield and purity, without needing immediate freezing (home collection and shipment the following day is possible) and is compatible with automation (Table 7). The Red tube also has the advantage of stabilizing both DNA and RNA, making it fit-for-purpose, not only for metagenomic but also metatranscriptomic applications. It is important to note that for comparative metagenomic analyses, only DNA samples from similarly stabilized stool and obtained using the same extraction method can be compared. Complete validation of the system Red tube/MSM I extraction in terms of intra-sample variability, aliquoting, and pre-processing stability will be examined in a separate study, along with a long-term stability study of this system at −80°C.
Footnotes
Acknowledgments
We thank Sarah MacKenzie, PhD, for medical writing assistance, Enterome for useful discussions, and Nadim Jose Ajami from Metanome for 16S rRNA gene amplicon sequencing and reporting.
Author Disclosure Statement
No conflicting financial interests exist.