
Abstract
In the past decade, the popularity and power of Tissue Microarray (TMA) technology has increased since it provides a method to detect diagnostic and prognostic markers in an array of clinical tissue specimens collected for translational research. TMAs allow for rapid and cost-effective analysis of hundreds of molecular markers at the nucleic acid and protein levels. This technology is particularly useful in the realization of the Human Protein Atlas Project, since it aims to create a reference database of non-redundant human proteins. In this context, it is important to assure the lack of cross-sample contamination due to the repeated use of the same needle in consecutive coring. Here we show that carry-over contamination from one tissue core to another does not occur, reinforcing the accuracy of the TMA technology in the simultaneous testing of multiple bio-samples.
Introduction
I
Despite its potential, there are several aspects of the TMA technology that must be considered to guarantee the reliability of the results: (i) ensuring the correct link between the tissue block and its spot on the array, and (ii) lack of cross-contamination during the coring, from sample preparation to analysis. In the first case, through the use of an ID checking system, the potential for mismatch between the donor block and its spots on the recipient TMA block is avoided. 11 However, cross-sample contamination is a more complex problem, since the transfer of tiny biological fragments can occur, affecting downstream DNA and RNA results.10,13 Cross-sample contamination may occur during core picking using the same needle for multiple specimens, without effective cleaning of the needle between samples. In this case, biological debris can be transferred from spot to spot.
Here we investigated the possible cross-sample contamination occurring during the construction of TMAs and cell microarrays (CMAs), where we assembled tissue fragments and cell pellets in two distinct arrays. Samples were also collected for DNA extraction and polymerase chain reaction (PCR) amplification for three SEL1L gene regions. SEL1L was chosen since it is commonly studied in the laboratory in which the investigation was conducted. 14 We show lack of contamination between tissue and cell fragments in two TMAs.
Materials and Methods
Cell and tissue paraffin blocks
Cross-sample contamination was investigated using cores from cell pellets and tissues. Three different cell lines were used: Panc1, Hek293, and Fibroblasts MGM18004E. The cell lines were kindly provided by the BioBank service of Integrated Systems Engineering S.r.l. (Milan, Italy). For artificial cell paraffin-embedded blocks, cells were harvested and processed according to La Spada et al, 2014. 15
Tissue paraffin blocks were obtained from the Anatomy and Pathology Department of Multimedica Hospital (Milan, Italy). In total, six different FFPETs were used as donor blocks: cholecyst, lung, ileum, prostate, ovary, and placenta. Sections of 5 μm from each tissue block were sliced using a microtome (RM2245, Leica Biosystems, Milan, Italy), and hematoxylin-eosin (H&E) stained to identify the appropriate areas to core.
TMA and CMA construction
Reference images of the histological slides with the specific area marked by the pathologist were digitally overlapped with the respective donor block. The semi-automatic tissue arrayer Galileo CK4500 (www.isenet.it) was used in this study. A 1 mm hollow needle diameter was used to sample cell free low-melting agarose (1.5%) (LMA) (Sigma, St. Louis, MO). Tissue and cell cores were then assembled in two different recipient paraffin blocks. In the first case, a core of LMA was picked after every single tissue and cell sample. From each different donor block, five cores were picked and the sampling order was: cholecyst, lung, ileum, prostate, ovary, placenta, Panc1, Hek293, and fibroblasts MGM18004E. The second array was assembled in the following sequence: 2 spots of LMA, 10 spots of each donor blocks (order: cholecyst, lung, ileum, prostate, ovary, placenta, Panc1, Hek293, and fibroblasts MGM18004E), and 3 control spots of LMA.
Up to 100 consecutive sections of 4 μm thickness were cut from each recipient block and mounted on microscope slides. One of every 30 slides and the last slide were H&E stained and scanned using a Scanscope FL scanner (Leica Biosystems, Milan, Italy).
Tissue and cell coring for nucleic acid extraction
Using the 1.0 mm diameter donor needle, 95 cores were picked from the donor blocks and placed into microfuge tubes by the Galileo CK4500 arrayer. The TMA software performed two operations: (1) linkage between the donor block and the respective tube corresponding to the array construction; and (2) selection of the region of interest on the donor block by digital overlapping of the slide image and the block area.
Nucleic acid extraction
DNA extraction from the paraffin-embedded cores required initial deparaffinization and rehydration by incubating in decreasing concentrations of alcohol and final digestion with Proteinase K. The silica-based procedure 16 was used to extract the DNA from two deparaffinized cores. Briefly, after rehydration the cores were incubated in 3 M guanidine thiocyanate, 20.0 mM EDTA pH 8, 10.0 mM Tris-HCl pH 6.8, 10 mg/mL DTT, 40 mg/mL Triton X-100, and 40 mg buffered silica matrix for 3 min at room temperature, and then centrifuged at 135 g for 60 sec at room temperature. The pellets were washed in 25% ethanol, 25% isopropanol, 100 mM NaCl, 10 mM Tris-HCl pH 8, followed by 100% ethanol. The pellets were air-dried and incubated in DNase/RNase free water for 3 min at 65°C. DNA concentration was determined using Nanodrop ND 1000 (Thermo Fisher Scientific, Milan, Italy) and yield was calculated by normalization to the original volume of 2 cores (=6.26 mm3×2=12.52 mm3).
Polymerase chain reaction analysis
The SEL1L promoter region and exons 9 and 11 were amplified by PCR. PCR was performed in a total volume of 30 μL with the Mastercycler instrument (Eppendorf AG, Milan, Italy). The reaction mixture contained 1 unit of Dream Taq polymerase and 1× Dream Taq PCR buffer (Thermo Fisher Scientific), 0.25 mM of each dNTPs (Euroclone), 0.9 μM forward primer and reverse primer, and 10 μL of DNA template. Positive control consisted in 130 ng of RR16 human cell line genomic DNA. The primer sequences and relative product sizes are listed in Table 1. The PCR conditions consisted of 5 min at 95°C, followed by 40 cycles at 95°C for 30 sec, 30 sec of annealing at the temperature reported in Table 1, and extension at 72°C for 1 min, followed by a final extension of 15 min at 72°C. All of the PCR products, volumes of 30 μL, were electrophoresed on 1.2% agarose gel, stained with ethidium bromide, and analyzed using a Luminescence Image Analyzer (ImageQuant LAS400, GE Healthcare, Milan, Italy).
Results
In this study, two different TMAs were constructed, in order to show lack of contamination between cores after 95 steps of consecutive coring.
The H&E staining in Figure 1A reveals the absence of tissue or cells in rows of the LMAs spots, columns 1, 3, 5, 7, 9, and 11, containing only agarose cores, but was positive in rows (a) cholecyst, (b) lung, (c) ileum, (d) prostate, (e) ovary, (f) placenta, (g) Panc1, (h) Hek293, and (i) fibroblasts MGM18004E. Figure 1B shows a close-up image of the cores 5-d, 10-b, and 6-h containing agarose, lung, and Hek293, respectively.
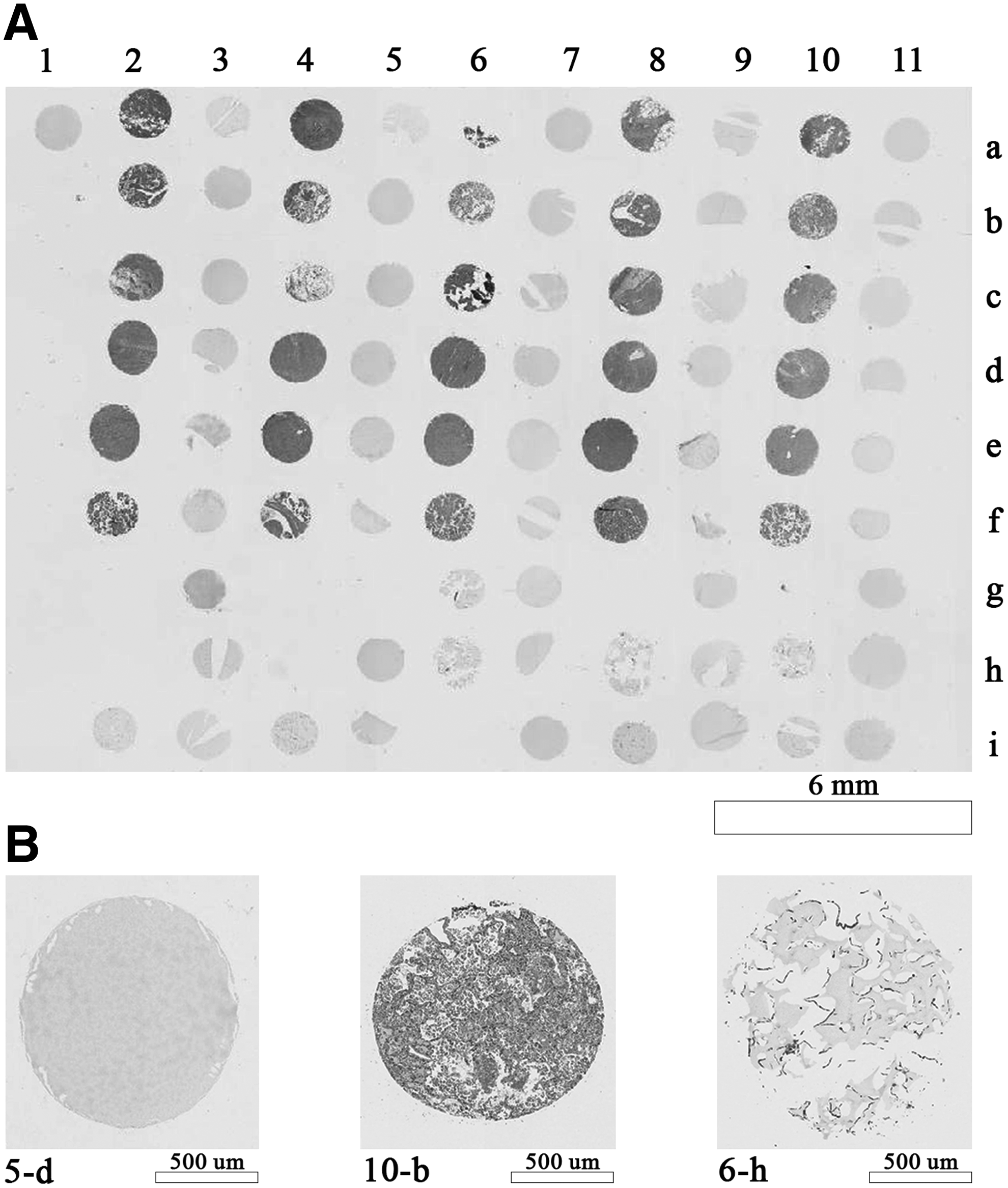
Figure 2B shows the H&E staining of a second array panel 2A reflecting: columns 1 and 11 containing two and three cell-free LMA cores respectively; columns 2, 3, 4, 5, 6, 7, 8, 9, and 10, containing cholecyst, lung, ileum, prostate, ovary, placenta, Panc1, Hek293, fibroblasts MGM18004E, respectively. Lack of cross-sample contamination is clearly demonstrated in the control lanes 1 and 11, showing no H&E staining.
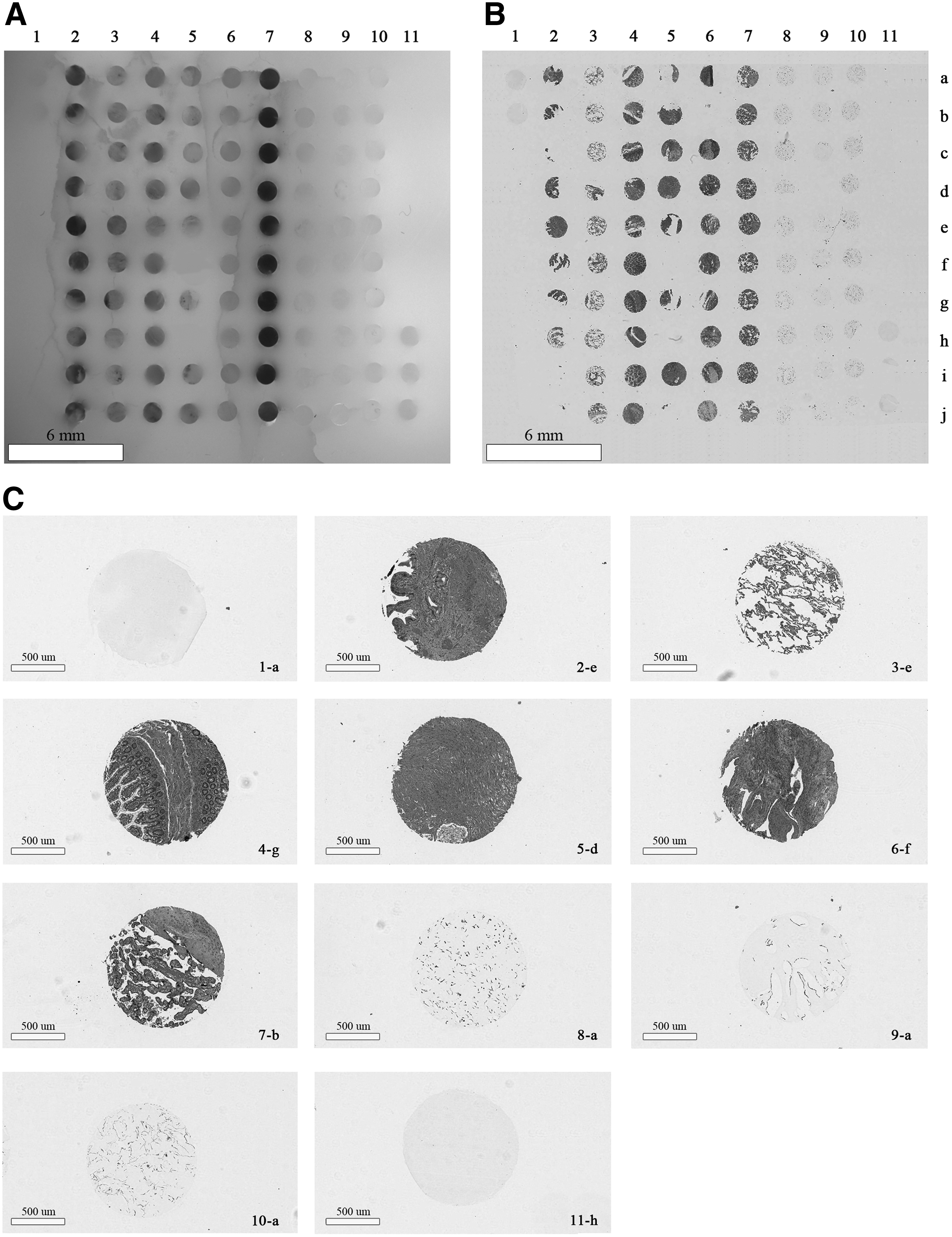
Figure 2C shows a magnified version of selected spots from Figure 2B. Definite staining was observed only in tissue (2-e, 3-e, 4-g, 5-d, 6-f, and 7-b) and cell (8-a, 9-a, and 10-a) cores, whereas the control columns 1-a and 11-h were empty, which is indicative of no cross-over of tissue fragments from the repeated use of the same needle.
DNA was extracted from all the cores including empty agarose samples in order to confirm the lack of sample contamination using PCR amplification and primers designed on the SEL1L gene. Table 2 shows the DNA yields from each core ranging from 76 ng/mm3 to 1140 ng/mm3, fibroblasts MGM18004E and placenta, respectively.
The yield was calculated divided for the volume of two cores: diameter 1 mm and length 4 mm, total volume 6,28 mm3.
PCR (Fig. 3) shows no DNA amplification product in the LMA cores, confirming no cross-sample carry-over occurring during TMA construction. Three sets of primers designed on the SEL1L gene were used corresponding to the minimal promoter region, exon 9 and exon 11. Human RR16 was used as the positive PCR control genomic DNA. The expected DNA amplification product of 189, 268, and 281 bp size, of promoter, exon 9, and exon 11, respectively, were clearly visible in all the TMA cores containing the tissue and cell samples. LMA cores revealed only annealed primers as expected.
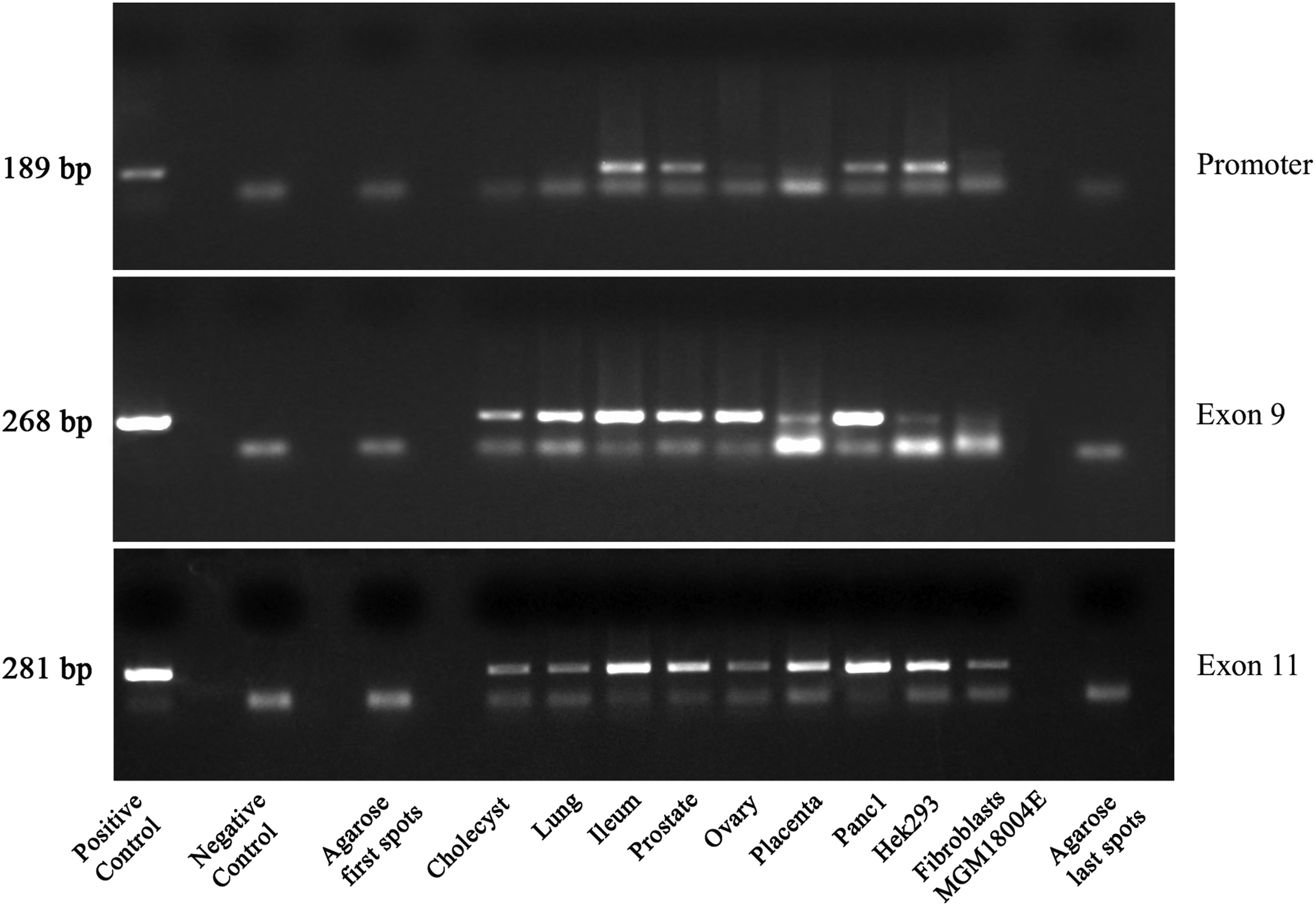
SEL1L promoter region, exon 9, and exon 11 gene amplification using DNA extracted from cholecyst, lung, ileum, prostate, ovary, placenta, Panc1, Hek293, fibroblasts MGM18004E, and cell-free agarose cores. Positive control genomic RR16 DNA, negative control was the reaction mixture without DNA template. PCR fragment size as established from previous publications from the laboratory staff.
Discussion
The TMA technology is a reliable and efficient tool for high–throughput analysis and it is very useful for diagnostic and research purposes.2–7 This technology can become fundamental in exploring the human proteome considering the information acquired from human genome efforts17–19 since it can provide a means to explore, in a rapid and cost-effective manner, the huge number of antibodies being generated from the non-redundant protein database. In addition, both proteomic and genomic data can be simultaneously obtained from the same specimen. 19 However, to meet this challenge and even more importantly its reliability in diagnostic and research applications, it is of paramount importance to ascertain the absence of cross-sample contamination, due to the use of the same needle in consecutive coring. To our knowledge, this particular issue, to date, has not yet been thoroughly reported.
In this study, we investigated this aspect and analyzed a series of tissues and cells using the Galileo CK4500 platform. Using cell-free agarose samples as negative control, H&E analyses revealed a complete lack of biological contamination between consecutive cores, as well as after 95 cores. We also examined possible contamination by extracting DNA from the paraffin-embedded cores and amplified different regions of the SEL1L gene by PCR.
Here we show the complete absence of cross-sample contamination using the TMA technology as evidenced by H&E staining of array slides, and by the very sensitive PCR amplification of the DNA extracted from the several cancer tissues and cell lines. The empty or incomplete core spots may reflect loss of the tissue or cell fragments due to defects in microtome and staining procedures. The different quantities of amplification product among the samples reflect the effects of formalin-fixed paraffin embedding procedures of the tissues known to damage DNA and affect downstream applications.20,21
Unequivocally, the results here reported indicate that the TMA technology, both with paraffin-embedded tissue and paraffin cell blocks, does not promote cross-sample contamination, and its use is safe as a high throughput proteomic and biomarker tool. The results here shown are limited to the Galileo platform, but through further studies they can be extended to all TMA instruments.
Footnotes
Acknowledgments
We express special thanks to Simona Baronchelli, Aikaterini Ntai, Tatiana Pellegrino, and Monica Cattaneo for helpful discussions and critical review of the manuscript.
Author Disclosure Statement
The authors declare no conflict of interest.